
Role of Human Antigen R (HuR) in the Regulation of Pulmonary ACE2 Expression


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Single Cell RNA-Sequencing Analysis
2.3. COPD Subjects
2.4. Multiplex Immunohistochemistry (mIHC)
2.5. Cell Culture
2.6. Western Blot
2.7. Quantitative RT-PCR (RT-qPCR)
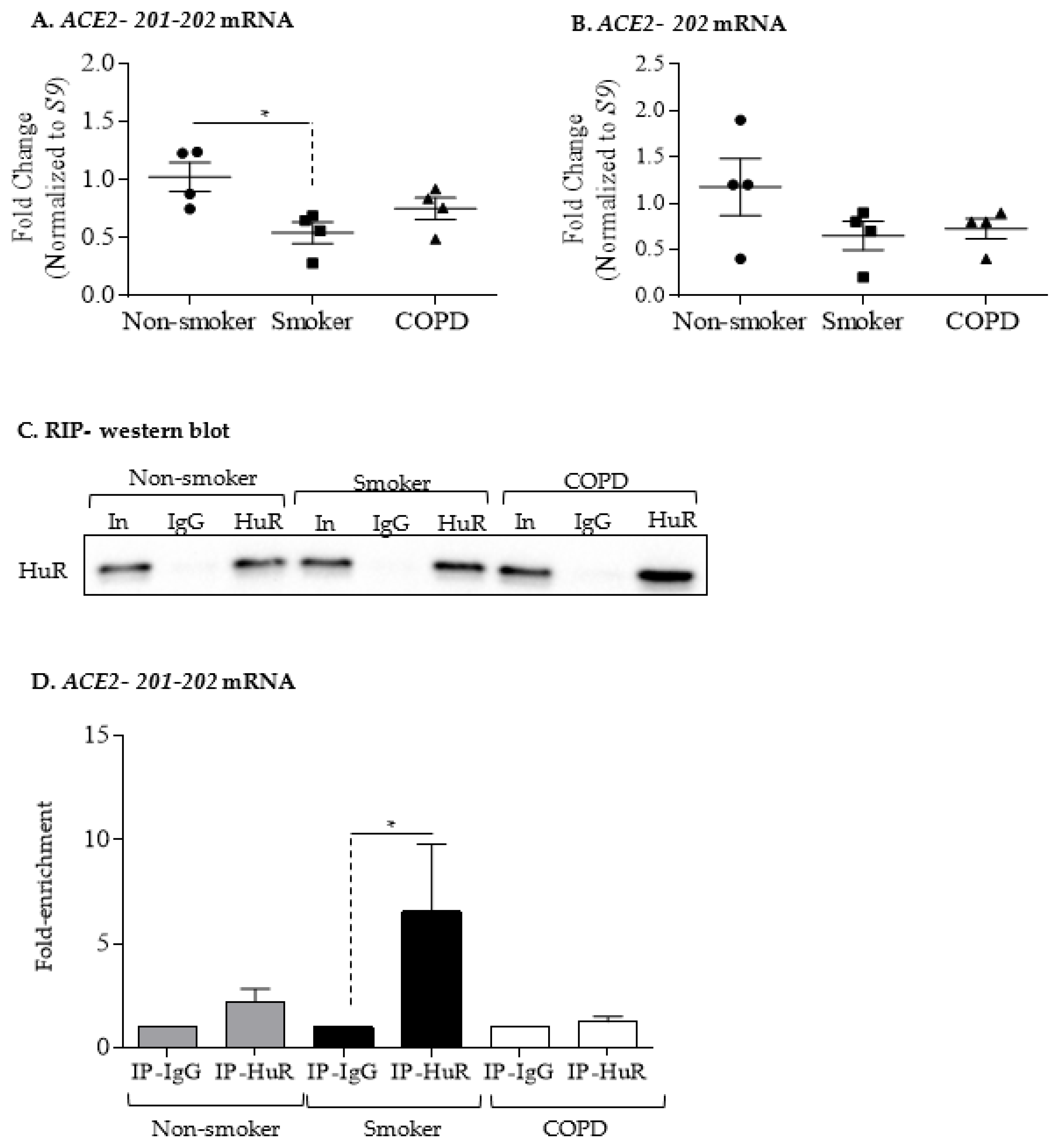
2.8. RNA Immunoprecipitation-qPCR (RIP-qPCR)
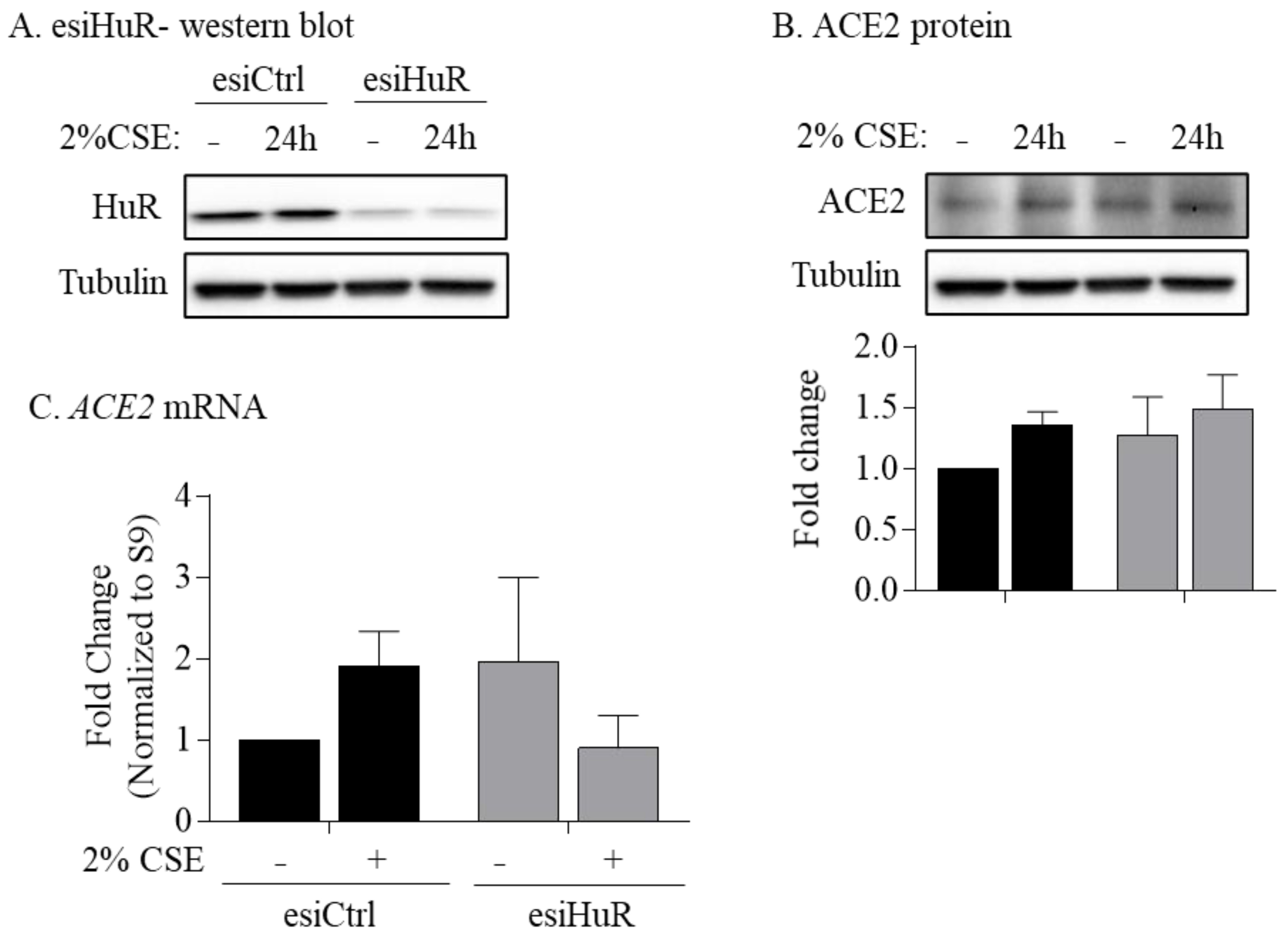
2.9. HuR-esiRNA Knockdown in Human Lung Fibroblasts
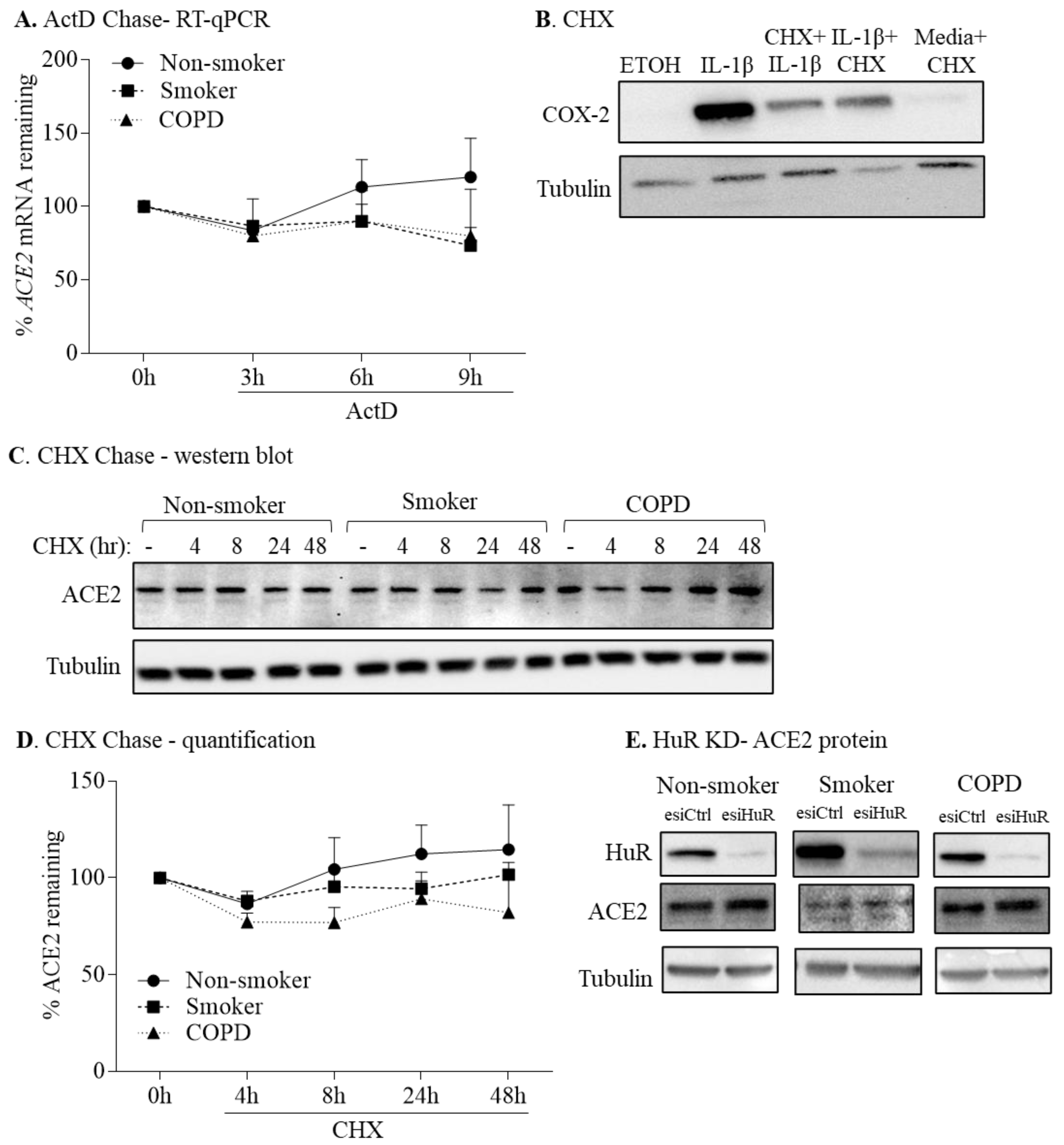
2.10. Determination of mRNA Stability
2.11. Determination of Protein Stability
2.12. Preparation of Cigarette Smoke Extract (CSE)
2.13. Imaging Flow Cytometry
2.14. Statistical Analysis
3. Results
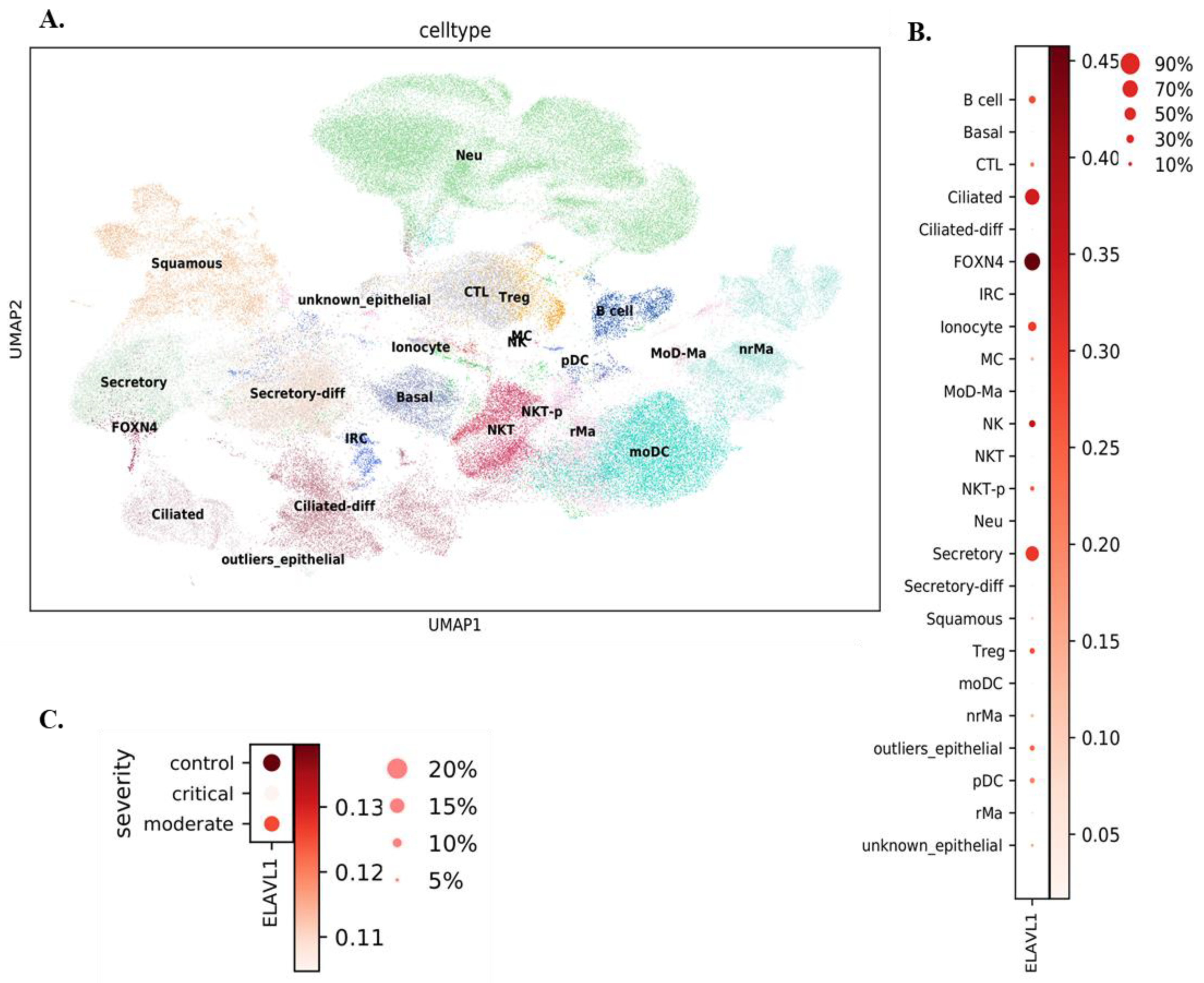
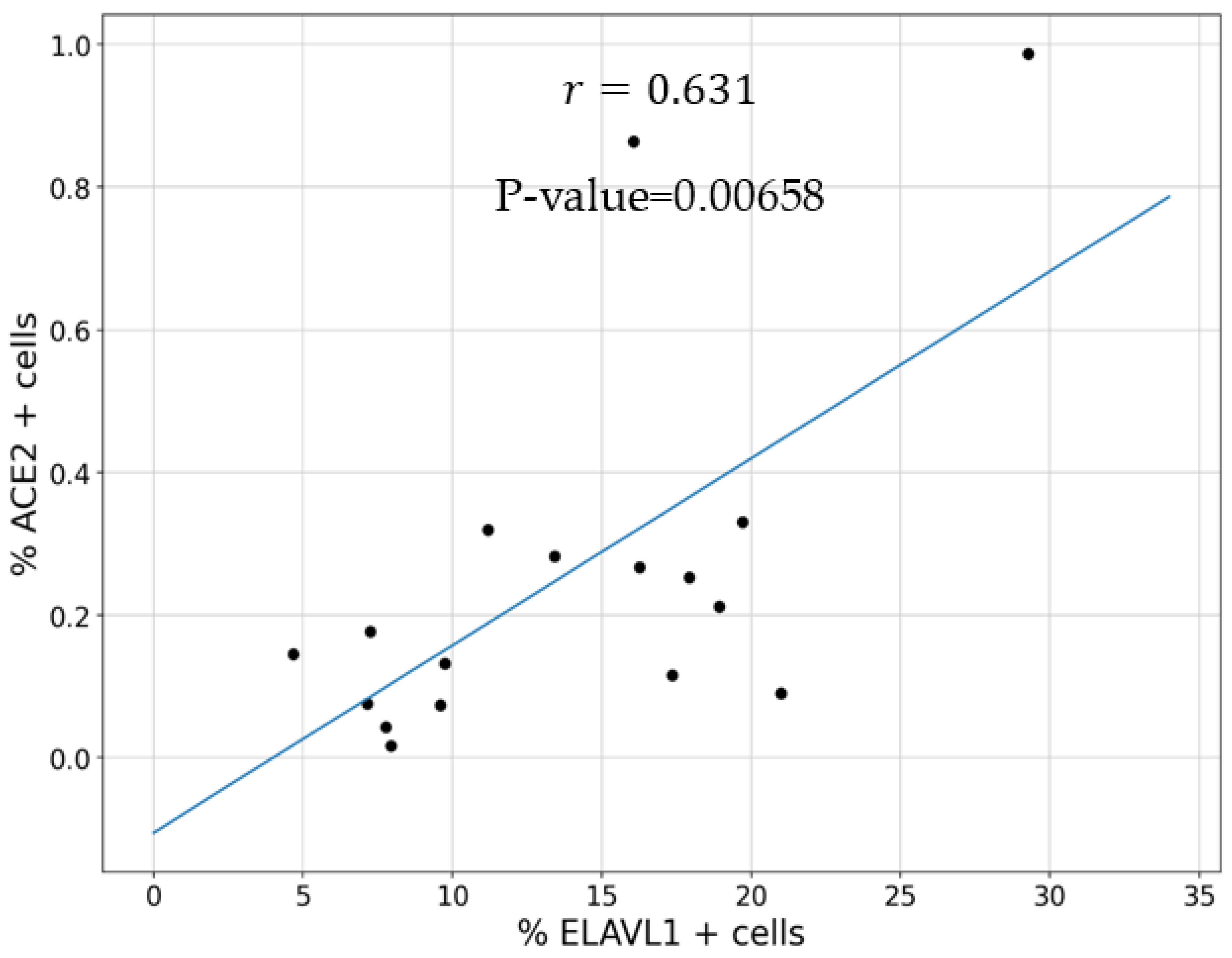
3.1. Expression of ELAVL1 and ACE2 in Cells of the Upper and Lower Respiratory System
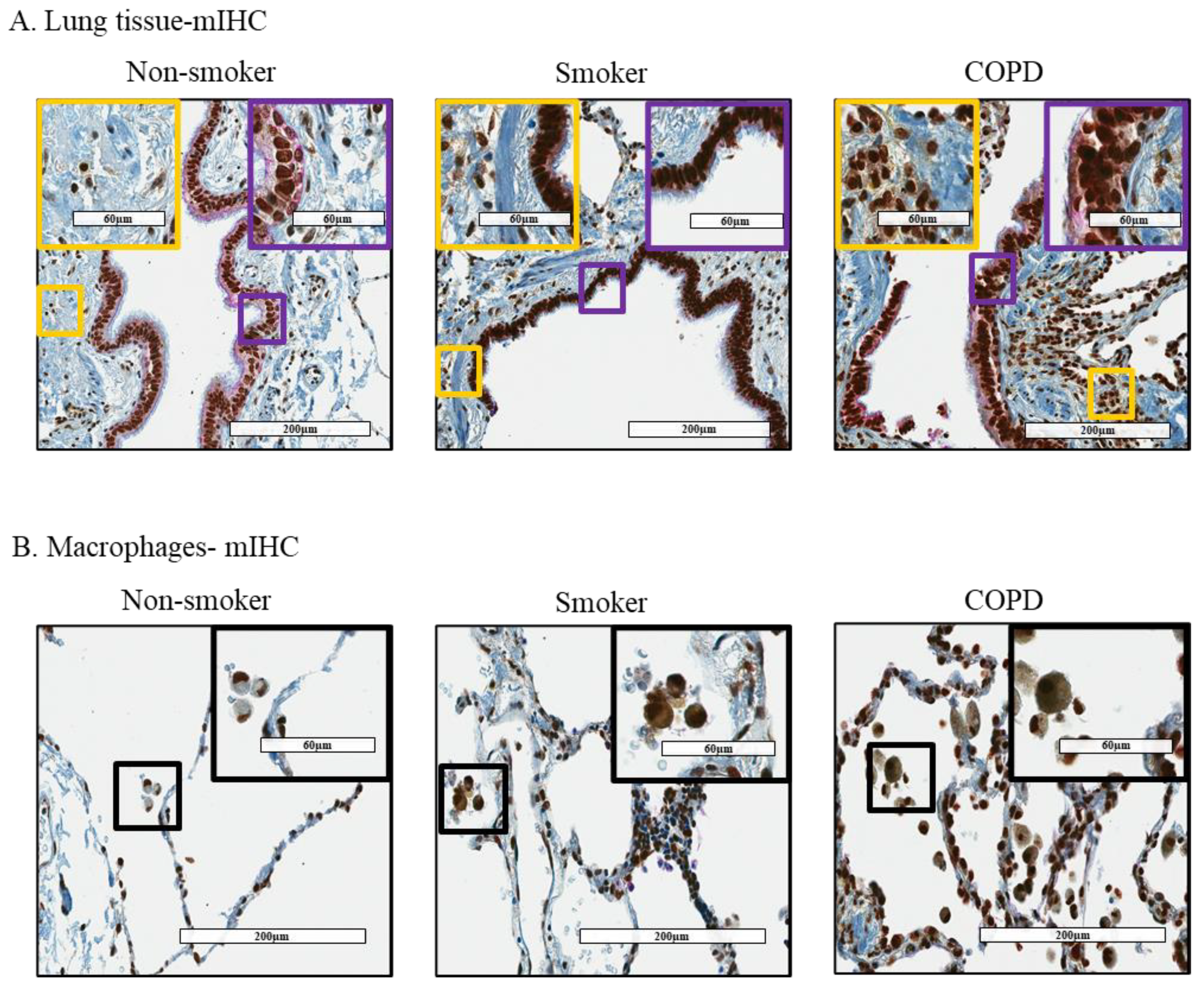
3.2. Cytoplasmic Localization of HuR Is Increased in Smoker and COPD Lung Cells
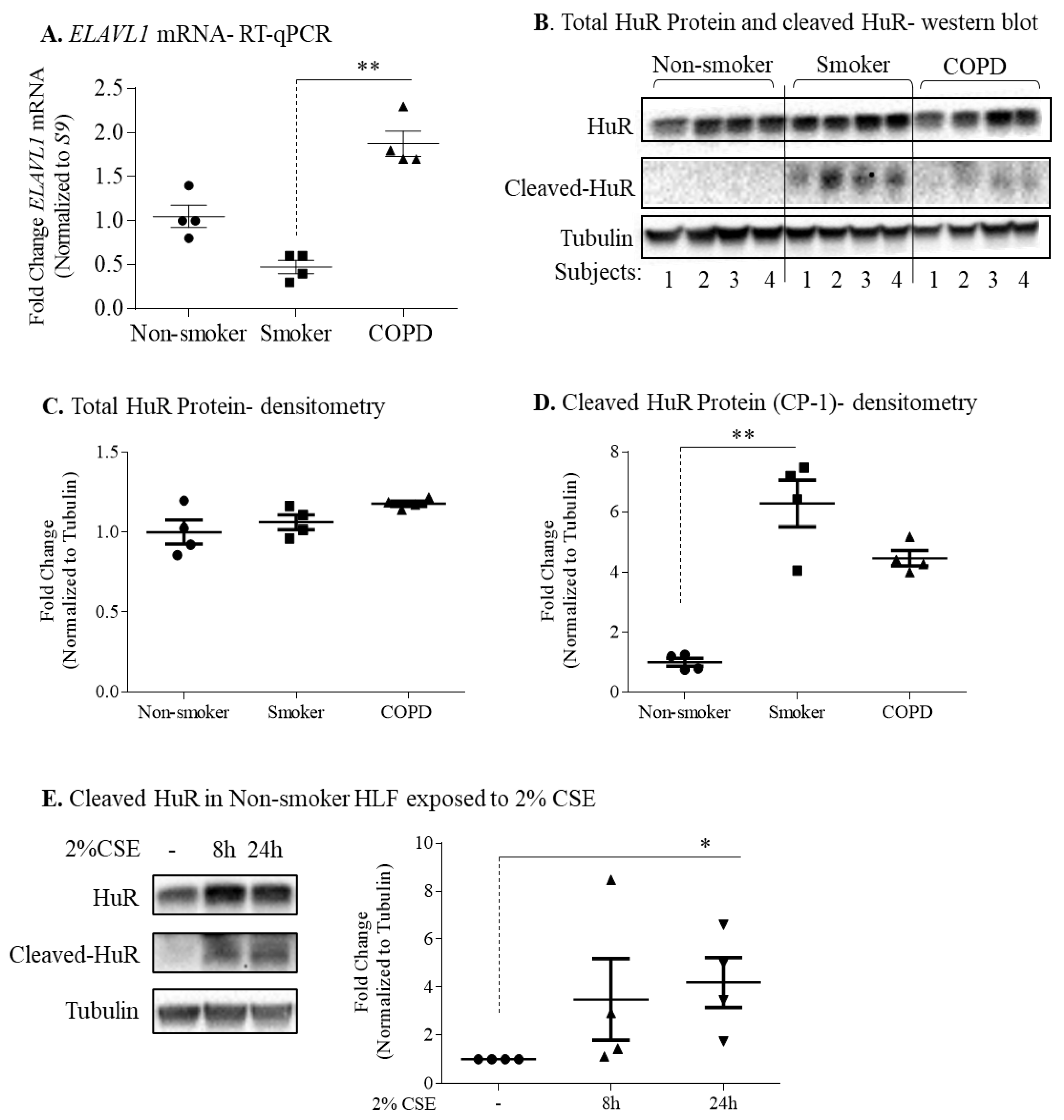
3.3. Protein Expression of HuR Is Similar between Non-Smoker, Smoker, and COPD-Derived Lung Fibroblasts
3.4. Increased ACE2 Protein in COPD-Derived Lung Fibroblasts Is Not Associated with Changes at the mRNA Level or Increased Binding to HuR
3.5. HuR Does Not Control ACE2 mRNA or Protein Stability
3.6. Non-Smoker and COPD-Derived Lung Fibroblasts Exposed to CSE Exhibit Increased Cytoplasmic HuR
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lange, P.; Celli, B.; Agusti, A.; Boje Jensen, G.; Divo, M.; Faner, R.; Guerra, S.; Marott, J.L.; Martinez, F.D.; Martinez-Camblor, P.; et al. Lung-Function Trajectories Leading to Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2015, 373, 111–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Report, W.H. Available online: https://www.who.int/en/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 20 December 2020).
- Global Strategy for the Diagnosis. Global Initiative for Chronic Obstructive Lung Disease (GOLD). 2020. Available online: https://goldcopd.org/ (accessed on 20 December 2020).
- Athanazio, R. Airway disease: Similarities and differences between asthma, COPD and bronchiectasis. Clinics 2012, 67, 1335–1343. [Google Scholar] [CrossRef]
- Attaway, A.A.; Zein, J.; Hatipoglu, U.S. SARS-CoV-2 infection in the COPD population is associated with increased healthcare utilization: An analysis of Cleveland clinic’s COVID-19 registry. EClinicalMedicine 2020, 26, 100515. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Aloufi, N.; Traboulsi, H.; Ding, J.; Fonseca, G.J.; Nair, P.; Huang, S.K.; Hussain, S.N.A.; Eidelman, D.H.; Baglole, C.J. Angiotensin-converting enzyme 2 expression in COPD and IPF fibroblasts: The forgotten cell in COVID-19. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 320, L152–L157. [Google Scholar] [CrossRef]
- Leung, J.M.; Yang, C.X.; Tam, A.; Shaipanich, T.; Hackett, T.L.; Singhera, G.K.; Dorscheid, D.R.; Sin, D.D. ACE-2 expression in the small airway epithelia of smokers and COPD patients: Implications for COVID-19. Eur. Respir. J. 2020, 55, 2000688. [Google Scholar] [CrossRef] [Green Version]
- Brake, S.J.; Barnsley, K.; Lu, W.; McAlinden, K.D.; Eapen, M.S.; Sohal, S.S. Smoking Upregulates Angiotensin-Converting Enzyme-2 Receptor: A Potential Adhesion Site for Novel Coronavirus SARS-CoV-2 (COVID-19). J. Clin. Med. 2020, 9, 841. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Rostami, M.R.; Leopold, P.L.; Mezey, J.G.; O’Beirne, S.L.; Strulovici-Barel, Y.; Crystal, R.G. Expression of the SARS-CoV-2 ACE2 Receptor in the Human Airway Epithelium. Am. J. Respir. Crit. Care Med. 2020, 202, 219–229. [Google Scholar] [CrossRef]
- Lumbers, E.R.; Delforce, S.J.; Pringle, K.G.; Smith, G.R. The Lung, the Heart, the Novel Coronavirus, and the Renin-Angiotensin System; The Need for Clinical Trials. Front. Med. 2020, 7, 248. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Penninger, J.M. Angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Cell. Mol. Life Sci. 2007, 64, 2006–2012. [Google Scholar] [CrossRef] [PubMed]
- Sparks, M.A.; Crowley, S.D.; Gurley, S.B.; Mirotsou, M.; Coffman, T.M. Classical Renin-Angiotensin system in kidney physiology. Compr. Physiol. 2014, 4, 1201–1228. [Google Scholar] [PubMed] [Green Version]
- Lelis, D.F.; Freitas, D.F.; Machado, A.S.; Crespo, T.S.; Santos, S.H.S. Angiotensin-(1-7), Adipokines and Inflammation. Metabolism 2019, 95, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.B.; Zhong, J.C.; Grant, M.B.; Oudit, G.Y. Role of the ACE2/Angiotensin 1-7 Axis of the Renin-Angiotensin System in Heart Failure. Circ. Res. 2016, 118, 1313–1326. [Google Scholar] [CrossRef] [Green Version]
- Engin, A.B.; Engin, E.D.; Engin, A. Two important controversial risk factors in SARS-CoV-2 infection: Obesity and smoking. Environ. Toxicol. Pharmacol. 2020, 78, 103411. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.C.; Sausville, E.L.; Girish, V.; Yuan, M.L.; Vasudevan, A.; John, K.M.; Sheltzer, J.M. Cigarette smoke exposure and inflammatory signaling increase the expression of the SARS-CoV-2 receptor ACE2 in the respiratory tract. Dev. Cell. 2020, 53, 514–529. [Google Scholar] [CrossRef]
- Jacobs, M.; Van Eeckhoutte, H.P.; Wijnant, S.R.A.; Janssens, W.; Joos, G.F.; Brusselle, G.G.; Bracke, K.R. Increased expression of ACE2, the SARS-CoV-2 entry receptor, in alveolar and bronchial epithelium of smokers and COPD subjects. Eur. Respir. J. 2020, 56, 2002378. [Google Scholar] [CrossRef]
- Shen, H.; Zhang, J.; Wang, C.; Jain, P.P.; Xiong, M.; Shi, X.; Lei, Y.; Chen, S.; Yin, Q.; Thistlethwaite, P.A.; et al. MDM2-Mediated Ubiquitination of Angiotensin-Converting Enzyme 2 Contributes to the Development of Pulmonary Arterial Hypertension. Circulation 2020, 142, 1190–1204. [Google Scholar] [CrossRef]
- Garcia-Maurino, S.M.; Rivero-Rodriguez, F.; Velazquez-Cruz, A.; Hernandez-Vellisca, M.; Diaz-Quintana, A.; De la Rosa, M.A.; Diaz-Moreno, I. RNA Binding Protein Regulation and Cross-Talk in the Control of AU-rich mRNA Fate. Front. Mol. Biosci. 2017, 4, 71. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Qu, H.; Hu, B.; Bi, C.; Li, M.; Wang, L.; Huang, X.; Zhang, M. Physiological cyclic stretch up-regulates angiotensin-converting enzyme 2 expression to reduce proliferation and migration of vascular smooth muscle cells. Biosci. Rep. 2020, 40, BSR20192012. [Google Scholar] [CrossRef]
- Srikantan, S.; Gorospe, M. HuR function in disease. Front. Biosci. Landmark Ed. 2012, 17, 189. [Google Scholar] [CrossRef] [Green Version]
- Grammatikakis, I.; Abdelmohsen, K.; Gorospe, M. Posttranslational control of HuR function. Wiley Interdiscip. Rev. RNA 2017, 8, e1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izquierdo, J.M. Hu antigen R (HuR) functions as an alternative pre-mRNA splicing regulator of Fas apoptosis-promoting receptor on exon definition. J. Biol. Chem. 2008, 283, 19077–19084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, C.M.; Steitz, J.A. HuR and mRNA stability. Cell. Mol. Life Sci. 2001, 58, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Zago, M.; Sheridan, J.A.; Nair, P.; Rico de Souza, A.; Gallouzi, I.E.; Rousseau, S.; Di Marco, S.; Hamid, Q.; Eidelman, D.H.; Baglole, C.J. Aryl hydrocarbon receptor-dependent retention of nuclear HuR suppresses cigarette smoke-induced cyclooxygenase-2 expression independent of DNA-binding. PLoS ONE 2013, 8, e74953. [Google Scholar] [CrossRef] [Green Version]
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; DeIuliis, G.; Januszyk, M.; et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1983. [Google Scholar] [CrossRef]
- Chua, R.L.; Lukassen, S.; Trump, S.; Hennig, B.P.; Wendisch, D.; Pott, F.; Debnath, O.; Thurmann, L.; Kurth, F.; Volker, M.T.; et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020, 38, 970–979. [Google Scholar] [CrossRef]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 2018, 19, 15. [Google Scholar] [CrossRef] [Green Version]
- Guerrina, N.; Traboulsi, H.; Rico de Souza, A.; Bosse, Y.; Thatcher, T.H.; Robichaud, A.; Ding, J.; Li, P.Z.; Simon, L.; Pareek, S.; et al. Aryl hydrocarbon receptor deficiency causes the development of chronic obstructive pulmonary disease through the integration of multiple pathogenic mechanisms. FASEB J. 2021, 35, e21376. [Google Scholar] [CrossRef] [PubMed]
- Baglole, C.J.; Reddy, S.Y.; Pollock, S.J.; Feldon, S.E.; Sime, P.J.; Smith, T.J.; Phipps, R.P. Isolation and phenotypic characterization of lung fibroblasts. Methods Mol. Med. 2005, 117, 115–127. [Google Scholar]
- Hecht, E.; Zago, M.; Sarill, M.; Rico de Souza, A.; Gomez, A.; Matthews, J.; Hamid, Q.; Eidelman, D.H.; Baglole, C.J. Aryl hydrocarbon receptor-dependent regulation of miR-196a expression controls lung fibroblast apoptosis but not proliferation. Toxicol. Appl. Pharmacol. 2014, 280, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Keene, J.D.; Komisarow, J.M.; Friedersdorf, M.B. RIP-Chip: The isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nat. Protoc. 2006, 1, 302–307. [Google Scholar] [CrossRef]
- Mubaid, S.; Ma, J.F.; Omer, A.; Ashour, K.; Lian, X.J.; Sanchez, B.J.; Robinson, S.; Cammas, A.; Dormoy-Raclet, V.; Di Marco, S.; et al. HuR counteracts miR-330 to promote STAT3 translation during inflammation-induced muscle wasting. Proc. Natl. Acad. Sci. USA 2019, 116, 17261–17270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyman, R.W.; Davidson, N. Kinetics of the in vitro inhibition of transcription by actinomycin. J. Mol. Biol. 1970, 50, 421–438. [Google Scholar] [CrossRef]
- Guerrina, N.; Aloufi, N.; Shi, F.; Prasade, K.; Mehrotra, C.; Traboulsi, H.; Matthews, J.; Eidelman, D.H.; Hamid, Q.; Baglole, C.J. The aryl hydrocarbon receptor reduces LC3II expression and controls endoplasmic reticulum stress. Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 320, L339–L355. [Google Scholar] [CrossRef]
- Schneider-Poetsch, T.; Ju, J.; Eyler, D.E.; Dang, Y.; Bhat, S.; Merrick, W.C.; Green, R.; Shen, B.; Liu, J.O. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat. Chem. Biol. 2010, 6, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Baglole, C.J.; Maggirwar, S.B.; Gasiewicz, T.A.; Thatcher, T.H.; Phipps, R.P.; Sime, P.J. The aryl hydrocarbon receptor attenuates tobacco smoke-induced cyclooxygenase-2 and prostaglandin production in lung fibroblasts through regulation of the NF-kappaB family member RelB. J. Biol. Chem. 2008, 283, 28944–28957. [Google Scholar] [CrossRef] [Green Version]
- Naderi, N.; Farias, R.; Abou Rjeili, M.; Mostafavi-Pour-Manshadi, S.M.; Krishnan, S.; Li, P.Z.; Baglole, C.J.; Bourbeau, J. Investigating the effect of pretreatment with azithromycin on inflammatory mediators in bronchial epithelial cells exposed to cigarette smoke. Exp. Lung Res. 2021, 47, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Mazroui, R.; Di Marco, S.; Clair, E.; von Roretz, C.; Tenenbaum, S.A.; Keene, J.D.; Saleh, M.; Gallouzi, I.E. Caspase-mediated cleavage of HuR in the cytoplasm contributes to pp32/PHAP-I regulation of apoptosis. J. Cell Biol. 2008, 180, 113–127. [Google Scholar] [CrossRef]
- Corley, M.; Burns, M.C.; Yeo, G.W. How RNA-Binding Proteins Interact with RNA: Molecules and Mechanisms. Mol. Cell 2020, 78, 9–29. [Google Scholar] [CrossRef]
- Dreyfuss, G.; Kim, V.N.; Kataoka, N. Messenger-RNA-binding proteins and the messages they carry. Nat. Rev. Mol. Cell. Biol. 2002, 3, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Moreno, M.; Jarvelin, A.I.; Castello, A. Unconventional RNA-binding proteins step into the virus-host battlefront. Wiley Interdiscip. Rev. RNA 2018, 9, e1498. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, N.; Lareau, C.A.; Keshishian, H.; Ganskih, S.; Schneider, C.; Hennig, T.; Melanson, R.; Werner, S.; Wei, Y.; Zimmer, M.; et al. The SARS-CoV-2 RNA-protein interactome in infected human cells. Nat. Microbiol. 2020, 6, 339–353. [Google Scholar] [CrossRef]
- Sokoloski, K.J.; Dickson, A.M.; Chaskey, E.L.; Garneau, N.L.; Wilusz, C.J.; Wilusz, J. Sindbis virus usurps the cellular HuR protein to stabilize its transcripts and promote productive infections in mammalian and mosquito cells. Cell Host Microbe 2010, 8, 196–207. [Google Scholar] [CrossRef] [Green Version]
- Spangberg, K.; Wiklund, L.; Schwartz, S. HuR, a protein implicated in oncogene and growth factor mRNA decay, binds to the 3′ ends of hepatitis C virus RNA of both polarities. Virology 2000, 274, 378–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cumming, S.A.; Chuen-Im, T.; Zhang, J.; Graham, S.V. The RNA stability regulator HuR regulates L1 protein expression in vivo in differentiating cervical epithelial cells. Virology 2009, 383, 142–149. [Google Scholar] [CrossRef] [Green Version]
- Barnhart, M.D.; Moon, S.L.; Emch, A.W.; Wilusz, C.J.; Wilusz, J. Changes in cellular mRNA stability, splicing, and polyadenylation through HuR protein sequestration by a cytoplasmic RNA virus. Cell Rep. 2013, 5, 909–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Gu, X.; Wu, N.; Zhang, P.; Liu, Y.; Jiang, S. Human antigen R enhances the epithelial-mesenchymal transition via regulation of ZEB-1 in the human airway epithelium. Respir. Res. 2018, 19, 109. [Google Scholar] [CrossRef] [Green Version]
- Vlahos, R.; Bozinovski, S. Role of alveolar macrophages in chronic obstructive pulmonary disease. Front. Immunol. 2014, 5, 435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, J.; Wang, Z.; Qu, Y.; Zhu, H.; Zhu, Q.; Tong, W.; Bao, L.; Lv, Q.; Cong, J.; Li, D.; et al. Distinct uptake, amplification, and release of SARS-CoV-2 by M1 and M2 alveolar macrophages. Cell Discov. 2021, 7, 24. [Google Scholar] [CrossRef]
- Barnes, P.J. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2016, 138, 16–27. [Google Scholar] [CrossRef] [Green Version]
- Ueland, T.; Holter, J.C.; Holten, A.R.; Muller, K.E.; Lind, A.; Bekken, G.K.; Dudman, S.; Aukrust, P.; Dyrhol-Riise, A.M.; Heggelund, L. Distinct and early increase in circulating MMP-9 in COVID-19 patients with respiratory failure. J. Infect. 2020, 81, e41–e43. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Modi, Y.; Yarovinsky, T.; Yu, J.; Collinge, M.; Kyriakides, T.; Zhu, Y.; Sessa, W.C.; Pardi, R.; Bender, J.R. Macrophage beta2 integrin-mediated, HuR-dependent stabilization of angiogenic factor-encoding mRNAs in inflammatory angiogenesis. Am. J. Pathol. 2012, 180, 1751–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Jarujaron, S.; Gurley, E.C.; Chen, L.; Ding, H.; Studer, E.; Pandak, W.M., Jr.; Hu, W.; Zou, T.; Wang, J.Y.; et al. HIV protease inhibitors increase TNF-alpha and IL-6 expression in macrophages: Involvement of the RNA-binding protein HuR. Atherosclerosis 2007, 195, e134–e143. [Google Scholar] [CrossRef]
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Y.; Guan, Z.; Li, H.; Ye, M.; Chen, X.; Shen, J.; Zhou, Y.; Shi, Z.L.; Zhou, P.; et al. SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal. Transduct. Target Ther. 2020, 5, 235. [Google Scholar] [CrossRef]
- Mahboubi, H.; Stochaj, U. Cytoplasmic stress granules: Dynamic modulators of cell signaling and disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 884–895. [Google Scholar] [CrossRef]
- White, E.S. Lung extracellular matrix and fibroblast function. Ann. Am. Thorac. Soc. 2015, 12 (Suppl. S1), S30–S33. [Google Scholar] [CrossRef] [Green Version]
- Buckley, C.D.; Pilling, D.; Lord, J.M.; Akbar, A.N.; Scheel-Toellner, D.; Salmon, M. Fibroblasts regulate the switch from acute resolving to chronic persistent inflammation. Trends Immunol. 2001, 22, 199–204. [Google Scholar] [CrossRef]
- Di Marco, S.; Mazroui, R.; Dallaire, P.; Chittur, S.; Tenenbaum, S.A.; Radzioch, D.; Marette, A.; Gallouzi, I.E. NF-kappa B-mediated MyoD decay during muscle wasting requires nitric oxide synthase mRNA stabilization, HuR protein, and nitric oxide release. Mol. Cell. Biol. 2005, 25, 6533–6545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doller, A.; Winkler, C.; Azrilian, I.; Schulz, S.; Hartmann, S.; Pfeilschifter, J.; Eberhardt, W. High-constitutive HuR phosphorylation at Ser 318 by PKC{delta} propagates tumor relevant functions in colon carcinoma cells. Carcinogenesis 2011, 32, 676–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, C.S.; Shi, Y.; Chang, S.Y.; Abdo, S.; Chenier, I.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.L.; Chan, J.S. Overexpression of heterogeneous nuclear ribonucleoprotein F stimulates renal Ace-2 gene expression and prevents TGF-beta1-induced kidney injury in a mouse model of diabetes. Diabetologia 2015, 58, 2443–2454. [Google Scholar] [CrossRef] [Green Version]
- Kawamata, T.; Tomari, Y. Making RISC. Trends Biochem. Sci. 2010, 35, 368–376. [Google Scholar] [CrossRef]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [Green Version]
- Lambert, D.W.; Lambert, L.A.; Clarke, N.E.; Hooper, N.M.; Porter, K.E.; Turner, A.J. Angiotensin-converting enzyme 2 is subject to post-transcriptional regulation by miR-421. Clin. Sci. 2014, 127, 243–249. [Google Scholar] [CrossRef]
- Izzotti, A.; Larghero, P.; Longobardi, M.; Cartiglia, C.; Camoirano, A.; Steele, V.E.; De Flora, S. Dose-responsiveness and persistence of microRNA expression alterations induced by cigarette smoke in mouse lung. Mutat. Res. 2011, 717, 9–16. [Google Scholar] [CrossRef]
- von Roretz, C.; Gallouzi, I.E. Protein kinase RNA/FADD/caspase-8 pathway mediates the proapoptotic activity of the RNA-binding protein human antigen R (HuR). J. Biol. Chem. 2010, 285, 16806–16813. [Google Scholar] [CrossRef] [Green Version]
- von Roretz, C.; Lian, X.J.; Macri, A.M.; Punjani, N.; Clair, E.; Drouin, O.; Dormoy-Raclet, V.; Ma, J.F.; Gallouzi, I.E. Apoptotic-induced cleavage shifts HuR from being a promoter of survival to an activator of caspase-mediated apoptosis. Cell Death Differ. 2013, 20, 154–168. [Google Scholar] [CrossRef] [Green Version]
- Chiappara, G.; Gjomarkaj, M.; Sciarrino, S.; Vitulo, P.; Pipitone, L.; Pace, E. Altered expression of p21, activated caspase-3, and PCNA in bronchiolar epithelium of smokers with and without chronic obstructive pulmonary disease. Exp. Lung Res. 2014, 40, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Thliveris, J.A.; Shaw, A.; Sowa, M.; Gilchrist, J.; Scott, J.E. Caspase 3 activity in isolated fetal rat lung fibroblasts and rat periodontal ligament fibroblasts: Cigarette smoke induced alterations. Tob. Induc. Dis. 2013, 11, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer Sequence | Reverse Primer Sequence |
|---|---|---|
| ELALV1 | AAC GCC TCC TCC GGC TGG TGC | GCG GTA GCC GTT CAG GCT GGC |
| ACE2-201-202 | AAC TGC TGC TCA GTC CAC CA | GAC CAT TTG TCC CCA GCA TT |
| ACE2-202 | CCC AGA GCA TGC CTG ATA GA | CCC ACT TCA GAG GGT GAA CA |
| S9 | CAG CTT CAT CTT GCC CTC A | CTG CTG ACG CTT GAT GAG AA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aloufi, N.; Haidar, Z.; Ding, J.; Nair, P.; Benedetti, A.; Eidelman, D.H.; Gallouzi, I.-E.; Di Marco, S.; Hussain, S.N.; Baglole, C.J. Role of Human Antigen R (HuR) in the Regulation of Pulmonary ACE2 Expression. Cells 2022, 11, 22. https://doi.org/10.3390/cells11010022
Aloufi N, Haidar Z, Ding J, Nair P, Benedetti A, Eidelman DH, Gallouzi I-E, Di Marco S, Hussain SN, Baglole CJ. Role of Human Antigen R (HuR) in the Regulation of Pulmonary ACE2 Expression. Cells. 2022; 11(1):22. https://doi.org/10.3390/cells11010022
Chicago/Turabian StyleAloufi, Noof, Zahraa Haidar, Jun Ding, Parameswaran Nair, Andrea Benedetti, David H. Eidelman, Imed-Eddine Gallouzi, Sergio Di Marco, Sabah N. Hussain, and Carolyn J. Baglole. 2022. "Role of Human Antigen R (HuR) in the Regulation of Pulmonary ACE2 Expression" Cells 11, no. 1: 22. https://doi.org/10.3390/cells11010022
APA StyleAloufi, N., Haidar, Z., Ding, J., Nair, P., Benedetti, A., Eidelman, D. H., Gallouzi, I. -E., Di Marco, S., Hussain, S. N., & Baglole, C. J. (2022). Role of Human Antigen R (HuR) in the Regulation of Pulmonary ACE2 Expression. Cells, 11(1), 22. https://doi.org/10.3390/cells11010022