
Heat Shock Protein 22 in Physiological and Pathological Hearts: Small Molecule, Large Potentials



Abstract
:1. Introduction
2. The Molecular Mechanisms of HSP22’s Cytoprotection in Cardiomyocytes
2.1. HSP22 Activates Multiple Cardiac Pro-Survival Signaling
2.2. HSP22 Chaperones Its Client Proteins and Facilitates Their Subcellular Redistribution
2.3. HSP22 Enhances Proteasome Activity and Participates in Energy Metabolism
2.4. HSP22 Participates in Cardiac Autophagy via BAG3
2.5. HSP22 Regulates Cardiac Reactive Oxygen Species (ROS) Production and Oxidative Stress
3. The Association of HSP22 in Heart Diseases
3.1. HSP22 in Human DCM
3.2. HSP 22 in Pressure Overload or Hypertensive Cardiac Hypertrophy and HF
3.3. HSP22 in Ischemic Heart Diseases
3.4. HSP22 in Age-Related Cardiomyopathy
3.5. HSP22 in Diabetic Heart Disease
4. Therapeutic Potential of HSP22 and Future Direction
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Stetler, R.A.; Gan, Y.; Zhang, W.; Liou, A.K.; Gao, Y.; Cao, G.; Chen, J. Heat shock proteins: Cellular and molecular mechanisms in the central nervous system. Prog. Neurobiol. 2010, 92, 184–211. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.W.; Kim, H.J.; Lim, J.H.; Lee, S.H. Heat Shock Proteins: Agents of Cancer Development and Therapeutic Targets in Anti-Cancer Therapy. Cells 2019, 9, 60. [Google Scholar] [CrossRef] [Green Version]
- Carra, S.; Alberti, S.; Arrigo, P.A.; Benesch, J.L.; Benjamin, I.J.; Boelens, W.; Bartelt-Kirbach, B.; Brundel, B.; Buchner, J.; Bukau, B.; et al. The growing world of small heat shock proteins: From structure to functions. Cell Stress Chaperones 2017, 22, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Jee, H. Size dependent classification of heat shock proteins: A mini-review. J. Exerc. Rehabil. 2016, 12, 255–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kappe, G.; Franck, E.; Verschuure, P.; Boelens, W.C.; Leunissen, J.A.; de Jong, W.W. The human genome encodes 10 alpha-crystallin-related small heat shock proteins: HspB1-10. Cell Stress Chaperones 2003, 8, 53–61. [Google Scholar] [CrossRef]
- Kappe, G.; Verschuure, P.; Philipsen, R.L.; Staalduinen, A.A.; Van de Boogaart, P.; Boelens, W.C.; De Jong, W.W. Characterization of two novel human small heat shock proteins: Protein kinase-related HspB8 and testis-specific HspB9. Biochim. Biophys. Acta 2001, 1520, 1–6. [Google Scholar] [CrossRef]
- Morrow, G.; Hightower, L.E.; Tanguay, R.M. Small heat shock proteins: Big folding machines. Cell Stress Chaperones 2015, 20, 207–212. [Google Scholar] [CrossRef] [Green Version]
- Benndorf, R.; Sun, X.; Gilmont, R.R.; Biederman, K.J.; Molloy, M.P.; Goodmurphy, C.W.; Cheng, H.; Andrews, P.C.; Welsh, M.J. HSP22, a new member of the small heat shock protein superfamily, interacts with mimic of phosphorylated HSP27 ((3D)HSP27). J. Biol. Chem. 2001, 276, 26753–26761. [Google Scholar] [CrossRef] [Green Version]
- Li, X.S.; Xu, Q.; Fu, X.Y.; Luo, W.S. Heat shock protein 22 overexpression is associated with the progression and prognosis in gastric cancer. J. Cancer Res. Clin. Oncol. 2014, 140, 1305–1313. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Fontaine, J.M.; Bartl, I.; Behnam, B.; Welsh, M.J.; Benndorf, R. Induction of Hsp22 (HspB8) by estrogen and the metalloestrogen cadmium in estrogen receptor-positive breast cancer cells. Cell Stress Chaperones 2007, 12, 307–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, M.; Matsushima-Nishiwaki, R.; Kuroyanagi, G.; Suzuki, N.; Takamatsu, R.; Furui, T.; Yoshimi, N.; Kozawa, O.; Morishige, K. Regulation by heat shock protein 22 (HSPB8) of transforming growth factor-alpha-induced ovary cancer cell migration. Arch. Biochem. Biophys. 2015, 571, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Matsushima-Nishiwaki, R.; Toyoda, H.; Takamatsu, R.; Yasuda, E.; Okuda, S.; Maeda, A.; Kaneoka, Y.; Yoshimi, N.; Kumada, T.; Kozawa, O. Heat shock protein 22 (HSPB8) reduces the migration of hepatocellular carcinoma cells through the suppression of the phosphoinositide 3-kinase (PI3K)/AKT pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1629–1639. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.V.; Kasakov, A.S.; Seit-Nebi, A.S.; Marston, S.B.; Gusev, N.B. Structure and properties of K141E mutant of small heat shock protein HSP22 (HspB8, H11) that is expressed in human neuromuscular disorders. Arch Biochem. Biophys. 2006, 454, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Chen, L.; Zhang, J.; Li, T.; Tang, J.; Xu, N.; Wang, X. Structure, function, property, and role in neurologic diseases and other diseases of the sHsp22. J. Neurosci. Res. 2007, 85, 2071–2079. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Tian, J.; Zhang, F.; Liu, A.; Xie, B.; Chen, Q. The protective effects of heat shock protein 22 in lung ischemia-reperfusion injury mice. Biochem. Biophys. Res. Commun. 2019, 512, 698–704. [Google Scholar] [CrossRef]
- Chowdary, T.K.; Raman, B.; Ramakrishna, T.; Rao, C.M. Mammalian Hsp22 is a heat-inducible small heat-shock protein with chaperone-like activity. Biochem. J. 2004, 381, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Depre, C.; Kim, S.J.; John, A.S.; Huang, Y.; Rimoldi, O.E.; Pepper, J.R.; Dreyfus, G.D.; Gaussin, V.; Pennell, D.J.; Vatner, D.E.; et al. Program of cell survival underlying human and experimental hibernating myocardium. Circ. Res. 2004, 95, 433–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depre, C.; Tomlinson, J.E.; Kudej, R.K.; Gaussin, V.; Thompson, E.; Kim, S.J.; Vatner, D.E.; Topper, J.N.; Vatner, S.F. Gene program for cardiac cell survival induced by transient ischemia in conscious pigs. Proc. Natl. Acad. Sci. USA 2001, 98, 9336–9341. [Google Scholar] [CrossRef] [Green Version]
- Hase, M.; Depre, C.; Vatner, S.F.; Sadoshima, J. H11 has dose-dependent and dual hypertrophic and proapoptotic functions in cardiac myocytes. Biochem. J. 2005, 388, 475–483. [Google Scholar] [CrossRef] [Green Version]
- Depre, C.; Wang, L.; Sui, X.; Qiu, H.; Hong, C.; Hedhli, N.; Ginion, A.; Shah, A.; Pelat, M.; Bertrand, L.; et al. H11 kinase prevents myocardial infarction by preemptive preconditioning of the heart. Circ. Res. 2006, 98, 280–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Lizano, P.; Zhao, X.; Sui, X.; Dhar, S.K.; Shen, Y.T.; Vatner, D.E.; Vatner, S.F.; Depre, C. Preemptive conditioning of the swine heart by H11 kinase/Hsp22 provides cardiac protection through inducible nitric oxide synthase. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1303–H1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sui, X.; Li, D.; Qiu, H.; Gaussin, V.; Depre, C. Activation of the bone morphogenetic protein receptor by H11kinase/Hsp22 promotes cardiac cell growth and survival. Circ. Res. 2009, 104, 887–895. [Google Scholar] [CrossRef] [Green Version]
- Laure, L.; Long, R.; Lizano, P.; Zini, R.; Berdeaux, A.; Depre, C.; Morin, D. Cardiac H11 kinase/Hsp22 stimulates oxidative phosphorylation and modulates mitochondrial reactive oxygen species production: Involvement of a nitric oxide-dependent mechanism. Free Radic. Biol. Med. 2012, 52, 2168–2176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Liang, Q.; Zhang, W.; Liao, M.; Wen, M.; Zhan, B.; Bao, H.; Cheng, X. HSP22 suppresses diabetes-induced endothelial injury by inhibiting mitochondrial reactive oxygen species formation. Redox Biol. 2019, 21, 101095. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Lizano, P.; Laure, L.; Sui, X.; Rashed, E.; Park, J.Y.; Hong, C.; Gao, S.; Holle, E.; Morin, D.; et al. H11 kinase/heat shock protein 22 deletion impairs both nuclear and mitochondrial functions of STAT3 and accelerates the transition into heart failure on cardiac overload. Circulation 2011, 124, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Sun, X.; Sun, X.; Lo, L.; Wang, C.; Xie, M.; Qin, G.; Qiu, H. Hsp22 Deficiency Induces Age-Dependent Cardiac Dilation and Dysfunction by Impairing Autophagy, Metabolism, and Oxidative Response. Antioxidants 2021, 10, 1550. [Google Scholar] [CrossRef] [PubMed]
- Voellmy, R. On mechanisms that control heat shock transcription factor activity in metazoan cells. Cell Stress Chaperones 2004, 9, 122–133. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, R.I. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008, 22, 1427–1438. [Google Scholar] [CrossRef] [Green Version]
- Knowlton, A.A.; Sun, L. Heat-shock factor-1, steroid hormones, and regulation of heat-shock protein expression in the heart. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H455–H464. [Google Scholar] [CrossRef]
- Eriksson, M.; Jokinen, E.; Sistonen, L.; Leppa, S. Heat shock factor 2 is activated during mouse heart development. Int. J. Dev. Biol. 2000, 44, 471–477. [Google Scholar]
- Fang, X.; Bogomolovas, J.; Wu, T.; Zhang, W.; Liu, C.; Veevers, J.; Stroud, M.J.; Zhang, Z.; Ma, X.; Mu, Y.; et al. Loss-of-function mutations in co-chaperone BAG3 destabilize small HSPs and cause cardiomyopathy. J. Clin. Investig. 2017, 127, 3189–3200. [Google Scholar] [CrossRef] [Green Version]
- Shemetov, A.A.; Seit-Nebi, A.S.; Gusev, N.B. Structure, properties, and functions of the human small heat-shock protein HSP22 (HspB8, H11, E2IG1): A critical review. J. Neurosci. Res. 2008, 86, 264–269. [Google Scholar] [CrossRef]
- Wu, W.; Lai, L.; Xie, M.; Qiu, H. Insights of heat shock protein 22 in the cardiac protection against ischemic oxidative stress. Redox Biol. 2020, 34, 101555. [Google Scholar] [CrossRef]
- Lizano, P.; Rashed, E.; Kang, H.; Dai, H.; Sui, X.; Yan, L.; Qiu, H.; Depre, C. The valosin-containing protein promotes cardiac survival through the inducible isoform of nitric oxide synthase. Cardiovasc. Res. 2013, 99, 685–693. [Google Scholar] [CrossRef] [Green Version]
- Rashed, E.; Lizano, P.; Dai, H.; Thomas, A.; Suzuki, C.K.; Depre, C.; Qiu, H. Heat shock protein 22 (Hsp22) regulates oxidative phosphorylation upon its mitochondrial translocation with the inducible nitric oxide synthase in mammalian heart. PLoS ONE 2015, 10, e0119537. [Google Scholar] [CrossRef]
- Depre, C.; Hase, M.; Gaussin, V.; Zajac, A.; Wang, L.; Hittinger, L.; Ghaleh, B.; Yu, X.; Kudej, R.K.; Wagner, T.; et al. H11 kinase is a novel mediator of myocardial hypertrophy in vivo. Circ. Res. 2002, 91, 1007–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedhli, N.; Wang, L.; Wang, Q.; Rashed, E.; Tian, Y.; Sui, X.; Madura, K.; Depre, C. Proteasome activation during cardiac hypertrophy by the chaperone H11 Kinase/Hsp22. Cardiovasc. Res. 2008, 77, 497–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarone, G.; Brancaccio, M. Keep your heart in shape: Molecular chaperone networks for treating heart disease. Cardiovasc. Res. 2014, 102, 346–361. [Google Scholar] [CrossRef] [Green Version]
- Arndt, V.; Dick, N.; Tawo, R.; Dreiseidler, M.; Wenzel, D.; Hesse, M.; Furst, D.O.; Saftig, P.; Saint, R.; Fleischmann, B.K.; et al. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr. Biol. 2010, 20, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Carra, S.; Seguin, S.J.; Landry, J. HspB8 and Bag3: A new chaperone complex targeting misfolded proteins to macroautophagy. Autophagy 2008, 4, 237–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulbricht, A.; Eppler, F.J.; Tapia, V.E.; van der Ven, P.F.; Hampe, N.; Hersch, N.; Vakeel, P.; Stadel, D.; Haas, A.; Saftig, P.; et al. Cellular mechanotransduction relies on tension-induced and chaperone-assisted autophagy. Curr. Biol. 2013, 23, 430–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villard, E.; Perret, C.; Gary, F.; Proust, C.; Dilanian, G.; Hengstenberg, C.; Ruppert, V.; Arbustini, E.; Wichter, T.; Germain, M.; et al. A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Eur. Heart J. 2011, 32, 1065–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintana, M.T.; Parry, T.L.; He, J.; Yates, C.C.; Sidorova, T.N.; Murray, K.T.; Bain, J.R.; Newgard, C.B.; Muehlbauer, M.J.; Eaton, S.C.; et al. Cardiomyocyte-Specific Human Bcl2-Associated Anthanogene 3 P209L Expression Induces Mitochondrial Fragmentation, Bcl2-Associated Anthanogene 3 Haploinsufficiency, and Activates p38 Signaling. Am. J. Pathol. 2016, 186, 1989–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morin, D.; Long, R.; Panel, M.; Laure, L.; Taranu, A.; Gueguen, C.; Pons, S.; Leoni, V.; Caccia, C.; Vatner, S.F.; et al. Hsp22 overexpression induces myocardial hypertrophy, senescence and reduced life span through enhanced oxidative stress. Free Radic. Biol. Med. 2019, 137, 194–200. [Google Scholar] [CrossRef]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kuhl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.R.; Carniel, E.; Mestroni, L. Cardiomyopathy, familial dilated. Orphanet J. Rare Dis. 2006, 1, 27. [Google Scholar] [CrossRef] [Green Version]
- Codd, M.B.; Sugrue, D.D.; Gersh, B.J.; Melton, L.J., 3rd. Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in Olmsted County, Minnesota, 1975–1984. Circulation 1989, 80, 564–572. [Google Scholar] [CrossRef] [Green Version]
- Sanbe, A.; Marunouchi, T.; Abe, T.; Tezuka, Y.; Okada, M.; Aoki, S.; Tsumura, H.; Yamauchi, J.; Tanonaka, K.; Nishigori, H.; et al. Phenotype of cardiomyopathy in cardiac-specific heat shock protein B8 K141N transgenic mouse. J. Biol. Chem. 2013, 288, 8910–8921. [Google Scholar] [CrossRef] [Green Version]
- Sanbe, A.; Yamauchi, J.; Miyamoto, Y.; Fujiwara, Y.; Murabe, M.; Tanoue, A. Interruption of CryAB-amyloid oligomer formation by HSP22. J. Biol. Chem. 2007, 282, 555–563. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, I.; Minamino, T. Physiological and pathological cardiac hypertrophy. J. Mol. Cell Cardiol. 2016, 97, 245–262. [Google Scholar] [CrossRef]
- Saheera, S.; Krishnamurthy, P. Cardiovascular Changes Associated with Hypertensive Heart Disease and Aging. Cell Transpl. 2020, 29. [Google Scholar] [CrossRef]
- Hori, M.; Nishida, K. Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovasc. Res. 2009, 81, 457–464. [Google Scholar] [CrossRef] [Green Version]
- Rashed, E.; Depre, C. Cardiac cell survival and reversibility of myocardial ischemia. Arch Mal. Coeur Vaiss 2006, 99, 1236–1243. [Google Scholar]
- Depre, C.; Vanoverschelde, J.L.; Taegtmeyer, H. Glucose for the heart. Circulation 1999, 99, 578–588. [Google Scholar] [CrossRef] [Green Version]
- Locke, M.; Tanguay, R.M. Diminished heat shock response in the aged myocardium. Cell Stress Chaperones 1996, 1, 251–260. [Google Scholar] [CrossRef]
- Morrow, G.; Samson, M.; Michaud, S.; Tanguay, R.M. Overexpression of the small mitochondrial Hsp22 extends Drosophila life span and increases resistance to oxidative stress. FASEB J. 2004, 18, 598–599. [Google Scholar] [CrossRef] [Green Version]
- Morrow, G.; Battistini, S.; Zhang, P.; Tanguay, R.M. Decreased lifespan in the absence of expression of the mitochondrial small heat shock protein Hsp22 in Drosophila. J. Biol. Chem. 2004, 279, 43382–43385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, D.H.; Wang, Z.V. Glucose Metabolism in Cardiac Hypertrophy and Heart Failure. J. Am. Heart Assoc. 2019, 8, e012673. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Song, J.; Hu, S. Metabolic remodeling of substrate utilization during heart failure progression. Heart Fail. Rev. 2019, 24, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, N.; Gomes-Ferreira, C.; Moura, C.; Roncon-Albuquerque, R., Jr.; Leite-Moreira, A.F.; Falcao-Pires, I. Worse cardiac remodeling in response to pressure overload in type 2 diabetes mellitus. Int. J. Cardiol. 2016, 217, 195–204. [Google Scholar] [CrossRef]
- Funk, S.D.; Yurdagul, A., Jr.; Orr, A.W. Hyperglycemia and endothelial dysfunction in atherosclerosis: Lessons from type 1 diabetes. Int. J. Vasc. Med. 2012, 2012, 569654. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zajac, A.; Hedhli, N.; Depre, C. Increased expression of H11 kinase stimulates glycogen synthesis in the heart. Mol. Cell Biochem. 2004, 265, 71–78. [Google Scholar] [CrossRef]
- Kirk, J.A.; Cheung, J.Y.; Feldman, A.M. Therapeutic targeting of BAG3: Considering its complexity in cancer and heart disease. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Myers, V.D.; McClung, J.M.; Wang, J.; Tahrir, F.G.; Gupta, M.K.; Gordon, J.; Kontos, C.H.; Khalili, K.; Cheung, J.Y.; Feldman, A.M. The Multifunctional Protein BAG3: A Novel Therapeutic Target in Cardiovascular Disease. JACC Basic Transl. Sci. 2018, 3, 122–131. [Google Scholar] [CrossRef]
- Chen, Q.; Xiang, J.; Gong, R.; Fang, H.Y.; Xu, C.C.; Zhang, H.Z.; Wu, Y.Q. Atorvastatin downregulates HSP22 expression in an atherosclerotic model in vitro and in vivo. Int. J. Mol. Med. 2019, 43, 821–829. [Google Scholar] [CrossRef] [Green Version]
- Gong, R.; Li, X.Y.; Chen, H.J.; Xu, C.C.; Fang, H.Y.; Xiang, J.; Wu, Y.Q. Role of heat shock protein 22 in the protective effect of geranylgeranylacetone in response to oxidized-LDL. Drug Des. Devel. Ther. 2019, 13, 2619–2632. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Sun, H.; Lu, J.; Li, X.; Chen, X.; Tao, D.; Huang, W.; Huang, B. Lifespan extension and elevated hsp gene expression in Drosophila caused by histone deacetylase inhibitors. J. Exp. Biol. 2005, 208, 697–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, D.; Lu, J.; Sun, H.; Zhao, Y.M.; Yuan, Z.G.; Li, X.X.; Huang, B.Q. Trichostatin A extends the lifespan of Drosophila melanogaster by elevating hsp22 expression. Acta Biochim. Biophys. Sin. 2004, 36, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Schriner, S.E.; Lee, K.; Truong, S.; Salvadora, K.T.; Maler, S.; Nam, A.; Lee, T.; Jafari, M. Extension of Drosophila lifespan by Rhodiola rosea through a mechanism independent from dietary restriction. PLoS ONE 2013, 8, e63886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanguy, J.; Pommerolle, L.; Garrido, C.; Kolb, M.; Bonniaud, P.; Goirand, F.; Bellaye, P.S. Extracellular Heat Shock Proteins as Therapeutic Targets and Biomarkers in Fibrosing Interstitial Lung Diseases. Int. J. Mol. Sci. 2021, 22, 9316. [Google Scholar] [CrossRef] [PubMed]


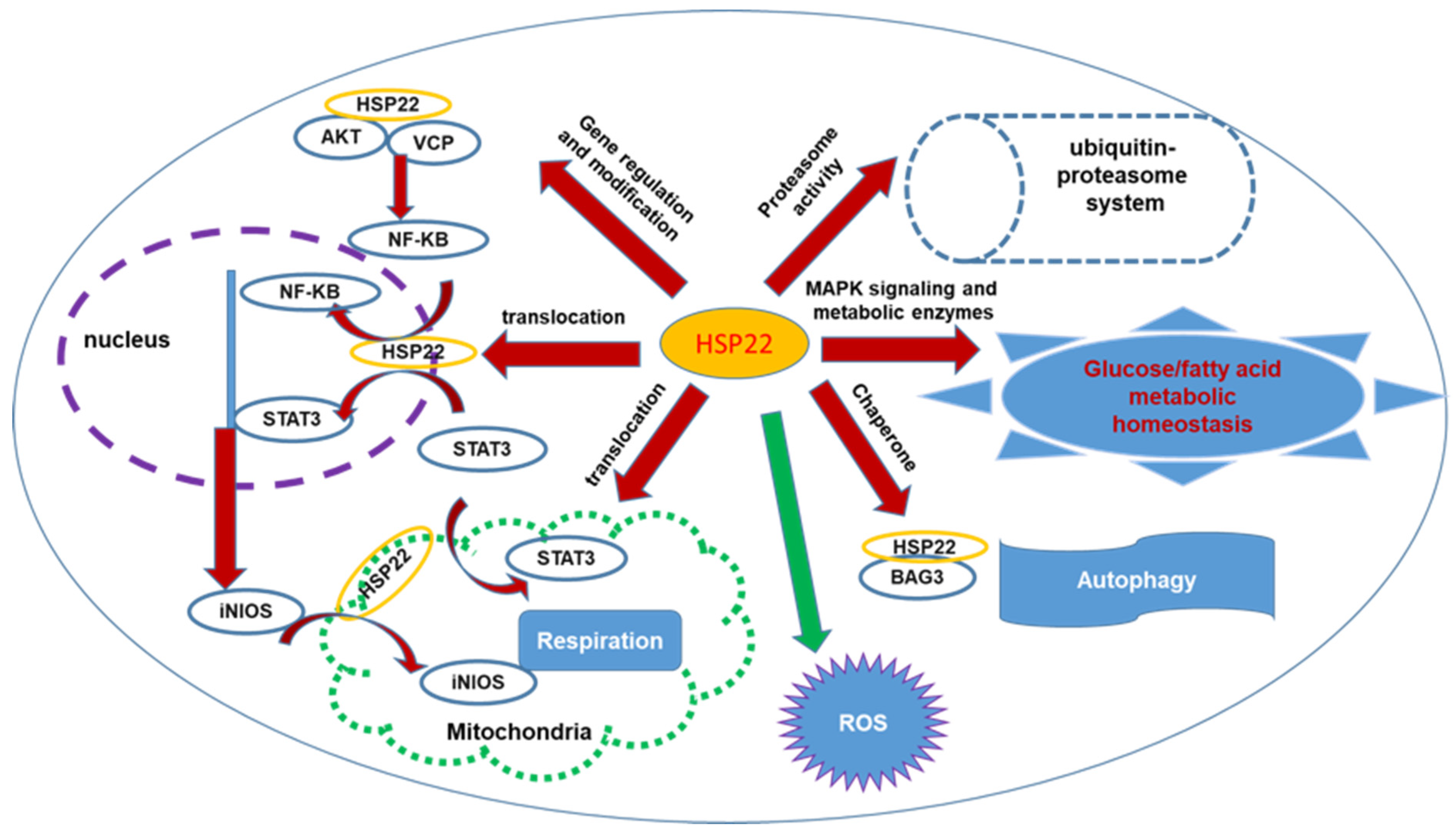{kind=link}
| Heart Diseases | HSP22 Expression and Function | Potential Therapeutic Targets |
|---|---|---|
| Dilated cardiomyopathy | Mutation of HSPB8(K141N) [48] HSP22 reduction with deficient BAG3 [26,42,43] | Increase HSP22 and BAG3 |
| Cardiac Hypertrophy | HSP22 upregulated in cardiac hypertrophy [36]; HSP22 induces cardiac hypertrophy by activating proteasome activity [37]; HSP22 protects the heart against HF by activating STAT3/Akt signaling pathway [14,25] | Proteasome inhibitor STAT3 inhibitor |
| Ischemic Heart Disease | HSP22 upregulated in ischemic heart HSP22 protects the heart against ischemia injury by activating P13K/Akt/VCP/NF-kB/STAT3/iNOS [20,33,35] | Pre-emptive conditioning of Hsp22 Enhance iNOS mitochondrial translocation |
| Age-related Cardiomyopathy | HSP22 upregulated due to the intrinsic stress HSP22 deletion leads to cardiac dysfunction with increasing age by interrupting cardiac autophagy and impairing FAO) [26] | Increase BAG3, Enhance FA metabolism |
| Diabetic Cardiomyopathy | HSP22 upregulated HSP22 protect hearts through inhibiting mtROS) [24] | Reducing mtROS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, X.; Siri, S.; Hurst, A.; Qiu, H. Heat Shock Protein 22 in Physiological and Pathological Hearts: Small Molecule, Large Potentials. Cells 2022, 11, 114. https://doi.org/10.3390/cells11010114
Sun X, Siri S, Hurst A, Qiu H. Heat Shock Protein 22 in Physiological and Pathological Hearts: Small Molecule, Large Potentials. Cells. 2022; 11(1):114. https://doi.org/10.3390/cells11010114
Chicago/Turabian StyleSun, Xiaonan, Sharadhi Siri, Amirah Hurst, and Hongyu Qiu. 2022. "Heat Shock Protein 22 in Physiological and Pathological Hearts: Small Molecule, Large Potentials" Cells 11, no. 1: 114. https://doi.org/10.3390/cells11010114
APA StyleSun, X., Siri, S., Hurst, A., & Qiu, H. (2022). Heat Shock Protein 22 in Physiological and Pathological Hearts: Small Molecule, Large Potentials. Cells, 11(1), 114. https://doi.org/10.3390/cells11010114