
High Throughput Small Molecule Screen for Reactivation of FMR1 in Fragile X Syndrome Human Neural Cells

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
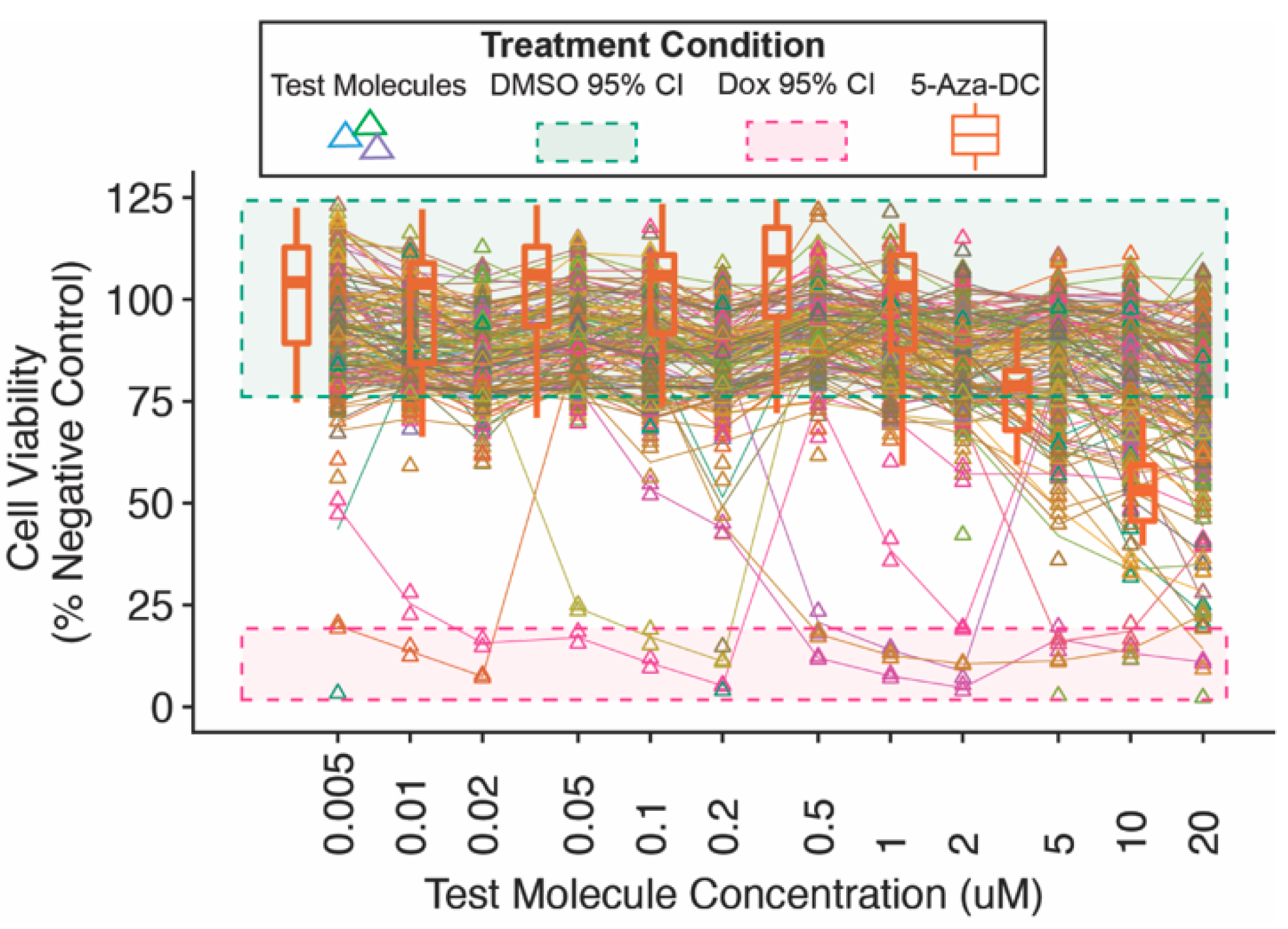{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
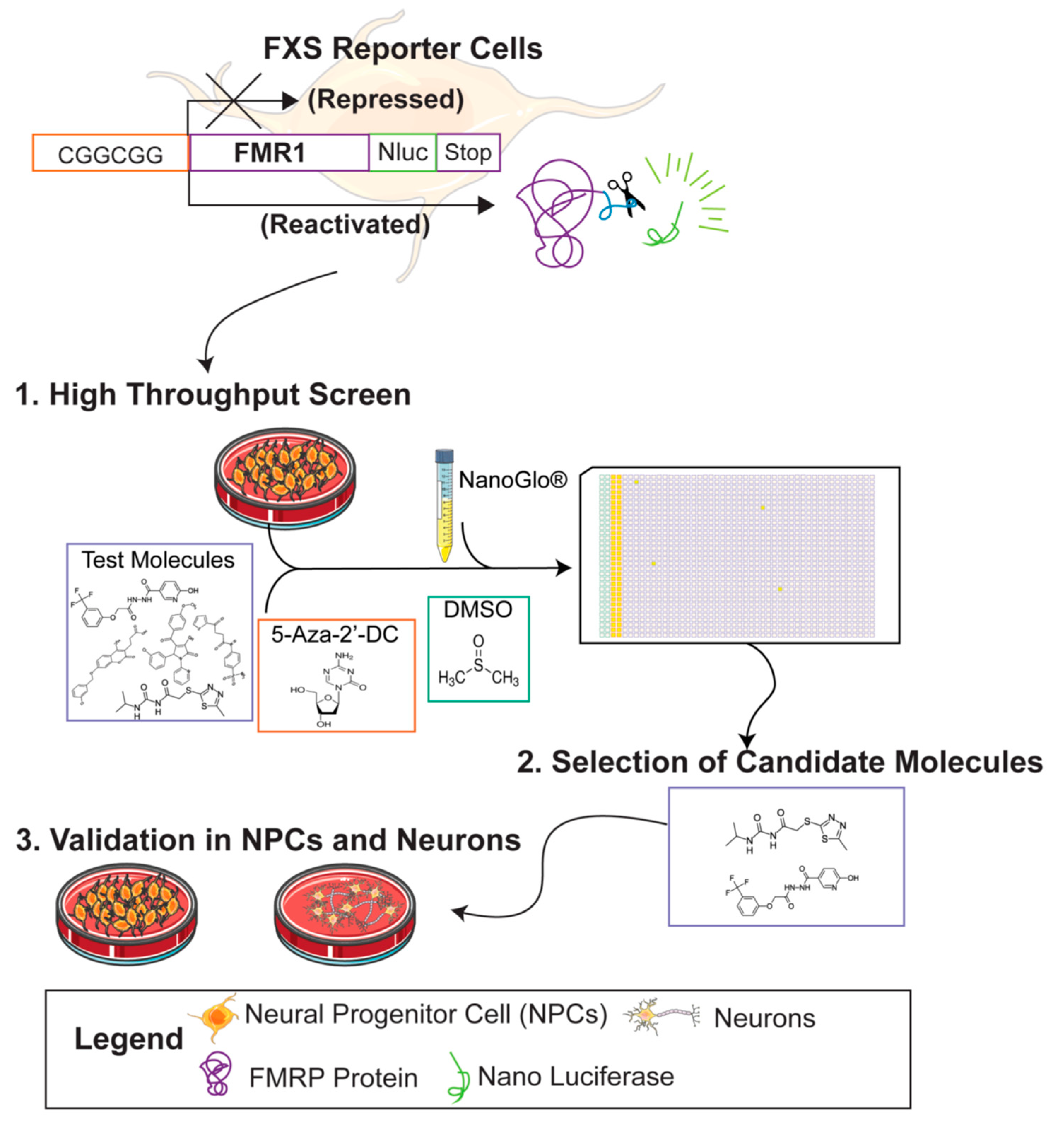
2.1. FMR1-Nano Luciferase Reporter Cell Lines
2.2. iPSC Neural Differentiation
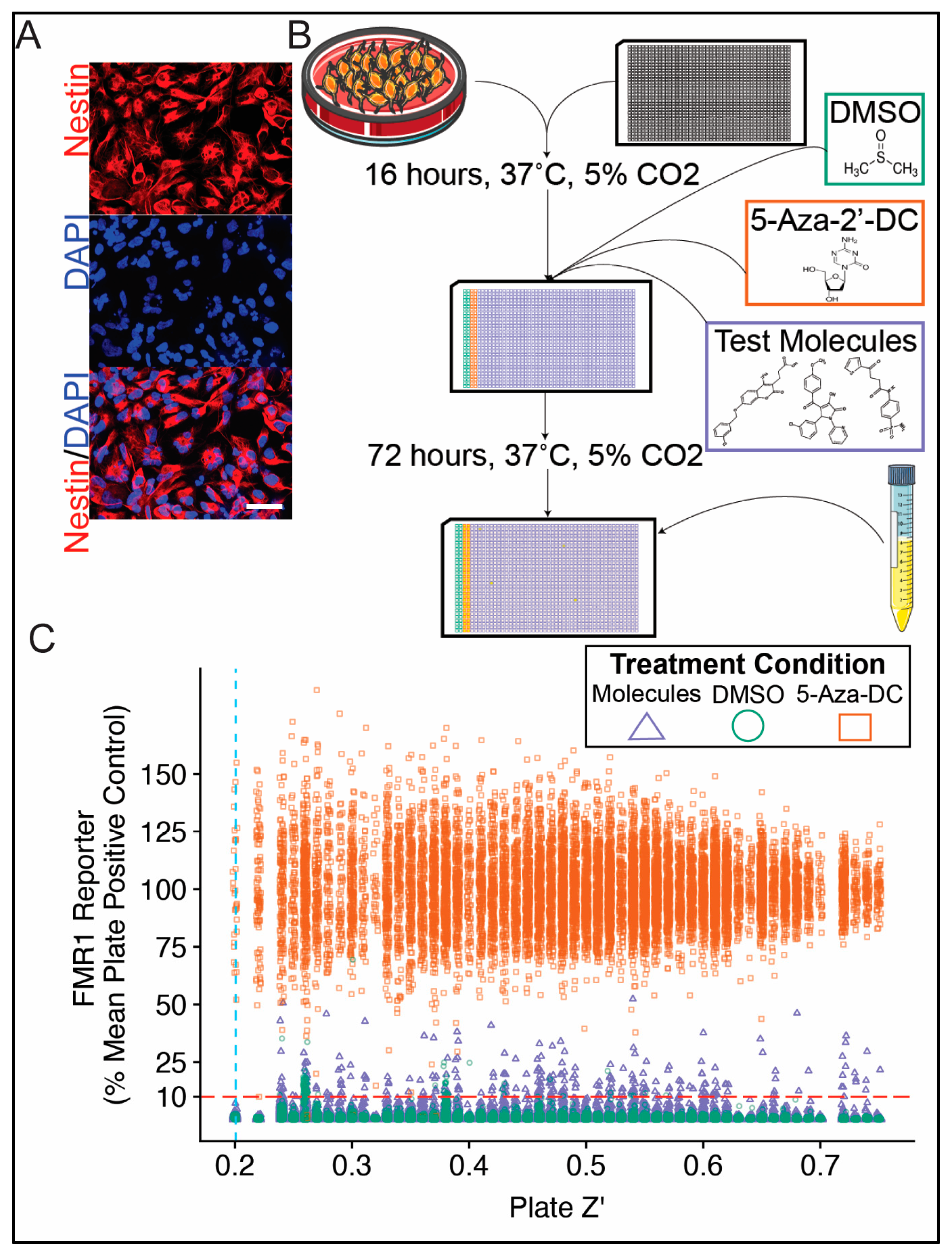
2.3. Immunocytochemistry and Fluorescence Imaging
2.4. High Throughput Drug Screen
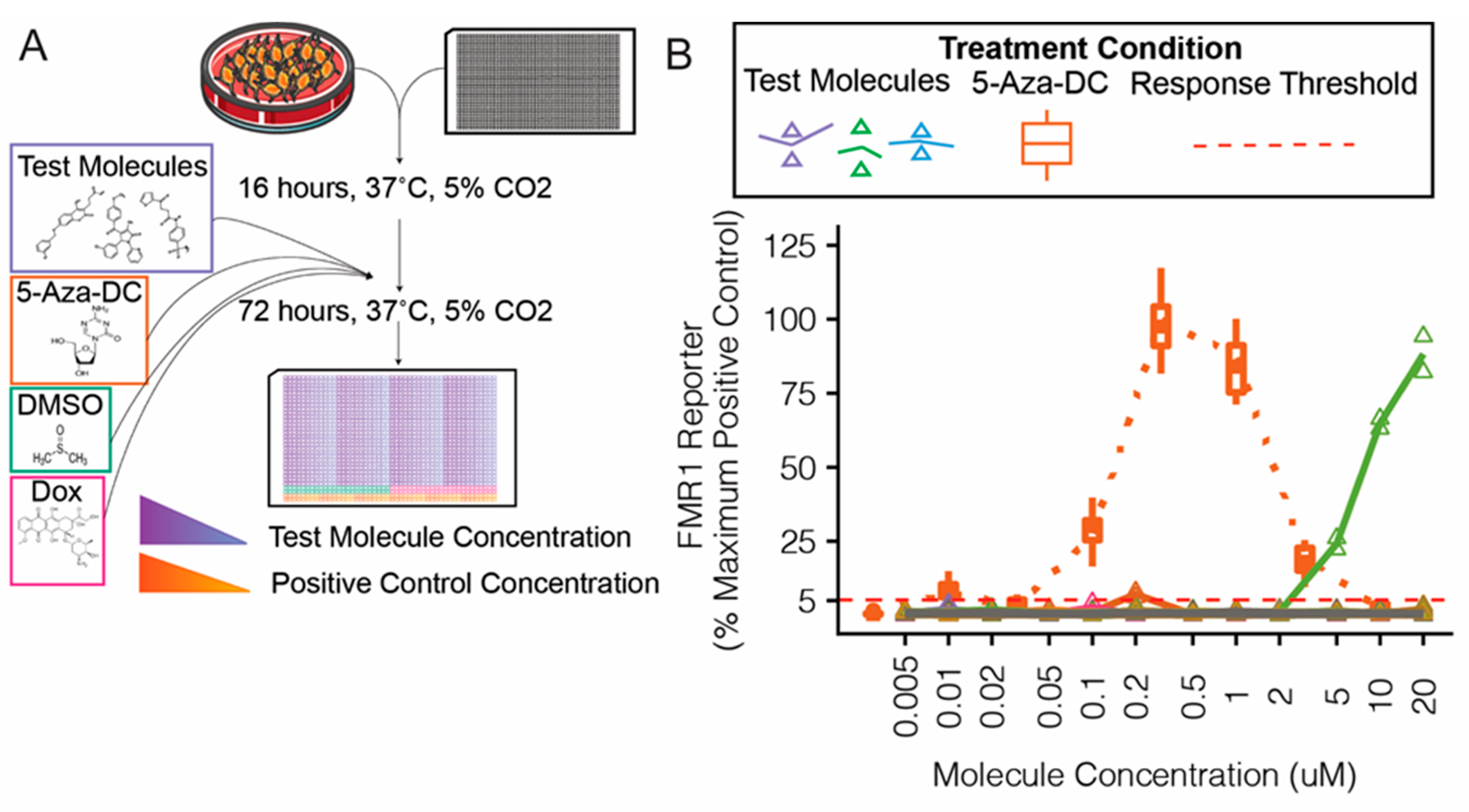
2.5. Cell Viability Assay
2.6. Small Molecules
2.7. Data Analysis
3. Results
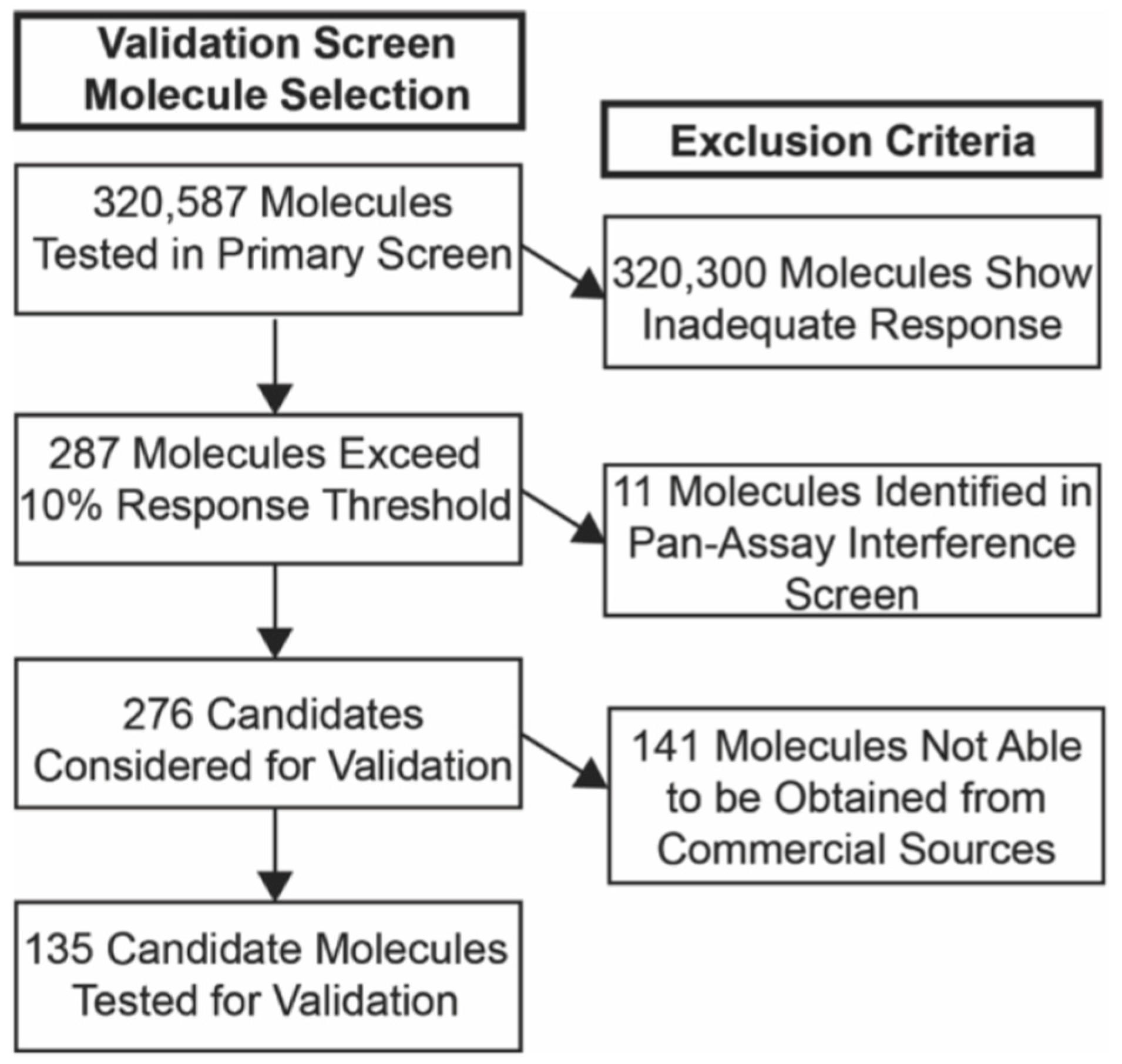
3.1. Primary Screen of Compounds for Reactivation at the FMR1 Locus in FXS NPCs
3.2. Selection of Candidate Molecules
3.3. Confirmatory Validation (Secondary) Screen for Candidate Molecules
4. Discussion
4.1. Summary and Limitations of Drug Screen
4.2. Validation Assays of Candidate Molecules
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coffee, B.; Keith, K.; Albizua, I.; Malone, T.; Mowrey, J.; Sherman, S.L.; Warren, S.T. Incidence of Fragile X Syndrome by Newborn Screening for Methylated FMR1 DNA. Am. J. Hum. Genet. 2009, 85, 503–514. [Google Scholar] [CrossRef] [Green Version]
- Bailey, D.B.; Raspa, M.; Olmsted, M.; Holiday, D.B. Co-occurring conditions associated with FMR1 gene variations: Findings from a national parent survey. Am. J. Med. Genet. Part A 2008, 146A, 2060–2069. [Google Scholar] [CrossRef] [PubMed]
- Sacco, P.; Capkun-Niggli, G.; Zhang, X.; Jose, R. The Economic Burden of Fragile X Syndrome: Healthcare Resource Utilization in the United States. Am. Health. Drug Benefits 2013, 6, 11. [Google Scholar]
- Pieretti, M.; Zhang, F.; Warren, S.T.; Oostra, B.A.; Caskey, C.T.; Nelson, D.L. Absence of Expression of the FMR-1 Gene in Fragile X Syndrome. Cell 1991, 66, 817–822. [Google Scholar] [CrossRef]
- Verkerk, A.; Pieretti, M.; Sutcliff, J.S.; Fu, Y.-H.; Kuhl, D.P.A.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.; et al. Identification of a Gene (FMR-1) Containing a CGG Repeat Coincident with a Breakpoint Cluster Region Exhibiting Length Variation in Fragile X Syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L.; et al. Fragile X syndrome. Nat. Rev. Dis. Primers 2017, 3, 17065. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.C.; Van Driesche, S.J.; Zhang, C.; Hung, K.Y.S.; Mele, A.; Fraser, C.E.; Stone, E.F.; Chen, C.; Fak, J.J.; Chi, S.W.; et al. FMRP Stalls Ribosomal Translocation on mRNAs Linked to Synaptic Function and Autism. Cell 2011, 146, 247–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curia, G.; Papouin, T.; Séguéla, P.; Avoli, M. Downregulation of Tonic GABAergic Inhibition in a Mouse Model of Fragile X Syndrome. Cereb. Cortex 2009, 19, 1515–1520. [Google Scholar] [CrossRef] [Green Version]
- Dölen, G.; Osterweil, E.; Rao, B.S.S.; Smith, G.B.; Auerbach, B.D.; Chattarji, S.; Bear, M.F. Correction of Fragile X Syndrome in Mice. Neuron 2007, 56, 955–962. [Google Scholar] [CrossRef] [Green Version]
- El Idrissi, A.; Ding, X.-H.; Scalia, J.; Trenkner, E.; Brown, W.T.; Dobkin, C. Decreased GABAA receptor expression in the seizure-prone fragile X mouse. Neurosci. Lett. 2005, 377, 141–146. [Google Scholar] [CrossRef]
- Huber, K.M.; Gallagher, S.M.; Warren, S.T.; Bear, M.F. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. USA 2002, 99, 7746–7750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilousova, T.V.; Dansie, L.; Ngo, M.; Aye, J.; Charles, J.R.; Ethell, D.W.; Ethell, I.M. Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model. J. Med. Genet. 2008, 46, 94–102. [Google Scholar] [CrossRef]
- Gantois, I.; Pop, A.S.; de Esch, C.E.F.; Buijsen, R.A.M.; Pooters, T.; Gomez-Mancilla, B.; Gasparini, F.; Oostra, B.A.; D’Hooge, R.; Willemsen, R. Chronic administration of AFQ056/Mavoglurant restores social behaviour in Fmr1 knockout mice. Behav. Brain Res. 2013, 239, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.L.; Pride, M.C.; Hayes, J.E.; Puhger, K.R.; Butler-Struben, H.M.; Baker, S.; Crawley, J.N. GABAB Receptor Agonist R-Baclofen Reverses Social Deficits and Reduces Repetitive Behavior in Two Mouse Models of Autism. Neuropsychopharmacology 2015, 40, 2228–2239. [Google Scholar] [CrossRef] [Green Version]
- Berry-Kravis, E.; Des Portes, V.; Hagerman, R.; Jacquemont, S.; Charles, P.; Visootsak, J.; Brinkman, M.; Rerat, K.; Koumaras, B.; Zhu, L.; et al. Mavoglurant in fragile X syndrome: Results of two randomized, double-blind, placebo-controlled trials. Sci. Transl. Med. 2016, 8, 321ra325. [Google Scholar] [CrossRef]
- Berry-Kravis, E.M.; Lindemann, L.; Jønch, A.E.; Apostol, G.; Bear, M.F.; Carpenter, R.L.; Crawley, J.N.; Curie, A.; Des Portes, V.; Hossain, F.; et al. Drug development for neurodevelopmental disorders: Lessons learned from fragile X syndrome. Nat. Rev. Drug Discov. 2017, 17, 280–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Group, F.S.; Youssef, E.A.; Berry-Kravis, E.; Czech, C.; Hagerman, R.J.; Hessl, D.; Wong, C.Y.; Rabbia, M.; Deptula, D.; John, A.; et al. Effect of the mGluR5-NAM Basimglurant on Behavior in Adolescents and Adults with Fragile X Syndrome in a Randomized, Double-Blind, Placebo-Controlled Trial: FragXis Phase 2 Results. Neuropsychopharmacology 2018, 43, 503–512. [Google Scholar] [CrossRef] [Green Version]
- Ligsay, A.; Van Dijck, A.; Nguyen, D.V.; Lozano, R.; Chen, Y.; Bickel, E.S.; Hessl, D.; Schneider, A.; Angkustsiri, K.; Tassone, F.; et al. A randomized double-blind, placebo-controlled trial of ganaxolone in children and adolescents with fragile X syndrome. J. Neurodev. Disord. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry-Kravis, E.; Horrigan, J.P.; Tartaglia, N.; Hagerman, R.; Kolevzon, A.; Erickson, C.A.; Hatti, S.; Snape, M.; Yaroshinsky, A.; Stoms, G.; et al. A Double-Blind, Randomized, Placebo-Controlled Clinical Study of Trofinetide in the Treatment of Fragile X Syndrome. Pediatric Neurol. 2020, 110, 30–41. [Google Scholar] [CrossRef]
- Berry-Kravis, E.M.; Harnett, M.D.; Reines, S.A.; Reese, M.A.; Ethridge, L.E.; Outterson, A.H.; Michalak, C.; Furman, J.; Gurney, M.E. Inhibition of phosphodiesterase-4D in adults with fragile X syndrome: A randomized, placebo-controlled, phase 2 clinical trial. Nat. Med. 2021, 27, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Erickson, C.A.; Kaufmann, W.E.; Budimirovic, D.B.; Lachiewicz, A.; Haas-Givler, B.; Miller, R.M.; Weber, J.D.; Abbeduto, L.; Hessl, D.; Hagerman, R.J.; et al. Best Practices in Fragile X Syndrome Treatment Development. Brain Sci. 2018, 8, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagerman, R.; Jacquemont, S.; Berry-Kravis, E.; Des Portes, V.; Stanfield, A.; Koumaras, B.; Rosenkranz, G.; Murgia, A.; Wolf, C.; Apostol, G.; et al. Mavoglurant in Fragile X Syndrome: Results of two open-label, extension trials in adults and adolescents. Sci. Rep. 2018, 8, 16970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luu, S.; Province, H.; Berry-Kravis, E.; Hagerman, R.; Hessl, D.; Vaidya, D.; Lozano, R.; Rosselot, H.; Erickson, C.; Kaufmann, W.E.; et al. Response to Placebo in Fragile X Syndrome Clinical Trials: An Initial Analysis. Brain Sci. 2020, 10, 629. [Google Scholar] [CrossRef] [PubMed]
- Yamasue, H.; Aran, A.; Berry-Kravis, E. Emerging pharmacological therapies in fragile X syndrome and autism. Curr. Opin. Neurol. 2019, 32, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Leigh, M.J.S.; Nguyen, D.V.; Mu, Y.; Winarni, T.I.; Schneider, A.; Chechi, T.; Polussa, J.; Doucet, P.; Tassone, F.; Rivera, S.M.; et al. A Randomized Double-Blind, Placebo-Controlled Trial of Minocycline in Children and Adolescents with Fragile X Syndrome. J. Dev. Behav. Pediatrics 2013, 34, 147–155. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Zhao, X. Human pluripotent stem cell models of Fragile X syndrome. Mol. Cell. Neurosci. 2016, 73, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbach, A.; Bar-Nur, O.; Daley, G.Q.; Benvenisty, N. Differential Modeling of Fragile X Syndrome by Human Embryonic Stem Cells and Induced Pluripotent Stem Cells. Cell Stem Cell 2010, 6, 407–411. [Google Scholar] [CrossRef] [Green Version]
- Doers, M.E.; Musser, M.T.; Nichol, R.; Berndt, E.R.; Baker, M.; Gomez, T.M.; Zhang, S.-C.; Abbeduto, L.; Bhattacharyya, A. iPSC-Derived Forebrain Neurons from FXS Individuals Show Defects in Initial Neurite Outgrowth. Stem Cells Dev. 2014, 23, 1777–1787. [Google Scholar] [CrossRef]
- Li, M.; Zhao, H.; Ananiev, G.E.; Musser, M.T.; Ness, K.H.; Maglaque, D.L.; Saha, K.; Bhattacharyya, A.; Zhao, X. Establishment of Reporter Lines for Detecting Fragile X Mental Retardation (FMR1) Gene Reactivation in Human Neural Cells: FMR1 Reporter Human iPSC Line. Stem Cells 2017, 35, 158–169. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, M.; Schuffenhauer, A.; Fruh, I.; Klein, J.; Thiemeyer, A.; Rigo, P.; Gomez-Mancilla, B.; Heidinger-Millot, V.; Bouwmeester, T.; Schopfer, U.; et al. High-Throughput Screening Using iPSC-Derived Neuronal Progenitors to Identify Compounds Counteracting Epigenetic Gene Silencing in Fragile X Syndrome. J. Biomol. Screen. 2015, 20, 1101–1111. [Google Scholar] [CrossRef] [Green Version]
- Vershkov, D.; Fainstein, N.; Suissa, S.; Golan-Lev, T.; Ben-Hur, T.; Benvenisty, N. FMR1 Reactivating Treatments in Fragile X iPSC-Derived Neural Progenitors In Vitro and In Vivo. Cell Rep. 2019, 26, 2531–2539. [Google Scholar] [CrossRef] [Green Version]
- Kumari, D.; Swaroop, M.; Southall, N.; Huang, W.; Zheng, W.; Usdin, K. High-Throughput Screening to Identify Compounds That Increase Fragile X Mental Retardation Protein Expression in Neural Stem Cells Differentiated From Fragile X Syndrome Patient-Derived Induced Pluripotent Stem Cells. Stem Cells Transl. Med. 2015, 4, 800–808. [Google Scholar] [CrossRef]
- Bar-Nur, O.; Caspi, I.; Benvenisty, N. Molecular analysis of FMR1 reactivation in fragile-X induced pluripotent stem cells and their neuronal derivatives. J. Mol. Cell Biol. 2012, 4, 180–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Momparler, R.L.; Samson, J.; Momparler, L.F.; Rivard, G.E. Cell cycle effects and cellular pharmacology of 5-aza-2′-deoxycytidine. Cancer Chemother Pharm. 1984, 13, 191–194. [Google Scholar] [CrossRef]
- Rocha, M.A.; Veronezi, G.M.B.; Felisbino, M.B.; Gatti, M.S.V.; Tamashiro, W.; Mello, M.L.S. Sodium valproate and 5-aza-2′-deoxycytidine differentially modulate DNA demethylation in G1 phase-arrested and proliferative HeLa cells. Sci. Rep. 2019, 9, 18236. [Google Scholar] [CrossRef]
- Li, M.; Hunt, J.F.V.S.; Bhattacharyya, A.; Zhao, X. One-Step Generation of Seamless Luciferase Gene Knockin Using CRISPR/Cas9 Genome Editing in Human Pluripotent Stem Cells. In Fragile-X Syndrome; Ben-Yosef, D., Mayshar, Y., Eds.; Springer: New York, NY, USA, 2019; Volume 1942, pp. 61–69. [Google Scholar]
- Hunt, J.F.V.S.; Li, M.; Zhao, X.; Bhattacharyya, A. Using Human Neural Progenitor Cell Models to Conduct Large-Scale Drug Screens for Neurological and Psychiatric Diseases. In Fragile-X Syndrome; Ben-Yosef, D., Mayshar, Y., Eds.; Springer: New York, NY, USA, 2019; Volume 1942, pp. 79–88. [Google Scholar]
- Li, M.; Shin, J.; Risgaard, R.D.; Parries, M.J.; Wang, J.; Chasman, D.; Liu, S.; Roy, S.; Bhattacharyya, A.; Zhao, X. Identification of FMR1-regulated molecular networks in human neurodevelopment. Genome Res. 2020, 30, 361–374. [Google Scholar] [CrossRef]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Foundation for Statistical, version 3.5.1; A Language and Environment for Statistical Computing Software; Team, R.C.R: Vienna, Austria, 2019.
- Dahlin, J.L.; Nissink, J.W.M.; Strasser, J.M.; Francis, S.; Higgins, L.; Zhou, H.; Zhang, Z.; Walters, M.A. PAINS in the Assay: Chemical Mechanisms of Assay Interference and Promiscuous Enzymatic Inhibition Observed during a Sulfhydryl-Scavenging HTS. J. Med. Chem. 2015, 58, 2091–2113. [Google Scholar] [CrossRef] [Green Version]
- Lagorce, D.; Bouslama, L.; Becot, J.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs4: Free ADME-tox filtering computations for chemical biology and early stages drug discovery. Bioinformatics 2017, 33, 3658–3660. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hunt, J.F.V.; Li, M.; Risgaard, R.; Ananiev, G.E.; Wildman, S.; Zhang, F.; Bugni, T.S.; Zhao, X.; Bhattacharyya, A. High Throughput Small Molecule Screen for Reactivation of FMR1 in Fragile X Syndrome Human Neural Cells. Cells 2022, 11, 69. https://doi.org/10.3390/cells11010069
Hunt JFV, Li M, Risgaard R, Ananiev GE, Wildman S, Zhang F, Bugni TS, Zhao X, Bhattacharyya A. High Throughput Small Molecule Screen for Reactivation of FMR1 in Fragile X Syndrome Human Neural Cells. Cells. 2022; 11(1):69. https://doi.org/10.3390/cells11010069
Chicago/Turabian StyleHunt, Jack F. V., Meng Li, Ryan Risgaard, Gene E. Ananiev, Scott Wildman, Fan Zhang, Tim S. Bugni, Xinyu Zhao, and Anita Bhattacharyya. 2022. "High Throughput Small Molecule Screen for Reactivation of FMR1 in Fragile X Syndrome Human Neural Cells" Cells 11, no. 1: 69. https://doi.org/10.3390/cells11010069
APA StyleHunt, J. F. V., Li, M., Risgaard, R., Ananiev, G. E., Wildman, S., Zhang, F., Bugni, T. S., Zhao, X., & Bhattacharyya, A. (2022). High Throughput Small Molecule Screen for Reactivation of FMR1 in Fragile X Syndrome Human Neural Cells. Cells, 11(1), 69. https://doi.org/10.3390/cells11010069