
GILZ as a Regulator of Cell Fate and Inflammation




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
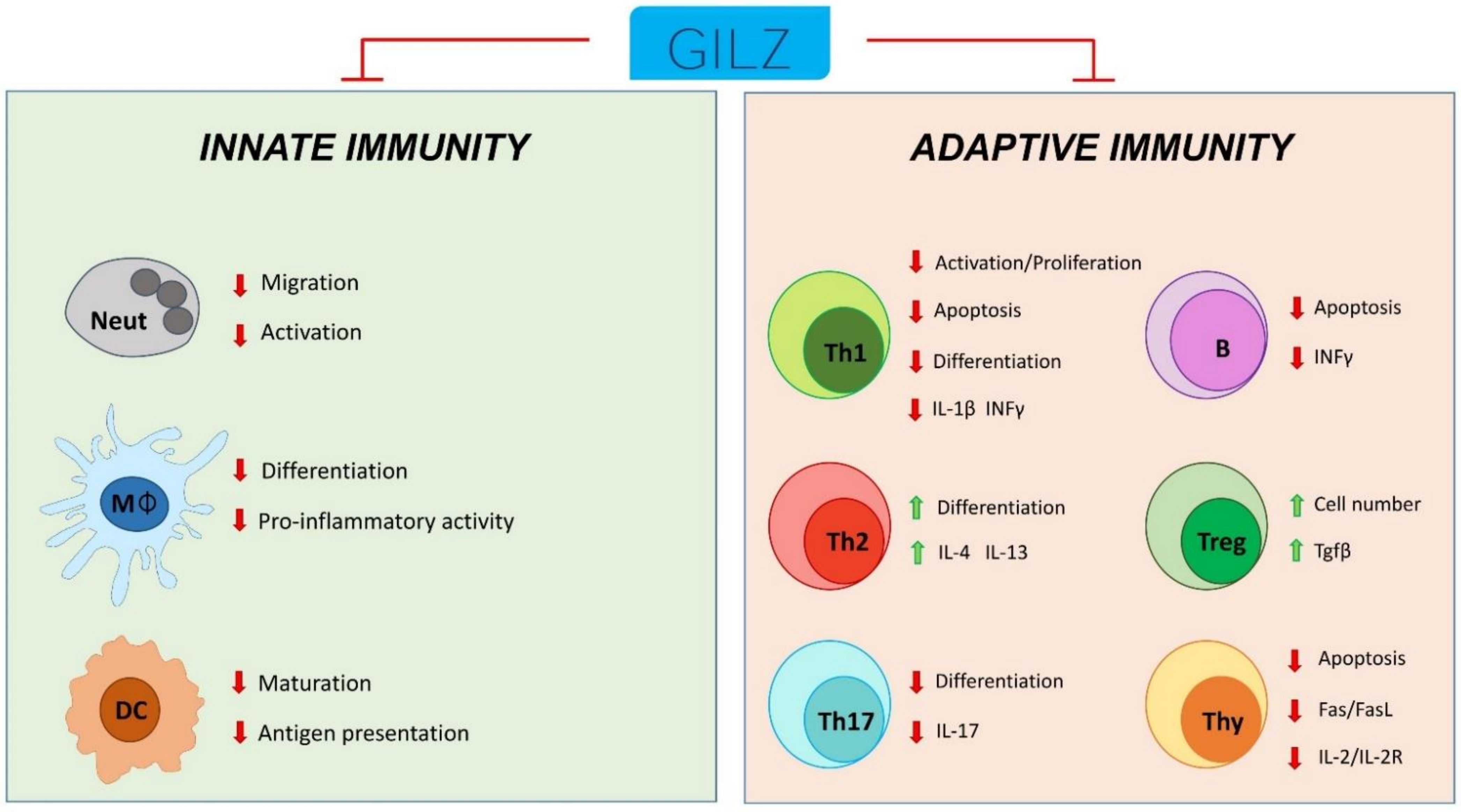
2. GILZ in Controlling Cell Growth: Effect on Apoptosis, Cell Proliferation and Differentiation
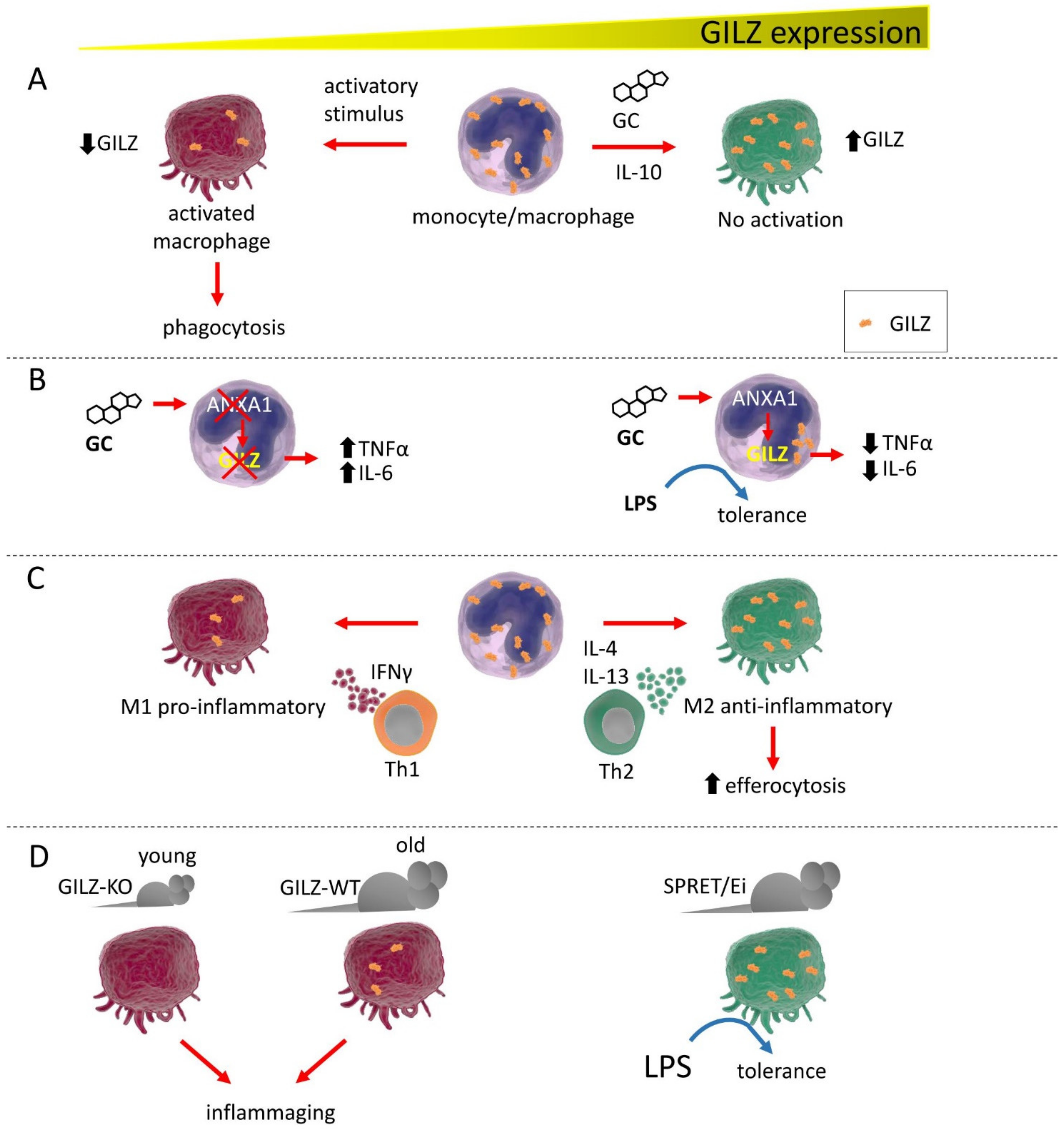
3. GILZ Function in Macrophages
4. GILZ Function in Neutrophils
5. GILZ Function in Dendritic Cells
6. GILZ Function in T Cells
7. GILZ Function in B Lymphocytes
8. GILZ in Various Cell Types
9. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Riccardi, C.; Cifone, M.G.; Migliorati, G. Glucocorticoid hormone-induced modulation of gene expression and regulation of T-cell death: Role of GITR and GILZ, two dexamethasone-induced genes. Cell Death Differ. 1999, 6, 1182–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Keeffe, G.W.; Gutierrez, H.; Pandolfi, P.P.; Riccardi, C.; Davies, A.M. NGF-promoted axon growth and target innervation requires GITRL-GITR signaling. Nat. Neurosci. 2008, 11, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Devgan, V.; Corrado, M.; Prabhu, N.S.; El-Deiry, W.S.; Riccardi, C.; Pandolfi, P.P.; Missero, C.; Dotto, G.P. Glucocorticoid-induced tumor necrosis factor receptor is a p21Cip1/WAF1 transcriptional target conferring resistance of keratinocytes to UV light-induced apoptosis. J. Biol. Chem. 2005, 280, 37725–37731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Adamio, F.; Zollo, O.; Moraca, R.; Ayroldi, E.; Bruscoli, S.; Bartoli, A.; Cannarile, L.; Migliorati, G.; Riccardi, C. A New Dexamethasone-Induced Gene of the Leucine Zipper Family Protects T Lymphocytes from TCR/CD3-Activated Cell Death. Immunity 1997, 7, 803–812. [Google Scholar] [CrossRef] [Green Version]
- Ronchetti, S.; Migliorati, G.; Riccardi, C. GILZ as a Mediator of the Anti-Inflammatory Effects of Glucocorticoids. Front. Endocrinol. 2015, 6, 170. [Google Scholar] [CrossRef] [Green Version]
- Cannarile, L.; Cuzzocrea, S.; Santucci, L.; Agostini, M.; Mazzon, E.; Esposito, E.; Muià, C.; Coppo, M.; di Paola, R.; Riccardi, C. Glucocorticoid-Induced Leucine Zipper Is Protective in Th1-Mediated Models of Colitis. Gastroenterology 2009, 136, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Delfino, D.V.; Agostini, M.; Spinicelli, S.; Vito, P.; Riccardi, C. Decrease of Bcl-xL and augmentation of thymocyte apoptosis in GILZ overexpressing transgenic mice. Blood 2004, 104, 4134–4141. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, M.C.; Di Marco, B.; Cifone, G.; Migliorati, G.; Riccardi, C.; Cifone, M.G. Dexamethasone-induced apoptosis of thymocytes: Role of glucocorticoid receptor–associated Src kinase and caspase-8 activation. Blood 2003, 101, 585–593. [Google Scholar] [CrossRef]
- Riccardi, C.; Zollo, O.; Nocentini, G.; Bruscoli, S.; Bartoli, A.; D’Adamio, F.; Cannarile, L.; Delfino, D.; Ayroldi, E.; Migliorati, G. Glucocorticoid hormones in the regulation of cell death. Therapie 2000, 55, 165–169. [Google Scholar]
- Cifone, M.G.; Migliorati, G.; Parroni, R.; Marchetti, C.; Millimaggi, D.; Santoni, A.; Riccardi, C. Dexamethasone-induced thymocyte apoptosis: Apoptotic signal involves the sequential activation of phosphoinositide-specific phospholipase C, acidic sphingomyelinase, and caspases. Blood 1999, 93, 2282–2296. [Google Scholar] [CrossRef]
- Ayroldi, E.; Zollo, O.; Macchiarulo, A.; Di Marco, B.; Marchetti, C.; Riccardi, C. Glucocorticoid-Induced Leucine Zipper Inhibits the Raf-Extracellular Signal-Regulated Kinase Pathway by Binding to Raf-1. Mol. Cell. Biol. 2002, 22, 7929–7941. [Google Scholar] [CrossRef] [Green Version]
- Ayroldi, E.; Zollo, O.; Bastianelli, A.; Marchetti, C.; Agostini, M.; Di Virgilio, R.; Riccardi, C. GILZ mediates the antiproliferative activity of glucocorticoids by negative regulation of Ras signaling. J. Clin. Investig. 2007, 117, 1605–1615. [Google Scholar] [CrossRef] [Green Version]
- Ayroldi, E.; Macchiarulo, A.; Riccardi, C. Targeting glucocorticoid side effects: Selective glucocorticoid receptor modulator or glucocorticoid-induced leucine zipper? A perspective. FASEB J. 2014, 28, 5055–5070. [Google Scholar] [CrossRef]
- Cannarile, L.; Fallarino, F.; Agostini, M.; Cuzzocrea, S.; Mazzon, E.; Vacca, C.; Genovese, T.; Migliorati, G.; Ayroldi, E.; Riccardi, C. Increased GILZ expression in transgenic mice up-regulates Th-2 lymphokines. Blood 2006, 107, 1039–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayroldi, E.; Migliorati, G.; Bruscoli, S.; Marchetti, C.; Zollo, O.; Cannarile, L.; D’Adamio, F.; Riccardi, C. Modulation of T-cell activation by the glucocorticoid-induced leucine zipper factor via inhibition of nuclear factor kappaB. Blood 2001, 98, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Cannarile, L.; Zollo, O.; D’Adamio, F.; Ayroldi, E.; Marchetti, C.; Tabilio, A.; Bruscoli, S.; Riccardi, C. Cloning, chromosomal assignment and tissue distribution of human GILZ, a glucocorticoid hormone-induced gene. Cell Death Differ. 2001, 8, 201–203. [Google Scholar] [CrossRef] [Green Version]
- Berrebi, D.; Bruscoli, S.; Cohen, N.; Foussat, A.; Migliorati, G.; Bouchet-Delbos, L.; Maillot, M.-C.; Portier, A.; Couderc, J.; Galanaud, P.; et al. Synthesis of glucocorticoid-induced leucine zipper (GILZ) by macrophages: An anti-inflammatory and immunosuppressive mechanism shared by glucocorticoids and IL-10. Blood J. Am. Soc. Hematol. 2003, 101, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Ricci, E.; Ronchetti, S.; Gabrielli, E.; Pericolini, E.; Gentili, M.; Roselletti, E.; Vecchiarelli, A.; Riccardi, C. GILZ restrains neutrophil activation by inhibiting the MAPK pathway. J. Leukoc. Biol. 2019, 105, 187–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheikh, M.H.; Solito, E. Annexin A1: Uncovering the Many Talents of an Old Protein. Int. J. Mol. Sci. 2018, 19, 1045. [Google Scholar] [CrossRef] [Green Version]
- Vago, J.P.; Tavares, L.P.; Riccardi, C.; Teixeira, M.M.; Sousa, L.P. Exploiting the pro-resolving actions of glucocorticoid-induced proteins Annexin A1 and GILZ in infectious diseases. Biomed. Pharmacother. 2021, 133, 111033. [Google Scholar] [CrossRef]
- Yang, Y.H.; Aeberli, D.; Dacumos, A.; Xue, J.R.; Morand, E. Annexin-1 Regulates Macrophage IL-6 and TNF via Glucocorticoid-Induced Leucine Zipper. J. Immunol. 2009, 183, 1435–1445. [Google Scholar] [CrossRef] [Green Version]
- Hoppstädter, J.; Kessler, S.M.; Bruscoli, S.; Huwer, H.; Riccardi, C.; Kiemer, A.K. Glucocorticoid-Induced Leucine Zipper: A Critical Factor in Macrophage Endotoxin Tolerance. J. Immunol. 2015, 194, 6057–6067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoppstädter, J.; Diesel, B.; Eifler, L.K.; Schmid, T.; Brüne, B.; Kiemer, A.K. Glucocorticoid-induced leucine zipper is downregulated in human alveolar macrophages upon Toll-like receptor activation. Eur. J. Immunol. 2012, 42, 1282–1293. [Google Scholar] [CrossRef]
- Hoppstädter, J.; Diesel, B.; Linnenberger, R.; Hachenthal, N.; Flamini, S.; Minet, M.; Leidinger, P.; Backes, C.; Grässer, F.; Meese, E.; et al. Amplified Host Defense by Toll-Like Receptor-Mediated Downregulation of the Glucocorticoid-Induced Leucine Zipper (GILZ) in Macrophages. Front. Immunol. 2019, 9, 3111. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, I.; Dejager, L.; Petta, I.; Vandevyver, S.; Puimège, L.; Mahieu, T.; Ballegeer, M.; Van Hauwermeiren, F.; Riccardi, C.; Vuylsteke, M.; et al. LPS resistance of SPRET/Ei mice is mediated by Gilz, encoded by the Tsc22d3 gene on the X chromosome. EMBO Mol. Med. 2013, 5, 456–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoppstädter, J.; Hachenthal, N.; Perez, J.V.V.; Lampe, S.; Astanina, K.; Kunze, M.M.; Bruscoli, S.; Riccardi, C.; Schmid, T.; Diesel, B.; et al. Induction of Glucocorticoid-induced Leucine Zipper (GILZ) Contributes to Anti-inflammatory Effects of the Natural Product Curcumin in Macrophages. J. Biol. Chem. 2016, 291, 22949–22960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vago, J.P.; Galvão, I.; Negreiros-Lima, G.L.; Teixeira, L.C.; Lima, K.M.; Sugimoto, M.A.; Moreira, I.Z.; Jones, S.A.; Lang, T.; Riccardi, C.; et al. Glucocorticoid-induced leucine zipper modulates macrophage polarization and apoptotic cell clearance. Pharmacol. Res. 2020, 158, 104842. [Google Scholar] [CrossRef]
- Ellouze, M.; Vigouroux, L.; Tcherakian, C.; Woerther, P.L.; Guguin, A.; Robert, O.; Surenaud, M.; Tran, T.; Calmette, J.; Barbin, T.; et al. Overexpression of GILZ in macrophages limits systemic inflammation while increasing bacterial clearance in sepsis in mice. Eur. J. Immunol. 2020, 50, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Perez, J.V.V.; Linnenberger, R.; Dembek, A.; Bruscoli, S.; Riccardi, C.; Schulz, M.H.; Meyer, M.R.; Kiemer, A.K.; Hoppstädter, J. Altered glucocorticoid metabolism represents a feature of macroph-aging. Aging Cell 2020, 19, e13156. [Google Scholar] [CrossRef]
- Espinasse, M.-A.; Pépin, A.; Virault-Rocroy, P.; Szely, N.; Chollet-Martin, S.; Pallardy, M.; Biola-Vidamment, A. Glucocorticoid-Induced Leucine Zipper Is Expressed in Human Neutrophils and Promotes Apoptosis through Mcl-1 Down-Regulation. J. Innate Immun. 2016, 8, 81–96. [Google Scholar] [CrossRef] [PubMed]
- Ricci, E.; Ronchetti, S.; Pericolini, E.; Gabrielli, E.; Cari, L.; Gentili, M.; Roselletti, E.; Migliorati, G.; Vecchiarelli, A.; Riccardi, C. Role of the glucocorticoid-induced leucine zipper gene in dexamethasone-induced inhibition of mouse neutrophil migration via control of annexin A1 expression. FASEB J. 2017, 31, 3054–3065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinasse, M.-A.; Hajage, D.; Montravers, P.; Piednoir, P.; Dufour, G.; Tubach, F.; Granger, V.; De Chaisemartin, L.; Noël, B.; Pallardy, M.; et al. Neutrophil expression of glucocorticoid-induced leucine zipper (GILZ) anti-inflammatory protein is associated with acute respiratory distress syndrome severity. Ann. Intensiv. Care 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baban, B.; Marchetti, C.; Khodadadi, H.; Malik, A.; Emami, G.; Lin, P.-C.; Arbab, A.S.; Riccardi, C.; Mozaffari, M.S. Glucocorticoid-Induced Leucine Zipper Promotes Neutrophil and T-Cell Polarization with Protective Effects in Acute Kidney Injury. J. Pharmacol. Exp. Ther. 2018, 367, 483–493. [Google Scholar] [CrossRef]
- Ronchetti, S.; Ricci, E.; Migliorati, G.; Gentili, M.; Riccardi, C. How Glucocorticoids Affect the Neutrophil Life. Int. J. Mol. Sci. 2018, 19, 4090. [Google Scholar] [CrossRef] [Green Version]
- Zimmer, A.; Bouley, J.; Le Mignon, M.; Pliquet, E.; Horiot, S.; Turfkruyer, M.; Baron-Bodo, V.; Horak, F.; Nony, E.; Louise, A.; et al. A regulatory dendritic cell signature correlates with the clinical efficacy of allergen-specific sublingual immunotherapy. J. Allergy Clin. Immunol. 2012, 129, 1020–1030. [Google Scholar] [CrossRef]
- Benkhoucha, M.; Molnarfi, N.; Dunand-Sauthier, I.; Merkler, D.; Schneiter, G.; Bruscoli, S.; Riccardi, C.; Tabata, Y.; Funakoshi, H.; Nakamura, T.; et al. Hepatocyte Growth Factor Limits Autoimmune Neuroinflammation via Glucocorticoid-Induced Leucine Zipper Expression in Dendritic Cells. J. Immunol. 2014, 193, 2743–2752. [Google Scholar] [CrossRef] [Green Version]
- Cohen, N.; Mouly, E.; Hamdi, H.; Maillot, M.-C.; Pallardy, M.; Godot, V.; Capel, F.; Balian, A.; Naveau, S.; Galanaud, P.; et al. GILZ expression in human dendritic cells redirects their maturation and prevents antigen-specific T lymphocyte response. Blood 2006, 107, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Hamdi, H.; Godot, V.; Maillot, M.-C.; Prejean, M.V.; Cohen, N.; Krzysiek, R.; Lemoine, F.M.; Zou, M.-H.; Emilie, D. Induction of antigen-specific regulatory T lymphocytes by human dendritic cells expressing the glucocorticoid-induced leucine zipper. Blood 2007, 110, 211–219. [Google Scholar] [CrossRef] [Green Version]
- Calmette, J.; Ellouze, M.; Tran, T.; Karaki, S.; Ronin, E.; Capel, F.; Pallardy, M.; Bachelerie, F.; Krzysiek, R.; Emilie, D.; et al. Glucocorticoid-Induced Leucine Zipper Enhanced Expression in Dendritic Cells Is Sufficient To Drive Regulatory T Cells Expansion In Vivo. J. Immunol. 2014, 193, 5863–5872. [Google Scholar] [CrossRef] [Green Version]
- Cathelin, D.; Met, Ö.; Svane, I.M. Silencing of the glucocorticoid-induced leucine zipper improves the immunogenicity of clinical-grade dendritic cells. Cytotherapy 2013, 15, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Karaki, S.; Garcia, G.; Tcherakian, C.; Capel, F.; Tran, T.; Pallardy, M.; Humbert, M.; Emilie, D.; Godot, V. Enhanced glucocorticoid-induced leucine zipper in dendritic cells induces allergen-specific regulatory CD4 + T-cells in respiratory allergies. Allergy 2014, 69, 624–631. [Google Scholar] [CrossRef]
- Calmette, J.; Bertrand, M.; Vétillard, M.; Ellouze, M.; Flint, S.; Nicolas, V.; Biola-Vidamment, A.; Pallardy, M.; Morand, E.; Bachelerie, F.; et al. Glucocorticoid-Induced Leucine Zipper Protein Controls Macropinocytosis in Dendritic Cells. J. Immunol. 2016, 197, 4247–4256. [Google Scholar] [CrossRef] [PubMed]
- Bereshchenko, O.; Coppo, M.; Bruscoli, S.; Biagioli, M.; Cimino, M.; Frammartino, T.; Sorcini, D.; Venanzi, A.; Di Sante, M.; Riccardi, C. GILZ promotes production of peripherally induced Treg cells and mediates the crosstalk between glucocorticoids and TGF-beta signaling. Cell Rep. 2014, 7, 464–475. [Google Scholar] [CrossRef] [Green Version]
- Bruscoli, S.; Velardi, E.; Di Sante, M.; Bereshchenko, O.; Venanzi, A.; Coppo, M.; Berno, V.; Mameli, M.G.; Colella, R.; Cavaliere, A.; et al. Long Glucocorticoid-induced Leucine Zipper (L-GILZ) Protein Interacts with Ras Protein Pathway and Contributes to Spermatogenesis Control. J. Biol. Chem. 2012, 287, 1242–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hommes, D.W.; Peppelenbosch, M.; Van Deventer, S.J.H. Mitogen activated protein (MAP) kinase signal transduction pathways and novel anti-inflammatory targets. Gut 2003, 52, 144–151. [Google Scholar] [CrossRef] [Green Version]
- Grugan, K.D.; Ma, C.; Singhal, S.; Krett, N.L.; Rosen, S.T. Dual regulation of glucocorticoid-induced leucine zipper (GILZ) by the glucocorticoid receptor and the PI3-kinase/AKT pathways in multiple myeloma. J. Steroid Biochem. Mol. Biol. 2008, 110, 244–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joha, S.; Nugues, A.L.; Hetuin, D.; Berthon, C.; Dezitter, X.; Dauphin, V.; Mahon, F.X.; Roche-Lestienne, C.; Preudhomme, C.; Quesnel, B.; et al. GILZ inhibits the mTORC2/AKT pathway in BCR-ABL(+) cells. Oncogene 2012, 31, 1419–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venanzi, A.; Di Sante, M.; Bruscoli, S.; Biagioli, M.; Sorcini, D.; Cimino, M.; Frammartino, T.; Bereshchenko, O.; Franconi, F.; Riccardi, C. Recombinant long-glucocorticoid-induced leucine zipper (L-GILZ) protein restores the control of proliferation in gilz KO spermatogonia. Eur. J. Pharm. Sci. 2014, 63, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Kasibhatla, S.; Brunner, T.; Genestier, L.; Echeverri, F.; Mahboubi, A.; Green, D.R. DNA damaging agents induce expression of Fas ligand and subsequent apoptosis in T lymphocytes via the activation of NF-kappa B and AP-1. Mol. Cell 1998, 1, 543–551. [Google Scholar] [CrossRef]
- Jain, J.; Loh, C.; Rao, A. Transcriptional regulation of the IL-2 gene. Curr. Opin. Immunol. 1995, 7, 333–342. [Google Scholar] [CrossRef]
- De Bosscher, K.; Vanden Berghe, W.; Haegeman, G. The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: Molecular mechanisms for gene repression. Endocr. Rev. 2003, 24, 488–522. [Google Scholar] [CrossRef] [Green Version]
- Asselin-Labat, M.-L.; David, M.; Biola-Vidamment, A.; Lecoeuche, D.; Zennaro, M.-C.; Bertoglio, J.; Pallardy, M. GILZ, a new target for the transcription factor FoxO3, protects T lymphocytes from interleukin-2 withdrawal–induced apoptosis. Blood 2004, 104, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Delfino, D.V.; Agostini, M.; Spinicelli, S.; Vacca, C.; Riccardi, C. Inhibited cell death, NF-kappaB activity and increased IL-10 in TCR-triggered thymocytes of transgenic mice overexpressing the glucocorticoid-induced protein GILZ. Int. Immunopharmacol. 2006, 6, 1126–1134. [Google Scholar] [CrossRef]
- Matsui, K.; Fine, A.; Zhu, B.; Marshak-Rothstein, A.; Ju, S.T. Identification of two NF-kappa B sites in mouse CD95 ligand (Fas ligand) promoter: Functional analysis in T cell hybridoma. J. Immunol. 1998, 161, 3469–3473. [Google Scholar]
- Lin, B.; Williams-Skipp, C.; Tao, Y.; Schleicher, M.S.; Cano, L.L.; Duke, R.C.; Scheinman, R.I. NF-kappaB functions as both a proapoptotic and antiapoptotic regulatory factor within a single cell type. Cell Death Differ. 1999, 6, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Lea, T.; Rasmussen, A.; Michaelsen, T. Differentiation antigens in human T-cell activation: Evidence that anti-VH antibodies react with a membrane structure on human T Lymphocytes distinct from the antigens detected by monoclonal antibodies of the OKT and Leu series. Cell. Immunol. 1983, 81, 209–218. [Google Scholar] [CrossRef]
- Tamura, T.; Nakano, H.; Nagase, H.; Morokata, T.; Igarashi, O.; Oshimi, Y.; Miyazaki, S.; Nariuchi, H. Early activation signal transduction pathways of Th1 and Th2 cell clones stimulated with anti-CD3. Roles of protein tyrosine kinases in the signal for IL-2 and IL-4 production. J. Immunol. 1995, 155, 4692–4701. [Google Scholar] [PubMed]
- Taves, M.D.; Ashwell, J.D. Glucocorticoids in T cell development, differentiation and function. Nat. Rev. Immunol. 2021, 21, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Esposito, E.; Bruscoli, S.; Mazzon, E.; Paterniti, I.; Coppo, M.; Velardi, E.; Cuzzocrea, S.; Riccardi, C. Glucocorticoid-Induced Leucine Zipper (GILZ) Over-Expression in T Lymphocytes Inhibits Inflammation and Tissue Damage in Spinal Cord Injury. Neurotherapeutics 2011, 9, 210–225. [Google Scholar] [CrossRef] [Green Version]
- Beaulieu, E.; Ngo, D.; Santos, L.; Yang, Y.H.; Smith, M.; Jorgensen, C.; Escriou, V.; Scherman, D.; Courties, G.; Apparailly, F.; et al. Glucocorticoid-induced leucine zipper is an endogenous antiinflammatory mediator in arthritis. Arthritis Rheum. 2010, 62, 2651–2661. [Google Scholar] [CrossRef] [PubMed]
- Luz-Crawford, P.; Tejedor, G.; Mausset-Bonnefont, A.L.; Beaulieu, E.; Morand, E.F.; Jorgensen, C.; Noël, D.; Djouad, F. Glucocorticoid-Induced Leucine Zipper Governs the Therapeutic Potential of Mesenchymal Stem Cells by Inducing a Switch From Pathogenic to Regulatory Th17 Cells in a Mouse Model of Collagen-Induced Arthritis. Arthritis Rheumatol. 2015, 67, 1514–1524. [Google Scholar] [CrossRef]
- Yosef, N.; Shalek, A.K.; Gaublomme, J.T.; Jin, H.; Lee, Y.; Awasthi, A.; Wu, C.; Karwacz, K.; Xiao, S.; Jorgolli, M.; et al. Dynamic regulatory network controlling TH17 cell differentiation. Nature 2013, 496, 461–468. [Google Scholar] [CrossRef]
- Qin, X.; Liu, J.Y.; Abdelsayed, R.; Shi, X.; Yu, J.C.; Mozaffari, M.S.; Baban, B. The status of glucocorticoid-induced leucine zipper protein in the salivary glands in Sjögren’s syndrome: Predictive and prognostic potentials. EPMA J. 2015, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.A.; Perera, D.N.; Fan, H.; Russ, B.E.; Harris, J.; Morand, E.F. GILZ regulates Th17 responses and restrains IL-17-mediated skin inflammation. J. Autoimmun. 2015, 61, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Libert, C.; Dejager, L. How Steroids Steer T Cells. Cell Rep. 2014, 7, 938–939. [Google Scholar] [CrossRef] [Green Version]
- Di Marco, B.; Massetti, M.; Bruscoli, S.; Macchiarulo, A.; Di Virgilio, R.; Velardi, E.; Donato, V.; Migliorati, G.; Riccardi, C. Glucocorticoid-induced leucine zipper (GILZ)/NF-kappaB interaction: Role of GILZ homo-dimerization and C-terminal domain. Nucleic Acids. Res. 2007, 35, 517–528. [Google Scholar] [CrossRef]
- Srinivasan, M.; Janardhanam, S. Novel p65 Binding Glucocorticoid-induced Leucine Zipper Peptide Suppresses Experimental Autoimmune Encephalomyelitis. J. Biol. Chem. 2011, 286, 44799–44810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baban, B.; Yin, L.; Qin, X.; Liu, J.Y.; Shi, X.; Mozaffari, M.S. The role of GILZ in modulation of adaptive immunity in a murine model of myocardial infarction. Exp. Mol. Pathol. 2017, 102, 408–414. [Google Scholar] [CrossRef]
- LeBien, T.W.; Tedder, T.F. B lymphocytes: How they develop and function. Blood 2008, 112, 1570–1580. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, W.J. To B or Not to B? Glucocorticoid Impact on B Lymphocyte Fate and Function. Endocrinology 2014, 155, 339–342. [Google Scholar] [CrossRef] [Green Version]
- Lund, F.E. Cytokine-producing B lymphocytes—Key regulators of immunity. Curr. Opin. Immunol. 2008, 20, 332–338. [Google Scholar] [CrossRef] [Green Version]
- Mauri, C. Regulation of immunity and autoimmunity by B cells. Curr. Opin. Immunol. 2010, 22, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Settipane, G.A.; Pudupakkam, R.K.; McGowan, J.H. Corticosteroid effect on immunoglobulins. J. Allergy Clin. Immunol. 1978, 62, 162–166. [Google Scholar] [CrossRef]
- Jabara, H.H.; Ahern, D.J.; Vercelli, D.; Geha, R.S. Hydrocortisone and IL-4 induce IgE isotype switching in human B cells. J. Immunol. 1991, 147, 1557–1560. [Google Scholar]
- Tokuyama, Y.; Tokuyama, H. Retinoic acid and steroid hormones regulate IgA production by LPS-stimulated murine spleen cells. Immunopharmacology 1994, 28, 145–151. [Google Scholar] [CrossRef]
- Igarashi, H.; Medina, K.L.; Yokota, T.; Rossi, M.I.D.; Sakaguchi, N.; Comp, P.C.; Kincade, P.W. Early lymphoid progenitors in mouse and man are highly sensitive to glucocorticoids. Int. Immunol. 2005, 17, 501–511. [Google Scholar] [CrossRef]
- Parrillo, J.E.; Fauci, A.S. Mechanisms of corticosteroid action on lymphocyte subpopulations. III. Differential effects of dexamethasone administration on subpopulations of effector cells mediating cellular cytotoxicity in man. Clin. Exp. Immunol. 1978, 31, 116–125. [Google Scholar]
- Garvy, B.A.; Telford, W.G.; King, L.E.; Fraker, P.J. Glucocorticoids and irradiation-induced apoptosis in normal murine bone marrow B-lineage lymphocytes as determined by flow cytometry. Immunology 1993, 79, 270–277. [Google Scholar]
- Lu, L.; Osmond, D.G. Apoptosis and its modulation during B lymphopoiesis in mouse bone marrow. Immunol. Rev. 2000, 175, 158–174. [Google Scholar] [CrossRef] [PubMed]
- Bruscoli, S.; Biagioli, M.; Sorcini, D.; Frammartino, T.; Cimino, M.; Sportoletti, P.; Mazzon, E.; Bereshchenko, O.; Riccardi, C. Lack of glucocorticoid-induced leucine zipper (GILZ) deregulates B-cell survival and results in B-cell lymphocytosis in mice. Blood 2015, 126, 1790–1801. [Google Scholar] [CrossRef] [Green Version]
- Glynne, R.; Akkaraju, S.; Healy, J.I.; Rayner, J.; Goodnow, C.C.; Mack, D.H. How self-tolerance and the immunosuppressive drug FK506 prevent B-cell mitogenesis. Nature 2000, 403, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Bruscoli, S.; Sorcini, D.; Flamini, S.; Gagliardi, A.; Adamo, F.; Ronchetti, S.; Migliorati, G.; Bereshchenko, O.; Riccardi, C. Glucocorticoid-Induced Leucine Zipper Inhibits Interferon-Gamma Production in B Cells and Suppresses Colitis in Mice. Front. Immunol. 2018, 9, 1720. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.A.; Toh, A.E.J.; Odobasic, D.; Oudin, M.-A.V.; Cheng, Q.; Lee, J.P.W.; White, S.J.; Russ, B.E.; Infantino, S.; Light, A.; et al. Glucocorticoid-induced leucine zipper (GILZ) inhibits B cell activation in systemic lupus erythematosus. Ann. Rheum. Dis. 2016, 75, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Romero, Y.; Vuandaba, M.; Suarez, P.; Grey, C.; Calvel, P.; Conne, B.; Pearce, D.; De Massy, B.; Hummler, E.; Nef, S. The Glucocorticoid-Induced Leucine Zipper (GILZ) Is Essential for Spermatogonial Survival and Spermatogenesis. Sex. Dev. 2012, 6, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Suarez, P.E.; Rodriguez, E.G.; Soundararajan, R.; Mérillat, A.-M.; Stehle, J.-C.; Rotman, S.; Roger, T.; Voirol, M.-J.; Wang, J.; Groß, O.; et al. The Glucocorticoid-Induced Leucine Zipper (Gilz/Tsc22d3-2) Gene Locus Plays a Crucial Role in Male Fertility. Mol. Endocrinol. 2012, 26, 1000–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngo, D.; Cheng, Q.; O′connor, A.E.; DeBoer, K.D.; Lo, C.Y.; Beaulieu, E.; De Seram, M.; Hobbs, R.M.; O′bryan, M.K.; Morand, E.F. Glucocorticoid-Induced Leucine Zipper (GILZ) Regulates Testicular FOXO1 Activity and Spermatogonial Stem Cell (SSC) Function. PLoS ONE 2013, 8, e59149. [Google Scholar] [CrossRef] [Green Version]
- Eddleston, J.; Herschbach, J.; Wagelie-Steffen, A.L.; Christiansen, S.C.; Zuraw, B.L. The anti-inflammatory effect of glucocorticoids is mediated by glucocorticoid-induced leucine zipper in epithelial cells. J. Allergy Clin. Immunol. 2007, 119, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, M.; Niu, C.; Luo, Z.; Dai, J.; Wang, L.; Liu, E.; Fu, Z. Dexamethasone Inhibits Repair of Human Airway Epithelial Cells Mediated by Glucocorticoid-Induced Leucine Zipper (GILZ). PLoS ONE 2013, 8, e60705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soundararajan, R.; Zhang, T.T.; Wang, J.; Vandewalle, A.; Pearce, D. A Novel Role for Glucocorticoid-induced Leucine Zipper Protein in Epithelial Sodium Channel-mediated Sodium Transport. J. Biol. Chem. 2005, 280, 39970–39981. [Google Scholar] [CrossRef] [Green Version]
- Rashmi, P.; Colussi, G.; Ng, M.; Wu, X.; Kidwai, A.; Pearce, D. Glucocorticoid-induced leucine zipper protein regulates sodium and potassium balance in the distal nephron. Kidney Int. 2017, 91, 1159–1177. [Google Scholar] [CrossRef] [PubMed]
- Malaise, O.; Relic, B.; Charlier, E.; Zeddou, M.; Neuville, S.; Deroyer, C.; Gillet, P.; Louis, E.; Malaise, M.G.; De Seny, D. Glucocorticoid-induced leucine zipper (GILZ) is involved in glucocorticoid-induced and mineralocorticoid-induced leptin production by osteoarthritis synovial fibroblasts. Arthritis Res. Ther. 2016, 18, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Q.; Fan, H.; Ngo, D.; Beaulieu, E.; Leung, P.; Lo, C.Y.; Burgess, R.; van der Zwan, Y.G.; White, S.J.; Khachigian, L.M.; et al. GILZ overexpression inhibits endothelial cell adhesive function through regulation of NF-kappaB and MAPK activity. J Immunol. 2013, 191, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, R.; Lei, B.; Jiang, C.; Xu, G. Glucocorticoid-Induced Leucine Zipper Suppresses ICAM-1 and MCP-1 Expression by Dephosphorylation of NF-kappaB p65 in Retinal Endothelial Cells. Investig. Ophthalmol. Vis. Sci. 2017, 58, 631–641. [Google Scholar] [CrossRef] [Green Version]
- Hahn, R.T.; Hoppstädter, J.; Hirschfelder, K.; Hachenthal, N.; Diesel, B.; Kessler, S.M.; Huwer, H.; Kiemer, A.K. Downregulation of the glucocorticoid-induced leucine zipper (GILZ) promotes vascular inflammation. Atherosclerosis 2014, 234, 391–400. [Google Scholar] [CrossRef]
- Aguilar, D.; Strom, J.; Chen, Q.M. Glucocorticoid Induced Leucine Zipper inhibits apoptosis of cardiomyocytes by doxorubicin. Toxicol. Appl. Pharmacol. 2014, 276, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappetta, D.; De Angelis, A.; Flamini, S.; Cozzolino, A.; Bereshchenko, O.; Ronchetti, S.; Cianflone, E.; Gagliardi, A.; Ricci, E.; Rafaniello, C.; et al. Deficit of glucocorticoid-induced leucine zipper amplifies angiotensin-induced cardiomyocyte hypertrophy and diastolic dysfunction. J. Cell. Mol. Med. 2021, 25, 217–228. [Google Scholar] [CrossRef]
- Bruscoli, S.; Donato, V.; Velardi, E.; Di Sante, M.; Migliorati, G.; Donato, R.; Riccardi, C. Glucocorticoid-induced Leucine Zipper (GILZ) and Long GILZ Inhibit Myogenic Differentiation and Mediate Anti-myogenic Effects of Glucocorticoids. J. Biol. Chem. 2010, 285, 10385–10396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoppstädter, J.; Perez, J.V.V.; Linnenberger, R.; Dahlem, C.; Legroux, T.M.; Hecksteden, A.; Tse, K.F.W.; Flamini, S.; Andreas, A.; Herrmann, J.; et al. The glucocorticoid-induced leucine zipper mediates statin-induced muscle damage. FASEB J. 2020, 34, 4684–4701. [Google Scholar] [CrossRef] [Green Version]
- Gu, R.; Lei, B.; Shu, Q.; Li, G.; Xu, G. Glucocorticoid-induced leucine zipper overexpression inhibits lipopolysaccharide-induced retinal inflammation in rats. Exp. Eye Res. 2017, 165, 151–163. [Google Scholar] [CrossRef]
- Gu, R.; Tang, W.; Lei, B.; Ding, X.; Jiang, C.; Xu, G. Glucocorticoid-Induced Leucine Zipper Protects the Retina From Light-Induced Retinal Degeneration by Inducing Bcl-xL in Rats. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3656–3668. [Google Scholar] [CrossRef] [Green Version]
- Jia, H.; Yu, Z.; Ge, X.; Chen, Z.; Huang, X.; Wei, Y. Glucocorticoid-induced leucine zipper protects noise-induced apoptosis in cochlear cells by inhibiting endoplasmic reticulum stress in rats. Med. Mol. Morphol. 2019, 53, 73–81. [Google Scholar] [CrossRef]
- Yachi, K.; Inoue, K.; Tanaka, H.; Yoshikawa, H.; Tohyama, M. Localization of glucocorticoid-induced leucine zipper (GILZ) expressing neurons in the central nervous system and its relationship to the stress response. Brain Res. 2007, 1159, 141–147. [Google Scholar] [CrossRef]
- Witek, E.; Hickman, D.; Lahiri, D.K.; Srinivasan, M. Glucocorticoid Induced Leucine Zipper in Lipopolysaccharide Induced Neuroinflammation. Front. Aging Neurosci. 2019, 10, 432. [Google Scholar] [CrossRef] [Green Version]
- Frodl, T.; Carballedo, A.; Hughes, M.M.; Saleh, K.; Fagan, A.J.; Skokauskas, N.; McLoughlin, D.; Meaney, J.F.; O’Keane, V.; Connor, T.J. Reduced expression of glucocorticoid-inducible genes GILZ and SGK-1: High IL-6 levels are associated with reduced hippocampal volumes in major depressive disorder. Transl. Psychiatry 2012, 2, e88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellestad, L.E.; Malkiewicz, S.A.; Guthrie, H.D.; Welch, G.R.; Porter, T.E. Expression and regulation of glucocorticoid-induced leucine zipper in the developing anterior pituitary gland. J. Mol. Endocrinol. 2009, 42, 171–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.; Shi, W.; Li, Q.; Song, B.; Wan, M.; Bai, S.; Cao, X. A glucocorticoid-induced leucine-zipper protein, GILZ, inhibits adipogenesis of mesenchymal cells. EMBO Rep. 2003, 4, 374–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, G.; Cao, J.; Yang, N.; Ding, K.; Fan, C.; Xiong, W.-C.; Hamrick, M.; Isales, C.M.; Shi, X.-M. Role of Glucocorticoid-induced Leucine Zipper (GILZ) in Bone Acquisition. J. Biol. Chem. 2014, 289, 19373–19382. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Yang, N.; Shi, X.-M. Regulation of Mesenchymal Stem Cell Osteogenic Differentiation by Glucocorticoid-induced Leucine Zipper (GILZ). J. Biol. Chem. 2008, 283, 4723–4729. [Google Scholar] [CrossRef] [Green Version]
- Perpetuini, A.C.; Giuliani, G.; Reggio, A.; Cerretani, M.; Santoriello, M.; Stefanelli, R.; Palma, A.; Vumbaca, S.; Harper, S.; Castagnoli, L.; et al. Janus effect of glucocorticoids on differentiation of muscle fibro/adipogenic progenitors. Sci. Rep. 2020, 10, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Hamdi, H.; Bigorgne, A.; Naveau, S.; Balian, A.; Bouchet-Delbos, L.; Cassard-Doulcier, A.M.; Maillot, M.C.; Durand-Gasselin, I.; Prevot, S.; Delaveaucoupet, J.; et al. Glucocorticoid-induced leucine zipper: A key protein in the sensitization of monocytes to lipopolysaccharide in alcoholic hepatitis. Hepatology 2007, 46, 1986–1992. [Google Scholar] [CrossRef]
- Flamini, S.; Sergeev, P.; de Barros, Z.V.; Mello, T.; Biagioli, M.; Paglialunga, M.; Fiorucci, C.; Prikazchikova, T.; Pagano, S.; Gagliardi, A.; et al. Glucocorticoid-induced leucine zipper regulates liver fibrosis by suppressing CCL2-mediated leukocyte recruitment. Cell Death Dis. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Robert, O.; Boujedidi, H.; Bigorgne, A.; Ferrere, G.; Voican, C.S.; Vettorazzi, S.; Tuckermann, J.P.; Bouchet-Delbos, L.; Tran, T.; Hemon, P.; et al. Decreased expression of the glucocorticoid receptor-GILZ pathway in Kupffer cells promotes liver inflammation in obese mice. J. Hepatol. 2016, 64, 916–924. [Google Scholar] [CrossRef]
- Ayroldi, E.; Petrillo, M.G.; Bastianelli, A.; Marchetti, M.C.; Ronchetti, S.; Nocentini, G.; Ricciotti, L.; Cannarile, L.; Riccardi, C. L-GILZ binds p53 and MDM2 and suppresses tumor growth through p53 activation in human cancer cells. Cell Death Differ. 2014, 22, 118–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayroldi, E.; Petrillo, M.G.; Marchetti, M.C.; Cannarile, L.; Ronchetti, S.; Ricci, E.; Cari, L.; Avenia, N.; Moretti, S.; Puxeddu, E.; et al. Long glucocorticoid-induced leucine zipper regulates human thyroid cancer cell proliferation. Cell Death Dis. 2018, 9, 305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchetti, M.C.; Cannarile, L.; Ronchetti, S.; Delfino, D.V.; Riccardi, C.; Ayroldi, E. L-GILZ binds and inhibits nuclear factor kappaB nuclear translocation in undifferentiated thyroid cancer cells. J. Chemother. 2020, 32, 263–267. [Google Scholar] [CrossRef]
- Ayroldi, E.; Cannarile, L.; Delfino, D.V.; Riccardi, C. A dual role for glucocorticoid-induced leucine zipper in glucocorticoid function: Tumor growth promotion or suppression? Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Lebson, L.; Wang, T.; Jiang, Q.; Whartenby, K.A. Induction of the glucocorticoid-induced leucine zipper gene limits the efficacy of dendritic cell vaccines. Cancer Gene Ther. 2011, 18, 563–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bruscoli, S.; Riccardi, C.; Ronchetti, S. GILZ as a Regulator of Cell Fate and Inflammation. Cells 2022, 11, 122. https://doi.org/10.3390/cells11010122
Bruscoli S, Riccardi C, Ronchetti S. GILZ as a Regulator of Cell Fate and Inflammation. Cells. 2022; 11(1):122. https://doi.org/10.3390/cells11010122
Chicago/Turabian StyleBruscoli, Stefano, Carlo Riccardi, and Simona Ronchetti. 2022. "GILZ as a Regulator of Cell Fate and Inflammation" Cells 11, no. 1: 122. https://doi.org/10.3390/cells11010122
APA StyleBruscoli, S., Riccardi, C., & Ronchetti, S. (2022). GILZ as a Regulator of Cell Fate and Inflammation. Cells, 11(1), 122. https://doi.org/10.3390/cells11010122