
Accelerated Growth, Differentiation, and Ploidy with Reduced Proliferation of Right Ventricular Cardiomyocytes in Children with Congenital Heart Defect Tetralogy of Fallot

 , , , , , , ,
, , , , , , ,
Abstract
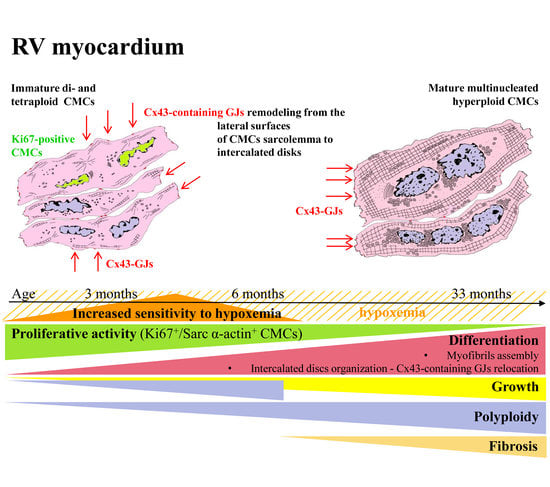
:
1. Introduction
2. Methods
2.1. Patients and Samples
2.2. Ethics Approval
2.3. CMCs Ploidy
2.4. Light Microscopy
2.5. Immunohistochemistry
2.6. Transmission Electron Microscopy (TEM)
2.7. Double Immunofluorescence
2.8. Statistical Analysis
3. Results
3.1. CMCs Ploidy
3.2. Morphometric Assessment of the CMCs Size
3.3. Immunohistochemical Study of the GJ Protein Cx43
3.4. Transmission Electron Microscopy (TEM)
3.5. Morphometric Assessment of the Proportion of Interstitial Tissue
3.6. Immunofluorescent Detection of Proliferating CMCs
4. Discussion
4.1. CMCs Ploidy
4.2. Morphometric Assessment of the Size of the CMCs
4.3. Immunohistochemical Detection of GJs Protein Cx43
4.4. TEM Study of the CMCs
4.5. Morphometric Assessment of Interstitial Tissue
4.6. Immunofluorescent Detection of Proliferating CMCs
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Study Limitations
Abbreviations
| CMC | Cardiomyocyte |
| TF | tetralogy of Fallot |
| RV | Right ventricle |
| LV | Left ventricle |
| GJ | gap junction |
| Cx43 | Connexin 43 |
| CHD | Congenital heart defect |
| TEM | Transmission electron microscopy |
References
- Rauch, R.; Hofbeck, M.; Zweier, C.; Koch, A.; Zink, S.; Trautmann, U.; Hoyer, J.; Kaulitz, R.; Singer, H.; Rauch, A. Comprehensive genotype-phenotype analysis in 230 patients with tetralogy of Fallot. J. Med. Genet. 2010, 47, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Scambler, P.J. The 22q11 deletion syndromes. Hum. Mol. Genet. 2000, 9, 2421–2426. [Google Scholar] [CrossRef] [Green Version]
- van Engelen, K.; Topf, A.; Keavney, B.D.; Goodship, J.A.; van der Velde, E.T.; Baars, M.J.; Snijder, S.; Moorman, A.F.; Postma, A.V.; Mulder, B.J. 22q11.2 Deletion Syndrome is under-recognised in adult patients with tetralogy of Fallot and pulmonary atresia. Heart 2010, 96, 621–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baban, A.; Postma, A.V.; Marini, M.; Trocchio, G.; Santilli, A.; Pelegrini, M.; Sirleto, P.; Lerone, M.; Albanese, S.B.; Barnett, P.; et al. Identification of TBX5 mutations in a series of 94 patients with Tetralogy of Fallot. Am. J. Med. Genet. A 2014, 164A, 3100–3107. [Google Scholar] [CrossRef] [PubMed]
- Mercer-Rosa, L.; Paridon, S.M.; Fogel, M.A.; Rychik, J.; Tanel, R.E.; Zhao, H.; Zhang, X.; Yang, W.; Shults, J.; Goldmuntz, E. 22q11.2 deletion status and disease burden in children and adolescents with tetralogy of Fallot. Circ. Cardiovasc. Genet. 2015, 8, 74–81. [Google Scholar] [CrossRef] [Green Version]
- Goldmuntz, E.; Geiger, E.; Benson, D.W. NKX2.5 mutations in patients with tetralogy of fallot. Circulation 2001, 104, 2565–2568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Lu, Y.; Chen, H.; Yin, M.; Yu, T.; Fu, Q. Investigation of somatic NKX2-5, GATA4 and HAND1 mutations in patients with tetralogy of Fallot. Pathology 2011, 43, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Kheirollahi, M.; Khosravi, F.; Ashouri, S.; Ahmadi, A. Existence of mutations in the homeodomain-encoding region of NKX2.5 gene in Iranian patients with tetralogy of Fallot. J. Res. Med. Sci. 2016, 21, 24. [Google Scholar] [CrossRef]
- Nemer, G.; Fadlalah, F.; Usta, J.; Nemer, M.; Dbaibo, G.; Obeid, M.; Bitar, F. A novel mutation in the GATA4 gene in patients with Tetralogy of Fallot. Hum. Mutat. 2006, 27, 293–294. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.Q.; Gharibeh, L.; Li, R.G.; Xin, Y.F.; Wang, J.; Liu, Z.M.; Qiu, X.B.; Xu, Y.J.; Xu, L.; Qu, X.K.; et al. GATA4 loss-of-function mutations underlie familial tetralogy of fallot. Hum. Mutat. 2013, 34, 1662–1671. [Google Scholar] [CrossRef]
- Wei, D.; Bao, H.; Liu, X.Y.; Zhou, N.; Wang, Q.; Li, R.G.; Xu, Y.J.; Yang, Y.Q. GATA5 loss-of-function mutations underlie tetralogy of fallot. Int. J. Med. Sci. 2013, 10, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Huo, Z.; Liu, X.; Zhang, Y.; Li, L.; Zhao, H.; Yan, B.; Liu, Y.; Yang, Y.; Chen, Y.H. A novel GATA6 mutation in patients with tetralogy of Fallot or atrial septal defect. J. Hum. Genet. 2010, 55, 662–667. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Luo, X.J.; Xin, Y.F.; Liu, Y.; Liu, Z.M.; Wang, Q.; Li, R.G.; Fang, W.Y.; Wang, X.Z.; Yang, Y.Q. Novel GATA6 mutations associated with congenital ventricular septal defect or tetralogy of fallot. DNA Cell Biol. 2012, 31, 1610–1617. [Google Scholar] [CrossRef] [Green Version]
- Bauer, R.C.; Laney, A.O.; Smith, R.; Gerfen, J.; Morrissette, J.J.; Woyciechowski, S.; Garbarini, J.; Loomes, K.M.; Krantz, I.D.; Urban, Z.; et al. Jagged1 (JAG1) mutations in patients with tetralogy of Fallot or pulmonic stenosis. Hum. Mutat. 2010, 31, 594–601. [Google Scholar] [CrossRef] [Green Version]
- Kola, S.; Koneti, N.R.; Golla, J.P.; Akka, J.; Gundimeda, S.D.; Mundluru, H.P. Mutational analysis of JAG1 gene in non-syndromic tetralogy of Fallot children. Clin. Chim. Acta 2011, 412, 2232–2236. [Google Scholar] [CrossRef]
- Ye, L.; Qiu, L.; Feng, B.; Jiang, C.; Huang, Y.; Zhang, H.; Zhang, H.; Hong, H.; Liu, J. Role of Blood Oxygen Saturation During Post-Natal Human Cardiomyocyte Cell Cycle Activities. JACC Basic Transl. Sci. 2020, 5, 447–460. [Google Scholar] [CrossRef]
- Ye, L.; Wang, S.; Xiao, Y.; Jiang, C.; Huang, Y.; Chen, H.; Zhang, H.; Zhang, H.; Liu, J.; Xu, Z.; et al. Pressure Overload Greatly Promotes Neonatal Right Ventricular Cardiomyocyte Proliferation: A New Model for the Study of Heart Regeneration. J. Am. Heart Assoc. 2020, 9, e015574. [Google Scholar] [CrossRef]
- Gu, J.; Chen, X.; Jin, Y.; Liu, M.; Xu, Q.; Liu, X.; Luo, Z.; Ling, S.; Liu, N.; Liu, S. A Neonatal Mouse Model for Pressure Overload: Myocardial Response Corresponds to Severity. Front. Cardiovasc. Med. 2021, 8, 660246. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Sun, S.; Zhu, H.; Xiao, Y.; Jiang, C.; Zhang, H.; Liu, J.; Ye, L.; Shen, J. Volume Overload Initiates an Immune Response in the Right Ventricle at the Neonatal Stage. Front. Cardiovasc. Med. 2021, 8, 772336. [Google Scholar] [CrossRef] [PubMed]
- Burton, P.B.; Raff, M.C.; Kerr, P.; Yacoub, M.H.; Barton, P.J. An intrinsic timer that controls cell-cycle withdrawal in cultured cardiac myocytes. Dev. Biol. 1999, 216, 659–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagahama, H.; Hatakeyama, S.; Nakayama, K.; Nagata, M.; Tomita, K.; Nakayama, K. Spatial and temporal expression patterns of the cyclin-dependent kinase (CDK) inhibitors p27Kip1 and p57Kip2 during mouse development. Anat. Embryol. 2001, 203, 77–87. [Google Scholar] [CrossRef]
- Hinrichsen, R.; Hansen, A.H.; Haunsø, S.; Busk, P.K. Phosphorylation of pRb by cyclin D kinase is necessary for development of cardiac hypertrophy. Cell Prolif. 2008, 41, 813–829. [Google Scholar] [CrossRef]
- Liu, Z.; Yue, S.; Chen, X.; Kubin, T.; Braun, T. Regulation of cardiomyocyte polyploidy and multinucleation by CyclinG1. Circ. Res. 2010, 106, 1498–1506. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, T.S.; Lee, S.T.; Wawrowsky, K.A.; Cheng, K.; Galang, G.; Malliaras, K.; Abraham, M.R.; Wang, C.; Marbán, E. Dedifferentiation and proliferation of mammalian cardiomyocytes. PLoS ONE 2010, 5, e12559. [Google Scholar] [CrossRef] [Green Version]
- Mahmoud, A.I.; Kocabas, F.; Muralidhar, S.A.; Kimura, W.; Koura, A.S.; Thet, S.; Porrello, E.R.; Sadek, H.A. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature 2013, 497, 249–253. [Google Scholar] [CrossRef]
- Ponnusamy, M.; Li, P.F.; Wang, K. Understanding cardiomyocyte proliferation: An insight into cell cycle activity. Cell. Mol. Life Sci. 2017, 74, 1019–1034. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, T.M.A.; Ang, Y.S.; Radzinsky, E.; Zhou, P.; Huang, Y.; Elfenbein, A.; Foley, A.; Magnitsky, S.; Srivastava, D. Regulation of Cell Cycle to Stimulate Adult Cardiomyocyte Proliferation and Cardiac Regeneration. Cell 2018, 173, 104–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Zhang, C.H.; Ammanamanchi, N.; Suresh, S.; Lewarchik, C.; Rao, K.; Uys, G.M.; Han, L.; Abrial, M.; Yimlamai, D.; et al. Control of cytokinesis by β-adrenergic receptors indicates an approach for regulating cardiomyocyte endowment. Sci. Transl. Med. 2019, 11, eaaw6419. [Google Scholar] [CrossRef]
- Jones, M.; Ferrans, V.J.; Morrow, A.G.; Roberts, W.C. Ultrastructure of crista supraventricularis muscle in patients with congenital heart diseases associated with right ventricular outflow tract obstruction. Circulation 1975, 51, 39–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodsky, W.Y.; Arefyeva, A.M.; Uryvaeva, I.V. Mitotic polyploidization of mouse heart myocytes during the first postnatal week. Cell Tissue Res. 1980, 210, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Anversa, P.; Olivetti, G.; Loud, A.V. Morphometric study of early postnatal development in the left and right ventricular myocardium of the rat. I. Hypertrophy, hyperplasia, and binucleation of myocytes. Circ. Res. 1980, 46, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Bogers, A.J.; van der Laarse, A.; Vliegen, H.W.; Quaegebeur, J.M.; Hollaar, L.; Egas, J.M.; Cornelisse, C.J.; Rohmer, J.; Huysmans, H.A. Assessment of hypertrophy in myocardial biopsies taken during correction of congenital heart disease. Thorac. Cardiovasc. Surg. 1988, 36, 137–140. [Google Scholar] [CrossRef] [PubMed]
- van der Laarse, A.; Hollaar, L.; Vliegen, H.W.; Egas, J.M.; Dijkshoorn, N.J.; Cornelisse, C.J.; Bogers, A.J.; Quaegebeur, J.M. Myocardial (iso)enzyme activities, DNA concentration and nuclear polyploidy in hearts of patients operated upon for congenital heart disease, and in normal and hypertrophic adult human hearts at autopsy. Eur. J. Clin. Investig. 1989, 19, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Soonpaa, M.H.; Kim, K.K.; Pajak, L.; Franklin, M.; Field, L.J. Cardiomyocyte DNA synthesis and binucleation during murine development. Am. J. Physiol. 1996, 271, H2183–H2189. [Google Scholar] [CrossRef]
- Li, F.; Wang, X.; Capasso, J.M.; Gerdes, A.M. Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J. Mol. Cell. Cardiol. 1996, 28, 1737–1746. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.; Cottrill, C.M.; O’Connor, W.N.; Salley, R. Right ventricular outflow muscle in tetralogy of Fallot: Histologic and immunohistochemical monoclonal antibody analysis. Cardiovasc. Pathol. 1996, 5, 121–131. [Google Scholar] [CrossRef]
- Egorova, I.F.; Serov, R.A.; Il’in, V.N.; Sharykin, A.S. Morphofunctional analysis of right ventricle cardiomyocytes in patients with Fallot’s tetrad during first year of life. Arkh. Patol. 2001, 63, 36–39. (In Russian) [Google Scholar] [PubMed]
- Kołcz, J.; Drukała, J.; Bzowska, M.; Rajwa, B.; Korohoda, W.; Malec, E. The expression of connexin 43 in children with Tetralogy of Fallot. Cell. Mol. Biol. Lett. 2005, 10, 287–303. [Google Scholar]
- Kołcz, J.; Rajwa, B.; Drukała, J.; Dobrucki, J.; Korohoda, W.; Malec, E. Three-dimensional visualization of connexin 43 on the human cardiomyocytes. Appl. Immunohistochem. Mol. Morphol. 2002, 10, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Shiraishi, I.; Takamatsu, T.; Hamaoka, K. Detection of TUNEL-positive cardiomyocytes and c-kit-positive progenitor cells in children with congenital heart disease. J. Mol. Cell. Cardiol. 2007, 43, 254–261. [Google Scholar] [CrossRef]
- Amir, G.; Ma, X.; Reddy, V.M.; Hanley, F.L.; Reinhartz, O.; Ramamoorthy, C.; Riemer, R.K. Dynamics of human myocardial progenitor cell populations in the neonatal period. Ann. Thorac. Surg. 2008, 86, 1311–1319. [Google Scholar] [CrossRef]
- Farah, M.C.; Castro, C.R.; Moreira, V.M.; Riso, A.; Lopes, A.A.; Aiello, V.D. The myocardium in tetralogy of Fallot: A histological and morphometric study. Arq. Bras. Cardiol. 2009, 92, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Farah, M.C.; Castro, C.R.; Moreira, V.M.; Binotto, M.A.; Guerra, V.C.; Riso, A.; Marcial, M.B.; Lopes, A.A.; Mathias, W., Jr.; Aiello, V.D. The impact of preexisting myocardial remodeling on ventricular function early after tetralogy of Fallot repair. J. Am. Soc. Echocardiogr. 2010, 23, 912–918. [Google Scholar] [CrossRef]
- Walsh, S.; Pontén, A.; Fleischmann, B.K.; Jovinge, S. Cardiomyocyte cell cycle control and growth estimation in vivo--an analysis based on cardiomyocyte nuclei. Cardiovasc. Res. 2010, 86, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Vijayan, K.; Colletti, E.J.; Harrington, D.A.; Matthiesen, T.S.; Simpson, D.; Goh, S.K.; Walker, B.L.; Almeida-Porada, G.; Wang, D.; et al. Characterization and functionality of cardiac progenitor cells in congenital heart patients. Circulation 2011, 123, 364–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, T.F.; Souza, G.K.; Simões, M.A.; Pabis, F.C.; Noronha, L. Immunohistochemical expression of cell differentiation and growth in neonate cardiomyocytes. Arq. Bras. Cardiol. 2012, 99, 797–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, S.R.; Hippenmeyer, S.; Saadat, L.V.; Luo, L.; Weissman, I.L.; Ardehali, R. Existing cardiomyocytes generate cardiomyocytes at a low rate after birth in mice. Proc. Natl. Acad. Sci. USA 2014, 111, 8850–8855. [Google Scholar] [CrossRef] [Green Version]
- Naqvi, N.; Li, M.; Calvert, J.W.; Tejada, T.; Lambert, J.P.; Wu, J.; Kesteven, S.H.; Holman, S.R.; Matsuda, T.; Lovelock, J.D.; et al. A proliferative burst during preadolescence establishes the final cardiomyocyte number. Cell 2014, 157, 795–807. [Google Scholar] [CrossRef] [Green Version]
- Alkass, K.; Panula, J.; Westman, M.; Wu, T.D.; Guerquin-Kern, J.L.; Bergmann, O. No Evidence for Cardiomyocyte Number Expansion in Preadolescent Mice. Cell 2015, 163, 1026–1036. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Qiu, L.; Zhang, H.; Chen, H.; Jiang, C.; Hong, H.; Liu, J. Cardiomyocytes in Young Infants With Congenital Heart Disease: A Three-Month Window of Proliferation. Sci. Rep. 2016, 6, 23188. [Google Scholar] [CrossRef]
- Huang, Y.; Hong, H.; Li, M.; Liu, J.; Jiang, C.; Zhang, H.; Ye, L.; Zheng, J. Age-Dependent Oxidative DNA Damage Does Not Correlate with Reduced Proliferation of Cardiomyocytes in Humans. PLoS ONE 2017, 12, e0170351. [Google Scholar] [CrossRef] [Green Version]
- van Amerongen, M.J.; Engel, F.B. Features of cardiomyocyte proliferation and its potential for cardiac regeneration. J. Cell. Mol. Med. 2008, 12, 2233–2244. [Google Scholar] [CrossRef] [Green Version]
- Paradis, A.N.; Gay, M.S.; Zhang, L. Binucleation of cardiomyocytes: The transition from a proliferative to a terminally differentiated state. Drug Discov. Today 2014, 19, 602–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rumyantsev, P.P. Interrelations of the proliferation and differentiation processes during cardiact myogenesis and regeneration. Int. Rev. Cytol. 1977, 51, 186–273. [Google Scholar]
- Kimura, W.; Xiao, F.; Canseco, D.C.; Muralidhar, S.; Thet, S.; Zhang, H.M.; Abderrahman, Y.; Chen, R.; Garcia, J.A.; Shelton, J.M.; et al. Hypoxia fate mapping identifies cycling cardiomyocytes in the adult heart. Nature 2015, 523, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Patterson, M.; Barske, L.; Van Handel, B.; Rau, C.D.; Gan, P.; Sharma, A.; Parikh, S.; Denholtz, M.; Huang, Y.; Yamaguchi, Y.; et al. Frequency of mononuclear diploid cardiomyocytes underlies natural variation in heart regeneration. Nat. Genet. 2017, 49, 1346–1353. [Google Scholar] [CrossRef]
- Leone, M.; Engel, F.B. Pseudo-bipolar spindle formation and cell division in postnatal binucleated cardiomyocytes. J. Mol. Cell. Cardiol. 2019, 134, 69–73. [Google Scholar] [CrossRef]
- Han, L.; Choudhury, S.; Mich-Basso, J.D.; Ammanamanchi, N.; Ganapathy, B.; Suresh, S.; Khaladkar, M.; Singh, J.; Maehr, R.; Zuppo, D.A.; et al. Lamin B2 Levels Regulate Polyploidization of Cardiomyocyte Nuclei and Myocardial Regeneration. Dev. Cell. 2020, 53, 42–59. [Google Scholar] [CrossRef] [PubMed]
- Hesse, M.; Bednarz, R.; Carls, E.; Becker, C.; Bondareva, O.; Lother, A.; Geisen, C.; Dreßen, M.; Krane, M.; Roell, W.; et al. Proximity to injury, but neither number of nuclei nor ploidy define pathological adaptation and plasticity in cardiomyocytes. J. Mol. Cell. Cardiol. 2021, 152, 95–104. [Google Scholar] [CrossRef]
- Yester, J.W.; Liu, H.; Gyngard, F.; Ammanamanchi, N.; Little, K.C.; Thomas, D.; Sullivan, M.L.G.; Lal, S.; Steinhauser, M.L.; Kühn, B. Use of stable isotope-tagged thymidine and multi-isotope imaging mass spectrometry (MIMS) for quantification of human cardiomyocyte division. Nat. Protoc. 2021, 16, 1995–2022. [Google Scholar] [CrossRef]
- Bergmann, O. Cardiomyocytes in congenital heart disease: Overcoming cytokinesis failure in tetralogy of Fallot. J. Thorac. Cardiovasc. Surg. 2021, 161, 1587–1590. [Google Scholar] [CrossRef]
- González-Rosa, J.M.; Sharpe, M.; Field, D.; Soonpaa, M.H.; Field, L.J.; Burns, C.E.; Burns, C.G. Myocardial Polyploidization Creates a Barrier to Heart Regeneration in Zebrafish. Dev. Cell 2018, 44, 433–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, R.S.; Richart, R.M. Nuclear DNA content of the normal and hypertrophied myocardium in the pediatric age group. J. Pediatr. 1973, 83, 445–450. [Google Scholar] [CrossRef]
- Kawai, S.; Okada, R.; Kitamura, K.; Suzuki, A.; Saito, S. A morphometrical study of myocardial disarray associated with right ventricular outflow tract obstruction. Jpn. Circ. J. 1984, 48, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Mitsuno, M.; Nakano, S.; Shimazaki, Y.; Taniguchi, K.; Kawamoto, T.; Kobayashi, J.; Matsuda, H.; Kawashima, Y. Fate of right ventricular hypertrophy in tetralogy of Fallot after corrective surgery. Am. J. Cardiol. 1993, 72, 694–698. [Google Scholar] [CrossRef]
- Schwartz, S.M.; Gordon, D.; Mosca, R.S.; Bove, E.L.; Heidelberger, K.P.; Kulik, T.J. Collagen content in normal, pressure, and pressure-volume overloaded developing human hearts. Am. J. Cardiol. 1996, 77, 734–738. [Google Scholar] [CrossRef]
- Kuruvilla, S.; Balakrishnan, K.; Parvathy, U. Right ventricular myocardium in Fallot’s tetralogy: A light microscopic, morphometric and ultrastructural study. Images Paediatr. Cardiol. 2004, 6, 1–30. [Google Scholar]
- Chowdhury, U.K.; Sathia, S.; Ray, R.; Singh, R.; Pradeep, K.K.; Venugopal, P. Histopathology of the right ventricular outflow tract and its relationship to clinical outcomes and arrhythmias in patients with tetralogy of Fallot. J. Thorac. Cardiovasc. Surg. 2006, 132, 270–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, S.; Osorio, J.C.; Duque, A.M.; Kaufman, B.D.; Phillips, A.B.; Chen, J.M.; Quaegebeur, J.; Mosca, R.S.; Mital, S. Failure of right ventricular adaptation in children with tetralogy of Fallot. Circulation 2006, 114. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Li, Y.; Cheng, T.O.; Wang, X.; Dong, N.; Nie, X.; Lu, Q.; Yang, Y.; He, L.; Li, L.; et al. The effect of right ventricular myocardial remodeling on ventricular function as assessed by two-dimensional speckle tracking echocardiography in patients with tetralogy of Fallot: A single center experience from China. Int. J. Cardiol. 2015, 178, 300–307. [Google Scholar] [CrossRef]
- Alpat, S.; Yilmaz, M.; Onder, S.; Sargon, M.F.; Guvener, M.; Dogan, R.; Demircin, M.; Pasaoglu, I. Histologic alterations in tetralogy of Fallot. J. Card Surg. 2017, 32, 38–44. [Google Scholar] [CrossRef] [Green Version]
- Pradegan, N.; Vida, V.L.; Geva, T.; Stellin, G.; White, M.T.; Sanders, S.P.; Padera, R.F. Myocardial histopathology in late-repaired and unrepaired adults with tetralogy of Fallot. Cardiovasc. Pathol. 2016, 25, 225–231. [Google Scholar] [CrossRef]
- Yekelchyk, M.; Guenther, S.; Preussner, J.; Braun, T. Mono- and multi-nucleated ventricular cardiomyocytes constitute a transcriptionally homogenous cell population. Basic Res. Cardiol. 2019, 114, 36. [Google Scholar] [CrossRef] [Green Version]
- Kellerman, S.; Moore, J.A.; Zierhut, W.; Zimmer, H.G.; Campbell, J.; Gerdes, A.M. Nuclear DNA content and nucleation patterns in rat cardiac myocytes from different models of cardiac hypertrophy. J. Mol. Cell. Cardiol. 1992, 24, 497–505. [Google Scholar] [CrossRef]
- Yücel, D.; Solinsky, J.; van Berlo, J.H. Isolation of Cardiomyocytes from Fixed Hearts for Immunocytochemistry and Ploidy Analysis. J. Vis. Exp. 2020, 164, e60938. [Google Scholar] [CrossRef]
- Kuwamura, Y.; Shono, M.; Hizawa, K. Cytofluorometric determination of nuclear DNA in heart muscles of patients with muscular dystrophy. Acta Pathol. Jpn. 1989, 39, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Mayhew, T.M.; Pharaoh, A.; Austin, A.; Fagan, D.G. Stereological estimates of nuclear number in human ventricular cardiomyocytes before and after birth obtained using physical disectors. J. Anat. 1997, 191, 107–115. [Google Scholar] [CrossRef]
- Takamatsu, T.; Nakanishi, K.; Fukuda, M.; Fujita, S. Cytofluorometric nuclear DNA-determinations in infant, adolescent, adult and aging human hearts. Histochemistry 1983, 77, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, V.Y.; Sarkisov, D.S.; Arefyeva, A.M.; Panova, N.W.; Gvasava, I.G. Polyploidy in cardiac myocytes of normal and hypertrophic human hearts; range of values. Virchows Arch. 1994, 424, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Adler, C.P.; Costabel, U. Cell number in human heart in atrophy, hypertrophy, and under the influence of cytostatics. Recent Adv. Stud. Cardiac. Struct. Metab. 1975, 6, 343–355. [Google Scholar] [PubMed]
- Mollova, M.; Bersell, K.; Walsh, S.; Savla, J.; Das, L.T.; Park, S.Y.; Silberstein, L.E.; Dos Remedios, C.G.; Graham, D.; Colan, S.; et al. Cardiomyocyte proliferation contributes to heart growth in young humans. Proc. Natl. Acad. Sci. USA 2013, 110, 1446–1451. [Google Scholar] [CrossRef] [Green Version]
- Adler, C.P.; Friedburg, H.; Herget, G.W.; Neuburger, M.; Schwalb, H. Variability of cardiomyocyte DNA content, ploidy level and nuclear number in mammalian hearts. Virchows Arch. 1996, 429, 159–164. [Google Scholar] [CrossRef]
- Schmid, G.; Pfitzer, P. Mitoses and binucleated cells in perinatal human hearts. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1985, 48, 59–67. [Google Scholar] [CrossRef]
- El Khoudary, S.R.; Fabio, A.; Yester, J.W.; Steinhauser, M.L.; Christopher, A.B.; Gyngard, F.; Adams, P.S.; Morell, V.O.; Viegas, M.; Da Silva, J.P.; et al. Design and rationale of a clinical trial to increase cardiomyocyte division in infants with tetralogy of Fallot. Int. J. Cardiol. 2021, 339, 36–42. [Google Scholar] [CrossRef]
- Gan, P.; Patterson, M.; Velasquez, A.; Wang, K.; Tian, D.; Windle, J.J.; Tao, G.; Judge, D.P.; Makita, T.; Park, T.J.; et al. Tnni3k alleles influence ventricular mononuclear diploid cardiomyocyte frequency. PLoS Genet. 2019, 15, e1008354. [Google Scholar] [CrossRef] [PubMed]
- Peters, N.S.; Severs, N.J.; Rothery, S.M.; Lincoln, C.; Yacoub, M.H.; Green, C.R. Spatiotemporal relation between gap junctions and fascia adherens junctions during postnatal development of human ventricular myocardium. Circulation 1994, 90, 713–725. [Google Scholar] [CrossRef] [Green Version]
- Salameh, A.; Haunschild, J.; Bräuchle, P.; Peim, O.; Seidel, T.; Reitmann, M.; Kostelka, M.; Bakhtiary, F.; Dhein, S.; Dähnert, I. On the role of the gap junction protein Cx43 (GJA1) in human cardiac malformations with Fallot-pathology. a study on paediatric cardiac specimen. PLoS ONE 2014, 9, e95344. [Google Scholar] [CrossRef] [PubMed]
- Sakashita, I.; Aoki, E.; Matsukawa, T.; Asano, K.I. Morphological and histochemical observations on right ventricular outflow tract of tetralogy of Fallot. With a brief reference to hemdynamic viewpoint I. Jpn. Heart J. 1969, 10, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Kajihara, H.; Taguchi, K.; Hara, H.H.; Iijima, S. Electron microscopic observation of human hypertrophied myocardium. Acta Pathol. Jpn. 1973, 23, 335–347. [Google Scholar] [CrossRef]
- Maron, B.J.; Ferrans, V.J.; Roberts, W.C. Ultrastructural features of degenerated cardiac muscle cells in patients with cardiac hypertrophy. Am. J. Pathol. 1975, 79, 387–434. [Google Scholar]
- Thiedemann, K.U.; Ferrans, V.J. Left atrial ultrastructure in mitral valvular disease. Am. J. Pathol. 1977, 89, 575–604. [Google Scholar] [CrossRef]
- Schaper, J.; Froede, R.; Hein, S.; Buck, A.; Hashizume, H.; Speiser, B.; Friedl, A.; Bleese, N. Impairment of the myocardial ultrastructure and changes of the cytoskeleton in dilated cardiomyopathy. Circulation 1991, 83, 504–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaper, J. Effects of multiple ischaemic events on human myocardium--an ultrastructural study. Eur. Heart J. 1988, 9, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, H.; Terasaki, F.; Hayashi, T.; Kitaura, Y.; Isomura, T.; Suma, H. Autophagic degeneration as a possible mechanism of myocardial cell death in dilated cardiomyopathy. Jpn. Circ. J. 2001, 65, 965–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hein, S.; Arnon, E.; Kostin, S.; Schönburg, M.; Elsässer, A.; Polyakova, V.; Bauer, E.P.; Klövekorn, W.P.; Schaper, J. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: Structural deterioration and compensatory mechanisms. Circulation 2003, 107, 984–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsässer, A.; Vogt, A.M.; Nef, H.; Kostin, S.; Möllmann, H.; Skwara, W.; Bode, C.; Hamm, C.; Schaper, J. Human hibernating myocardium is jeopardized by apoptotic and autophagic cell death. J. Am. Coll. Cardiol. 2004, 43, 2191–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takemura, G.; Miyata, S.; Kawase, Y.; Okada, H.; Maruyama, R.; Fujiwara, H. Autophagic degeneration and death of cardiomyocytes in heart failure. Autophagy 2006, 2, 212–214. [Google Scholar] [CrossRef] [Green Version]
- Fidziańska, A.; Bilińska, Z.T.; Walczak, E.; Witkowski, A.; Chojnowska, L. Autophagy in transition from hypertrophic cardiomyopathy to heart failure. J. Electron. Microsc. 2010, 59, 181–183. [Google Scholar] [CrossRef]
- Saito, T.; Asai, K.; Sato, S.; Hayashi, M.; Adachi, A.; Sasaki, Y.; Takano, H.; Mizuno, K.; Shimizu, W. Autophagic vacuoles in cardiomyocytes of dilated cardiomyopathy with initially decompensated heart failure predict improved prognosis. Autophagy 2016, 12, 579–587. [Google Scholar] [CrossRef]
- Kassiotis, C.; Ballal, K.; Wellnitz, K.; Vela, D.; Gong, M.; Salazar, R.; Frazier, O.H.; Taegtmeyer, H. Markers of autophagy are downregulated in failing human heart after mechanical unloading. Circulation 2009, 120, S191–S197. [Google Scholar] [CrossRef] [Green Version]
- Nakai, A.; Yamaguchi, O.; Takeda, T.; Higuchi, Y.; Hikoso, S.; Taniike, M.; Omiya, S.; Mizote, I.; Matsumura, Y.; Asahi, M.; et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007, 13, 619–624. [Google Scholar] [CrossRef]
- Macmahon, H.E. Hyperplasia and Regeneration of the Myocardium in Infants and in Children. Am. J. Pathol. 1937, 13, 845–854. [Google Scholar]
- Urbanek, K.; Quaini, F.; Tasca, G.; Torella, D.; Castaldo, C.; Nadal-Ginard, B.; Leri, A.; Kajstura, J.; Quaini, E.; Anversa, P. Intense myocyte formation from cardiac stem cells in human cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2003, 100, 10440–10445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quaini, F.; Cigola, E.; Lagrasta, C.; Saccani, G.; Quaini, E.; Rossi, C.; Olivetti, G.; Anversa, P. End-stage cardiac failure in humans is coupled with the induction of proliferating cell nuclear antigen and nuclear mitotic division in ventricular myocytes. Circ. Res. 1994, 75, 1050–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltrami, A.P.; Urbanek, K.; Kajstura, J.; Yan, S.M.; Finato, N.; Bussani, R.; Nadal-Ginard, B.; Silvestri, F.; Leri, A.; Beltrami, C.A.; et al. Evidence that human cardiac myocytes divide after myocardial infarction. N. Engl. J. Med. 2001, 344, 1750–1757. [Google Scholar] [CrossRef]
- Chimenti, C.; Kajstura, J.; Torella, D.; Urbanek, K.; Heleniak, H.; Colussi, C.; Di Meglio, F.; Nadal-Ginard, B.; Frustaci, A.; Leri, A.; et al. Senescence and death of primitive cells and myocytes lead to premature cardiac aging and heart failure. Circ. Res. 2003, 93, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Sukhacheva, T.V.; Chudinovskikh, Y.A.; Eremeeva, M.V.; Serov, R.A.; Bockeria, L.A. Proliferative Potential of Cardiomyocytes in Hypertrophic Cardiomyopathy: Correlation with Myocardial Remodeling. Bull. Exp. Biol. Med. 2016, 162, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, V.Y.; Uryvaeva, I.V. Genome Multiplication in Growth and Development. Biology of Polyploid and Polytene Cells. Developmental and Cell Biology; Cambridge University Press: Cambridge, MA, USA, 1985; 305p. [Google Scholar]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Hill, J.A.; Richardson, J.A.; Olson, E.N.; Sadek, H.A. Transient regenerative potential of the neonatal mouse heart. Science 2011, 331, 1078–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haubner, B.J.; Adamowicz-Brice, M.; Khadayate, S.; Tiefenthaler, V.; Metzler, B.; Aitman, T.; Penninger, J.M. Complete cardiac regeneration in a mouse model of myocardial infarction. Aging 2012, 4, 966–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aurora, A.B.; Porrello, E.R.; Tan, W.; Mahmoud, A.I.; Hill, J.A.; Bassel-Duby, R.; Sadek, H.A.; Olson, E.N. Macrophages are required for neonatal heart regeneration. J. Clin. Investig. 2014, 124, 1382–1392. [Google Scholar] [CrossRef] [Green Version]
- Lavine, K.J.; Epelman, S.; Uchida, K.; Weber, K.J.; Nichols, C.G.; Schilling, J.D.; Ornitz, D.M.; Randolph, G.J.; Mann, D.L. Distinct macrophage lineages contribute to.o disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc. Natl. Acad. Sci. USA 2014, 111, 16029–16034. [Google Scholar] [CrossRef] [Green Version]
- Sampaio-Pinto, V.; Rodrigues, S.C.; Laundos, T.L.; Silva, E.D.; Vasques-Nóvoa, F.; Silva, A.C.; Cerqueira, R.J.; Resende, T.P.; Pianca, N.; Leite-Moreira, A.; et al. Neonatal Apex Resection Triggers Cardiomyocyte Proliferation, Neovascularization and Functional Recovery Despite Local Fibrosis. Stem Cell Rep. 2018, 10, 860–874. [Google Scholar] [CrossRef]
- Zebrowski, D.C.; Jensen, C.H.; Becker, R.; Ferrazzi, F.; Baun, C.; Hvidsten, S.; Sheikh, S.P.; Polizzotti, B.D.; Andersen, D.C.; Engel, F.B. Cardiac injury of the newborn mammalian heart accelerates cardiomyocyte terminal differentiation. Sci. Rep. 2017, 7, 8362. [Google Scholar] [CrossRef] [Green Version]
- Sedmera, D.; Thompson, R.P.; Kolar, F. Effect of increased pressure loading on heart growth in neonatal rats. J. Mol. Cell. Cardiol. 2003, 35, 301–309. [Google Scholar] [CrossRef]
- Neffgen, J.F.; Korecky, B. Cellular hyperplasia and hypertrophy in cardiomegalies induced by anemia in young and adult rats. Circ. Res. 1972, 30, 104–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivetti, G.; Quaini, F.; Lagrasta, C.; Ricci, R.; Tiberti, G.; Capasso, J.M.; Anversa, P. Myocyte cellular hypertrophy and hyperplasia contribute to ventricular wall remodeling in anemia-induced cardiac hypertrophy in rats. Am. J. Pathol. 1992, 141, 227–239. [Google Scholar] [PubMed]
- Soonpaa, M.H.; Field, L.J. Assessment of cardiomyocyte DNA synthesis in normal and injured adult mouse hearts. Am. J. Physiol. 1997, 272, H220–H226. [Google Scholar] [CrossRef]
- Guski, H.; Kranz, D. The effect of exercise on cell proliferation in the non-ischemic heart after myocardial infarction. Exp. Pathol. 1982, 21, 54–58. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Borges, A.; Liew, C.C. Telomerase activity during cardiac development. J. Mol. Cell. Cardiol. 1997, 29, 2717–2724. [Google Scholar] [CrossRef]
- Aix, E.; Gutiérrez-Gutiérrez, Ó.; Sánchez-Ferrer, C.; Aguado, T.; Flores, I. Postnatal telomere dysfunction induces cardiomyocyte cell-cycle arrest through p21 activation. J. Cell Biol. 2016, 213, 571–583. [Google Scholar] [CrossRef] [Green Version]
- Richardson, G.D.; Breault, D.; Horrocks, G.; Cormack, S.; Hole, N.; Owens, W.A. Telomerase expression in the mammalian heart. FASEB J. 2012, 26, 4832–4840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, H.; Taffet, G.E.; Youker, K.A.; Entman, M.L.; Overbeek, P.A.; Michael, L.H.; Schneider, M.D. Telomerase reverse transcriptase promotes cardiac muscle cell proliferation, hypertrophy, and survival. Proc. Natl. Acad. Sci. USA 2001, 98, 10308–10313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campa, V.M.; Gutiérrez-Lanza, R.; Cerignoli, F.; Díaz-Trelles, R.; Nelson, B.; Tsuji, T.; Barcova, M.; Jiang, W.; Mercola, M. Notch activates cell cycle reentry and progression in quiescent cardiomyocytes. J. Cell Biol. 2008, 183, 129–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monroe, T.O.; Hill, M.C.; Morikawa, Y.; Leach, J.P.; Heallen, T.; Cao, S.; Krijger, P.H.L.; de Laat, W.; Wehrens, X.H.T.; Rodney, G.G.; et al. YAP Partially Reprograms Chromatin Accessibility to Directly Induce Adult Cardiogenesis In Vivo. Dev. Cell 2019, 48, 765–779.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilsbach, R.; Preissl, S.; Grüning, B.A.; Schnick, T.; Burger, L.; Benes, V.; Würch, A.; Bönisch, U.; Günther, S.; Backofen, R.; et al. Dynamic DNA methylation orchestrates cardiomyocyte development, maturation and disease. Nat. Commun. 2014, 5, 5288. [Google Scholar] [CrossRef] [Green Version]
- Rommel, C.; Hein, L. Four Dimensions of the Cardiac Myocyte Epigenome: From Fetal to Adult Heart. Curr. Cardiol. Rep. 2020, 22, 26. [Google Scholar] [CrossRef] [Green Version]
- Gilsbach, R.; Schwaderer, M.; Preissl, S.; Grüning, B.A.; Kranzhöfer, D.; Schneider, P.; Nührenberg, T.G.; Mulero-Navarro, S.; Weichenhan, D.; Braun, C.; et al. Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat. Commun. 2018, 9, 391. [Google Scholar] [CrossRef] [Green Version]
- Sheng, W.; Wang, H.; Ma, X.; Qian, Y.; Zhang, P.; Wu, Y.; Zheng, F.; Chen, L.; Huang, G.; Ma, D. LINE-1 methylation status and its association with tetralogy of fallot in infants. BMC Med. Genom. 2012, 5, 20. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Kong, Q.; Li, Z.; Xu, M.; Cai, Z.; Zhao, C. Association between the promoter methylation of the TBX20 gene and tetralogy of fallot. Scand. Cardiovasc. J. 2018, 52, 287–291. [Google Scholar] [CrossRef]
- Gong, J.; Sheng, W.; Ma, D.; Huang, G.; Liu, F. DNA methylation status of TBX20 in patients with tetralogy of Fallot. BMC Med. Genom. 2019, 12, 75. [Google Scholar] [CrossRef] [Green Version]
- Oliveira-Carvalho, V.; da Silva, M.M.; Guimarães, G.V.; Bacal, F.; Bocchi, E.A. MicroRNAs: New players in heart failure. Mol. Biol. Rep. 2013, 40, 2663–2670. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Liu, H.; Cretoiu, D.; Toader, D.O.; Suciu, N.; Shi, J.; Shen, S.; Bei, Y.; Sluijter, J.P.; Das, S.; et al. miR-31a-5p promotes postnatal cardiomyocyte proliferation by targeting RhoBTB1. Exp. Mol. Med. 2017, 49, e386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakao, K.; Minobe, W.; Roden, R.; Bristow, M.R.; Leinwand, L.A. Myosin heavy chain gene expression in human heart failure. J. Clin. Investig. 1997, 100, 2362–2370. [Google Scholar] [CrossRef]
- Liu, X.; Pu, W.; He, L.; Li, Y.; Zhao, H.; Li, Y.; Liu, K.; Huang, X.; Weng, W.; Wang, Q.D.; et al. Cell proliferation fate mapping reveals regional cardiomyocyte cell-cycle activity in subendocardial muscle of left ventricle. Nat. Commun. 2021, 12, 5784. [Google Scholar] [CrossRef]
- Nakada, Y.; Canseco, D.C.; Thet, S.; Abdisalaam, S.; Asaithamby, A.; Santos, C.X.; Shah, A.M.; Zhang, H.; Faber, J.E.; Kinter, M.T.; et al. Hypoxia induces heart regeneration in adult mice. Nature 2017, 541, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Dowell, R.T.; McManus, R.E., 3rd. Pressure-induced cardiac enlargement in neonatal and adult rats. Left ventricular functional characteristics and evidence of cardiac muscle cell proliferation in the neonate. Circ. Res. 1978, 42, 303–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberpriller, J.O.; Ferrans, V.J.; Carroll, R.J. DNA synthesis in rat atrial myocytes as a response to left ventricular infarction. An autoradiographic study of enzymatically dissociated myocytes. J. Mol. Cell. Cardiol. 1984, 16, 1119–1126. [Google Scholar] [CrossRef]
- Kajstura, J.; Zhang, X.; Reiss, K.; Szoke, E.; Li, P.; Lagrasta, C.; Cheng, W.; Darzynkiewicz, Z.; Olivetti, G.; Anversa, P. Myocyt Liu X e cellular hyperplasia and myocyte cellular hypertrophy contribute to chronic ventricular remodeling in coronary artery narrowing-induced cardiomyopathy in rats. Circ. Res. 1994, 74, 383–400. [Google Scholar] [CrossRef] [Green Version]
- Unno, K.; Oikonomopoulos, A.; Fujikawa, Y.; Okuno, Y.; Narita, S.; Kato, T.; Hayashida, R.; Kondo, K.; Shibata, R.; Murohara, T.; et al. Alteration in ventricular pressure stimulates cardiac repair and remodeling. J. Mol. Cell. Cardiol. 2019, 133, 174–187. [Google Scholar] [CrossRef]
- Jonker, S.S.; Giraud, M.K.; Giraud, G.D.; Chattergoon, N.N.; Louey, S.; Davis, L.E.; Faber, J.J.; Thornburg, K.L. Cardiomyocyte enlargement, proliferation and maturation during chronic fetal anaemia in sheep. Exp. Physiol. 2010, 95, 131–139. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Parameters | M ± SD (Min–Max) |
|---|---|
| Age, months (min–max) | 8.4 ± 4.4 (3.0–33.5) |
| Left ventricular ejection fraction (LVFE), % | 68.7 ± 2.6 (62–79) |
| The end-diastolic volume of the left ventricle, mL | 45.7 ± 8.8 (33–70) |
| The systolic pressure gradient between the RV and pulmonary artery, mm Hg | 85.9 ± 9.8 (68–110) |
| Sat O2, % | 91.9 ± 11.5 (40–101.7) |
| Hemoglobin, g/L | 133.1 ± 16.1 (100–162) |
| Hematocrit, % | 39.2 ± 4,4 (30–49.5) |
| Nakata index, mm/m2 | 375.2 ± 105.3 (152.6–696) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sukhacheva, T.V.; Serov, R.A.; Nizyaeva, N.V.; Burov, A.A.; Pavlovich, S.V.; Podurovskaya, Y.L.; Samsonova, M.V.; Chernyaev, A.L.; Shchegolev, A.I.; Kim, A.I.; et al. Accelerated Growth, Differentiation, and Ploidy with Reduced Proliferation of Right Ventricular Cardiomyocytes in Children with Congenital Heart Defect Tetralogy of Fallot. Cells 2022, 11, 175. https://doi.org/10.3390/cells11010175
Sukhacheva TV, Serov RA, Nizyaeva NV, Burov AA, Pavlovich SV, Podurovskaya YL, Samsonova MV, Chernyaev AL, Shchegolev AI, Kim AI, et al. Accelerated Growth, Differentiation, and Ploidy with Reduced Proliferation of Right Ventricular Cardiomyocytes in Children with Congenital Heart Defect Tetralogy of Fallot. Cells. 2022; 11(1):175. https://doi.org/10.3390/cells11010175
Chicago/Turabian StyleSukhacheva, Tatyana V., Roman A. Serov, Natalia V. Nizyaeva, Artem A. Burov, Stanislav V. Pavlovich, Yulia L. Podurovskaya, Maria V. Samsonova, Andrey L. Chernyaev, Aleksandr I. Shchegolev, Alexei I. Kim, and et al. 2022. "Accelerated Growth, Differentiation, and Ploidy with Reduced Proliferation of Right Ventricular Cardiomyocytes in Children with Congenital Heart Defect Tetralogy of Fallot" Cells 11, no. 1: 175. https://doi.org/10.3390/cells11010175
APA StyleSukhacheva, T. V., Serov, R. A., Nizyaeva, N. V., Burov, A. A., Pavlovich, S. V., Podurovskaya, Y. L., Samsonova, M. V., Chernyaev, A. L., Shchegolev, A. I., Kim, A. I., Bockeria, L. A., & Sukhikh, G. T. (2022). Accelerated Growth, Differentiation, and Ploidy with Reduced Proliferation of Right Ventricular Cardiomyocytes in Children with Congenital Heart Defect Tetralogy of Fallot. Cells, 11(1), 175. https://doi.org/10.3390/cells11010175