
Impact of STING Inflammatory Signaling during Intracellular Bacterial Infections

 , , , and
, , , and 
Abstract
:1. Introduction
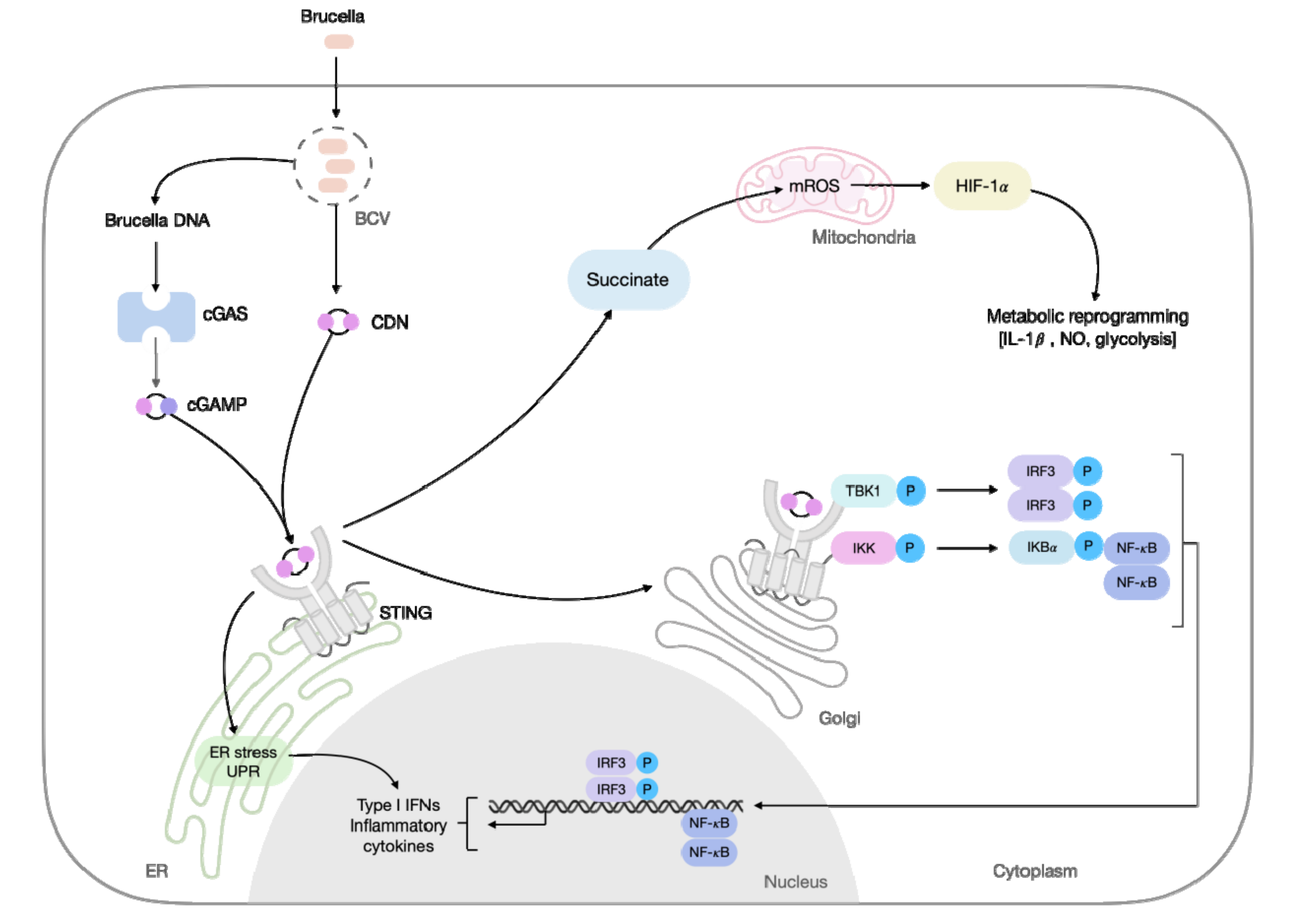
2. Basis of STING Activation
3. STING and Endoplasmic Reticulum Stress
4. Macrophage Polarization: STING Activation as an Inflammatory Inducer
5. STING Role during Intracellular Bacterial Infections
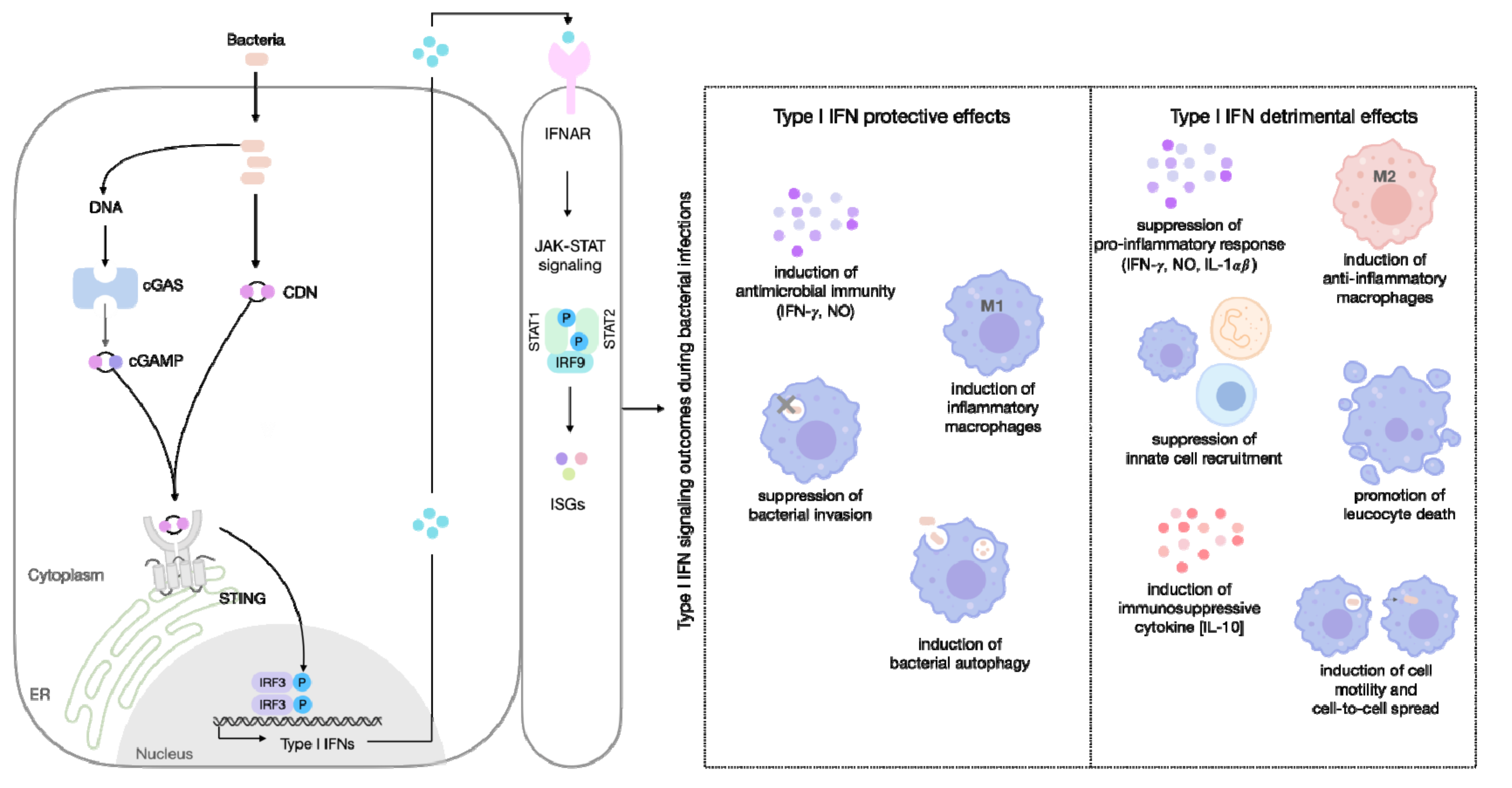
6. The Dichotomy of Type I IFN Responses: Host Resistance versus Susceptibility
6.1. Type I IFN-Inducing Protection in Bacterial Infection
6.2. Detrimental Role of Type I IFN during Bacterial Infection
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kranzusch, P.J.; Wilson, S.C.; Lee, A.S.; Berger, J.M.; Doudna, J.A.; Vance, R.E. Ancient Origin of cGAS-STING Reveals Mechanism of Universal 2′,3′ cGAMP Signaling. Mol. Cell 2015, 59, 891–903. [Google Scholar] [CrossRef] [Green Version]
- Medzhitov, R. Recognition of microorganisms and activation of the immune response. Nature 2007, 449, 819–826. [Google Scholar] [CrossRef]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef]
- Boxx, G.M.; Cheng, G. The Roles of Type I Interferon in Bacterial Infection. Cell Host Microbe 2016, 19, 760–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, G.N. STING: Infection, inflammation and cancer. Nat. Rev. Immunol. 2015, 15, 760–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marinho, F.V.; Benmerzoug, S.; Oliveira, S.C.; Ryffel, B.; Quesniaux, V.F.J. The Emerging Roles of STING in Bacterial Infections. Trends Microbiol. 2017, 25, 906–918. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V. A STING to inflammation and autoimmunity. J. Leukoc. Biol. 2019, 106, 171–185. [Google Scholar] [CrossRef]
- Burdette, D.L.; Monroe, K.M.; Sotelo-Troha, K.; Iwig, J.S.; Eckert, B.; Hyodo, M.; Hayakawa, Y.; Vance, R.E. STING is a direct innate immune sensor of cyclic di-GMP. Nature 2011, 478, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Woodward, J.J.; Iavarone, A.T.; Portnoy, D.A. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 2010, 328, 1703–1705. [Google Scholar] [CrossRef] [Green Version]
- Krasteva, P.V.; Sondermann, H. Versatile modes of cellular regulation via cyclic dinucleotides. Nat. Chem. Biol. 2017, 13, 350–359. [Google Scholar] [CrossRef]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Rohl, I.; Hopfner, K.P.; Ludwig, J.; Hornung, V. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diner, E.J.; Burdette, D.L.; Wilson, S.C.; Monroe, K.M.; Kellenberger, C.A.; Hyodo, M.; Hayakawa, Y.; Hammond, M.C.; Vance, R.E. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep. 2013, 3, 1355–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.D.; Wu, J.; Gao, D.; Wang, H.; Sun, L.; Chen, Z.J. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 2013, 341, 1390–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, G.; Ma, Z.; Damania, B. cGAS and STING: At the intersection of DNA and RNA virus-sensing networks. PLoS Pathog. 2018, 14, e1007148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, D.; Wu, J.; Wu, Y.T.; Du, F.; Aroh, C.; Yan, N.; Sun, L.; Chen, Z.J. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 2013, 341, 903–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, W.O.; Butler, N.S.; Lindner, S.E.; Akilesh, H.M.; Sather, D.N.; Kappe, S.H.; Hamerman, J.A.; Gale, M., Jr.; Liles, W.C.; Pepper, M. cGAS-mediated control of blood-stage malaria promotes Plasmodium-specific germinal center responses. JCI Insight 2018, 3, e94142. [Google Scholar] [CrossRef] [Green Version]
- Souza, C.; Sanches, R.C.O.; Assis, N.R.G.; Marinho, F.V.; Mambelli, F.S.; Morais, S.B.; Gimenez, E.G.T.; Guimaraes, E.S.; Castro, T.B.R.; Oliveira, S.C. The role of the adaptor molecule STING during Schistosoma mansoni infection. Sci. Rep. 2020, 10, 7901. [Google Scholar] [CrossRef]
- Hopfner, K.P.; Hornung, V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef]
- Luecke, S.; Holleufer, A.; Christensen, M.H.; Jonsson, K.L.; Boni, G.A.; Sorensen, L.K.; Johannsen, M.; Jakobsen, M.R.; Hartmann, R.; Paludan, S.R. cGAS is activated by DNA in a length-dependent manner. EMBO Rep. 2017, 18, 1707–1715. [Google Scholar] [CrossRef]
- Barnett, K.C.; Coronas-Serna, J.M.; Zhou, W.; Ernandes, M.J.; Cao, A.; Kranzusch, P.J.; Kagan, J.C. Phosphoinositide Interactions Position cGAS at the Plasma Membrane to Ensure Efficient Distinction between Self- and Viral DNA. Cell 2019, 176, 1432–1446.e1411. [Google Scholar] [CrossRef] [Green Version]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Shi, H.; Wu, J.; Zhang, X.; Sun, L.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol. Cell 2013, 51, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Burdette, D.L.; Vance, R.E. STING and the innate immune response to nucleic acids in the cytosol. Nat. Immunol. 2013, 14, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef] [PubMed]
- Shang, G.; Zhang, C.; Chen, Z.J.; Bai, X.C.; Zhang, X. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature 2019, 567, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, E.; Mukai, K.; Saito, K.; Arai, H.; Taguchi, T. The binding of TBK1 to STING requires exocytic membrane traffic from the ER. Biochem. Biophys. Res. Commun. 2018, 503, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Shang, G.; Gui, X.; Zhang, X.; Bai, X.C.; Chen, Z.J. Structural basis of STING binding with and phosphorylation by TBK1. Nature 2019, 567, 394–398. [Google Scholar] [CrossRef]
- Balka, K.R.; Louis, C.; Saunders, T.L.; Smith, A.M.; Calleja, D.J.; D’Silva, D.B.; Moghaddas, F.; Tailler, M.; Lawlor, K.E.; Zhan, Y.; et al. TBK1 and IKKepsilon Act Redundantly to Mediate STING-Induced NF-kappaB Responses in Myeloid Cells. Cell Rep. 2020, 31, 107492. [Google Scholar] [CrossRef]
- Pomerantz, J.L.; Baltimore, D. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. 1999, 18, 6694–6704. [Google Scholar] [CrossRef] [PubMed]
- Gui, X.; Yang, H.; Li, T.; Tan, X.; Shi, P.; Li, M.; Du, F.; Chen, Z.J. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 2019, 567, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Gulen, M.F.; Koch, U.; Haag, S.M.; Schuler, F.; Apetoh, L.; Villunger, A.; Radtke, F.; Ablasser, A. Signalling strength determines proapoptotic functions of STING. Nat. Commun. 2017, 8, 427. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, G.R.; Darnell, J.E., Jr. The JAK-STAT pathway at twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.; Barber, G.N. Self-DNA, STING-dependent signaling and the origins of autoinflammatory disease. Curr. Opin. Immunol. 2014, 31, 121–126. [Google Scholar] [CrossRef]
- Hebert, D.N.; Molinari, M. In and out of the ER: Protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 2007, 87, 1377–1408. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Grootjans, J.; Kaser, A.; Kaufman, R.J.; Blumberg, R.S. The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 2016, 16, 469–484. [Google Scholar] [CrossRef] [Green Version]
- Janssens, S.; Pulendran, B.; Lambrecht, B.N. Emerging functions of the unfolded protein response in immunity. Nat. Immunol. 2014, 15, 910–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iracheta-Vellve, A.; Petrasek, J.; Gyongyosi, B.; Satishchandran, A.; Lowe, P.; Kodys, K.; Catalano, D.; Calenda, C.D.; Kurt-Jones, E.A.; Fitzgerald, K.A.; et al. Endoplasmic Reticulum Stress-induced Hepatocellular Death Pathways Mediate Liver Injury and Fibrosis via Stimulator of Interferon Genes. J. Biol. Chem. 2016, 291, 26794–26805. [Google Scholar] [CrossRef] [Green Version]
- Petrasek, J.; Iracheta-Vellve, A.; Csak, T.; Satishchandran, A.; Kodys, K.; Kurt-Jones, E.A.; Fitzgerald, K.A.; Szabo, G. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc. Natl. Acad. Sci. USA 2013, 110, 16544–16549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.A. STING, the Endoplasmic Reticulum, and Mitochondria: Is Three a Crowd or a Conversation? Front. Immunol. 2020, 11, 611347. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zhao, D.; Sreevatsan, S.; Liu, C.; Yang, W.; Song, Z.; Yang, L.; Barrow, P.; Zhou, X. Mycobacterium bovis Induces Endoplasmic Reticulum Stress Mediated-Apoptosis by Activating IRF3 in a Murine Macrophage Cell Line. Front. Cell. Infect. Microbiol. 2016, 6, 182. [Google Scholar] [CrossRef] [Green Version]
- Moretti, J.; Roy, S.; Bozec, D.; Martinez, J.; Chapman, J.R.; Ueberheide, B.; Lamming, D.W.; Chen, Z.J.; Horng, T.; Yeretssian, G.; et al. STING Senses Microbial Viability to Orchestrate Stress-Mediated Autophagy of the Endoplasmic Reticulum. Cell 2017, 171, 809–823.e813. [Google Scholar] [CrossRef]
- Guimaraes, E.S.; Gomes, M.T.R.; Campos, P.C.; Mansur, D.S.; Dos Santos, A.A.; Harms, J.; Splitter, G.; Smith, J.A.; Barber, G.N.; Oliveira, S.C. Brucella abortus Cyclic Dinucleotides Trigger STING-Dependent Unfolded Protein Response That Favors Bacterial Replication. J. Immunol. 2019, 202, 2671–2681. [Google Scholar] [CrossRef] [PubMed]
- Benoit, M.; Desnues, B.; Mege, J.L. Macrophage polarization in bacterial infections. J. Immunol. 2008, 181, 3733–3739. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, D.G.; Huang, L.; VanderVen, B.C. Immunometabolism at the interface between macrophages and pathogens. Nat. Rev. Immunol. 2019, 19, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Downey, C.M.; Aghaei, M.; Schwendener, R.A.; Jirik, F.R. DMXAA causes tumor site-specific vascular disruption in murine non-small cell lung cancer, and like the endogenous non-canonical cyclic dinucleotide STING agonist, 2′3′-cGAMP, induces M2 macrophage repolarization. PLoS ONE 2014, 9, e99988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, F.J.; Gomez, M.I.; Wetzel, D.M.; Memmi, G.; O’Seaghdha, M.; Soong, G.; Schindler, C.; Prince, A. Staphylococcus aureus activates type I IFN signaling in mice and humans through the Xr repeated sequences of protein A. J. Clin. Investig. 2009, 119, 1931–1939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa Franco, M.M.; Marim, F.; Guimaraes, E.S.; Assis, N.R.G.; Cerqueira, D.M.; Alves-Silva, J.; Harms, J.; Splitter, G.; Smith, J.; Kanneganti, T.D.; et al. Brucella abortus Triggers a cGAS-Independent STING Pathway to Induce Host Protection That Involves Guanylate-Binding Proteins and Inflammasome Activation. J. Immunol. 2018, 200, 607–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, M.T.R.; Guimarães, E.S.; Marinho, F.V.; Macedo, I.; Aguiar, E.; Barber, G.N.; Moraes-Vieira, P.M.M.; Alves-Filho, J.C.; Oliveira, S.C. STING regulates metabolic reprogramming in macrophages via HIF-1α during Brucella infection. PLoS Pathog. 2021, 17, e1009597. [Google Scholar] [CrossRef]
- Olson, G.S.; Murray, T.A.; Jahn, A.N.; Mai, D.; Diercks, A.H.; Gold, E.S.; Aderem, A. Type I interferon decreases macrophage energy metabolism during mycobacterial infection. Cell Rep. 2021, 35, 109195. [Google Scholar] [CrossRef]
- Benmerzoug, S.; Bounab, B.; Rose, S.; Gosset, D.; Biet, F.; Cochard, T.; Xavier, A.; Rouxel, N.; Fauconnier, L.; Horsnell, W.G.C.; et al. Sterile Lung Inflammation Induced by Silica Exacerbates Mycobacterium tuberculosis Infection via STING-Dependent Type 2 Immunity. Cell Rep. 2019, 27, 2649–2664.e2645. [Google Scholar] [CrossRef] [Green Version]
- Van Dis, E.; Sogi, K.M.; Rae, C.S.; Sivick, K.E.; Surh, N.H.; Leong, M.L.; Kanne, D.B.; Metchette, K.; Leong, J.J.; Bruml, J.R.; et al. STING-Activating Adjuvants Elicit a Th17 Immune Response and Protect against Mycobacterium tuberculosis Infection. Cell Rep. 2018, 23, 1435–1447. [Google Scholar] [CrossRef]
- Dey, R.J.; Dey, B.; Singh, A.K.; Praharaj, M.; Bishai, W. Bacillus Calmette-Guérin Overexpressing an Endogenous Stimulator of Interferon Genes Agonist Provides Enhanced Protection Against Pulmonary Tuberculosis. J. Infect. Dis. 2020, 221, 1048–1056. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.K.; Praharaj, M.; Lombardo, K.A.; Yoshida, T.; Matoso, A.; Baras, A.S.; Zhao, L.; Prasad, P.; Powell, J.D.; Kates, M.; et al. Recombinant BCG overexpressing a STING agonist elicits trained immunity and improved antitumor efficacy in non-muscle invasive bladder cancer. Urol. Oncol. Semin. Orig. Investig. 2020, 38, 899. [Google Scholar] [CrossRef]
- Lippmann, J.; Muller, H.C.; Naujoks, J.; Tabeling, C.; Shin, S.; Witzenrath, M.; Hellwig, K.; Kirschning, C.J.; Taylor, G.A.; Barchet, W.; et al. Dissection of a type I interferon pathway in controlling bacterial intracellular infection in mice. Cell. Microbiol. 2011, 13, 1668–1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul-Sater, A.A.; Grajkowski, A.; Erdjument-Bromage, H.; Plumlee, C.; Levi, A.; Schreiber, M.T.; Lee, C.; Shuman, H.; Beaucage, S.L.; Schindler, C. The overlapping host responses to bacterial cyclic dinucleotides. Microbes Infect. 2012, 14, 188–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Moreno, J.S.; Hamann, L.; Shah, J.A.; Verbon, A.; Mockenhaupt, F.P.; Puzianowska-Kuznicka, M.; Naujoks, J.; Sander, L.E.; Witzenrath, M.; Cambier, J.C.; et al. The common HAQ STING variant impairs cGAS-dependent antibacterial responses and is associated with susceptibility to Legionnaires’ disease in humans. PLoS Pathog. 2018, 14, e1006829. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, R.; Tschismarov, R.; Meissner, F.; Prabakaran, T.; Krissanaprasit, A.; Farahani, E.; Zhang, B.C.; Assil, S.; Martin, A.; Bertrams, W.; et al. Intracellular bacteria engage a STING-TBK1-MVB12b pathway to enable paracrine cGAS-STING signalling. Nat. Microbiol. 2019, 4, 701–713. [Google Scholar] [CrossRef]
- Cao, Y.; Guan, K.; He, X.; Wei, C.; Zheng, Z.; Zhang, Y.; Ma, S.; Zhong, H.; Shi, W. Yersinia YopJ negatively regulates IRF3-mediated antibacterial response through disruption of STING-mediated cytosolic DNA signaling. Biochim. Biophys. Acta 2016, 1863, 3148–3159. [Google Scholar] [CrossRef] [PubMed]
- Ku, J.W.K.; Chen, Y.; Lim, B.J.W.; Gasser, S.; Crasta, K.C.; Gan, Y.H. Bacterial-induced cell fusion is a danger signal triggering cGAS-STING pathway via micronuclei formation. Proc. Natl. Acad. Sci. USA 2020, 117, 15923–15934. [Google Scholar] [CrossRef]
- Webster, S.J.; Brode, S.; Ellis, L.; Fitzmaurice, T.J.; Elder, M.J.; Gekara, N.O.; Tourlomousis, P.; Bryant, C.; Clare, S.; Chee, R.; et al. Detection of a microbial metabolite by STING regulates inflammasome activation in response to Chlamydia trachomatis infection. PLoS Pathog. 2017, 13, e1006383. [Google Scholar] [CrossRef]
- Sixt, B.S.; Bastidas, R.J.; Finethy, R.; Baxter, R.M.; Carpenter, V.K.; Kroemer, G.; Coers, J.; Valdivia, R.H. The Chlamydia trachomatis Inclusion Membrane Protein CpoS Counteracts STING-Mediated Cellular Surveillance and Suicide Programs. Cell Host Microbe 2017, 21, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.; Li, Z. The STING pathway in response to chlamydial infection. Microb. Pathog. 2020, 140, 103950. [Google Scholar] [CrossRef]
- Louie, A.; Bhandula, V.; Portnoy, D.A. Secretion of c-di-AMP by Listeria monocytogenes Leads to a STING-Dependent Antibacterial Response during Enterocolitis. Infect. Immun. 2020, 88, e00407-20. [Google Scholar] [CrossRef]
- Park, S.M.; Omatsu, T.; Zhao, Y.; Yoshida, N.; Shah, P.; Zagani, R.; Reinecker, H.C. T cell fate following Salmonella infection is determined by a STING-IRF1 signaling axis in mice. Commun. Biol. 2019, 2, 464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerqueira, D.M.; Gomes, M.T.R.; Silva, A.L.N.; Rungue, M.; Assis, N.R.G.; Guimaraes, E.S.; Morais, S.B.; Broz, P.; Zamboni, D.S.; Oliveira, S.C. Guanylate-binding protein 5 licenses caspase-11 for Gasdermin-D mediated host resistance to Brucella abortus infection. PLoS Pathog. 2018, 14, e1007519. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Harms, J.S.; Liu, Y.; Eickhoff, J.; Tan, J.W.; Hu, T.; Cai, F.; Guimaraes, E.; Oliveira, S.C.; Dahl, R.; et al. Brucella suppress STING expression via miR-24 to enhance infection. PLoS Pathog. 2020, 16, e1009020. [Google Scholar] [CrossRef] [PubMed]
- Marinho, F.V.; Benmerzoug, S.; Rose, S.; Campos, P.C.; Marques, J.T.; Bafica, A.; Barber, G.; Ryffel, B.; Oliveira, S.C.; Quesniaux, V.F.J. The cGAS/STING Pathway Is Important for Dendritic Cell Activation but Is Not Essential to Induce Protective Immunity against Mycobacterium tuberculosis Infection. J. Innate Immun. 2018, 10, 239–252. [Google Scholar] [CrossRef]
- Li, Q.; Liu, C.; Yue, R.; El-Ashram, S.; Wang, J.; He, X.; Zhao, D.; Zhou, X.; Xu, L. cGAS/STING/TBK1/IRF3 Signaling Pathway Activates BMDCs Maturation following Mycobacterium bovis Infection. Int. J. Mol. Sci. 2019, 20, 895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, S.; Shen, H.; Lian, Q.; Jin, W.; Zhang, R.; Lin, X.; Gu, W.; Sun, X.; Meng, G.; Tian, Z.; et al. Deficiency of the AIM2-ASC Signal Uncovers the STING-Driven Overreactive Response of Type I IFN and Reciprocal Depression of Protective IFN-gamma Immunity in Mycobacterial Infection. J. Immunol. 2018, 200, 1016–1026. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, W.; Dong, C.; Xiong, S. Mycobacterium tuberculosis MmsA (Rv0753c) Interacts with STING and Blunts the Type I Interferon Response. mBio 2020, 11, e03254-19. [Google Scholar] [CrossRef] [PubMed]
- Ruangkiattikul, N.; Nerlich, A.; Abdissa, K.; Lienenklaus, S.; Suwandi, A.; Janze, N.; Laarmann, K.; Spanier, J.; Kalinke, U.; Weiss, S.; et al. cGAS-STING-TBK1-IRF3/7 induced interferon-beta contributes to the clearing of non tuberculous mycobacterial infection in mice. Virulence 2017, 8, 1303–1315. [Google Scholar] [CrossRef] [Green Version]
- Decker, T.; Muller, M.; Stockinger, S. The yin and yang of type I interferon activity in bacterial infection. Nat. Rev. Immunol. 2005, 5, 675–687. [Google Scholar] [CrossRef]
- De la Maza, L.M.; Peterson, E.M.; Goebel, J.M.; Fennie, C.W.; Czarniecki, C.W. Interferon-induced inhibition of Chlamydia trachomatis: Dissociation from antiviral and antiproliferative effects. Infect. Immun. 1985, 47, 719–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazar, J.; Gillmore, J.D.; Gordon, F.B. Effect of Interferon and Interferon Inducers on Infections with a Nonviral Intracellular Microorganism, Chlamydia trachomatis. Infect. Immun. 1971, 3, 825–832. [Google Scholar] [CrossRef] [Green Version]
- Rodel, J.; Assefa, S.; Prochnau, D.; Woytas, M.; Hartmann, M.; Groh, A.; Straube, E. Interferon-beta induction by Chlamydia pneumoniae in human smooth muscle cells. FEMS Immunol. Med. Microbiol. 2001, 32, 9–15. [Google Scholar] [CrossRef]
- Opitz, B.; Vinzing, M.; van Laak, V.; Schmeck, B.; Heine, G.; Gunther, S.; Preissner, R.; Slevogt, H.; N’Guessan, P.D.; Eitel, J.; et al. Legionella pneumophila induces IFNbeta in lung epithelial cells via IPS-1 and IRF3, which also control bacterial replication. J. Biol. Chem. 2006, 281, 36173–36179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plumlee, C.R.; Lee, C.; Beg, A.A.; Decker, T.; Shuman, H.A.; Schindler, C. Interferons direct an effective innate response to Legionella pneumophila infection. J. Biol. Chem. 2009, 284, 30058–30066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiavoni, G.; Mauri, C.; Carlei, D.; Belardelli, F.; Pastoris, M.C.; Proietti, E. Type I IFN protects permissive macrophages from Legionella pneumophila infection through an IFN-gamma-independent pathway. J. Immunol. 2004, 173, 1266–1275. [Google Scholar] [CrossRef] [Green Version]
- Ang, D.K.; Oates, C.V.; Schuelein, R.; Kelly, M.; Sansom, F.M.; Bourges, D.; Boon, L.; Hertzog, P.J.; Hartland, E.L.; van Driel, I.R. Cutting edge: Pulmonary Legionella pneumophila is controlled by plasmacytoid dendritic cells but not type I IFN. J. Immunol. 2010, 184, 5429–5433. [Google Scholar] [CrossRef] [Green Version]
- Monroe, K.M.; McWhirter, S.M.; Vance, R.E. Identification of host cytosolic sensors and bacterial factors regulating the type I interferon response to Legionella pneumophila. PLoS Pathog. 2009, 5, e1000665. [Google Scholar] [CrossRef]
- Niesel, D.W.; Hess, C.B.; Cho, Y.J.; Klimpel, K.D.; Klimpel, G.R. Natural and recombinant interferons inhibit epithelial cell invasion by Shigella spp. Infect. Immun. 1986, 52, 828–833. [Google Scholar] [CrossRef] [Green Version]
- Helbig, K.J.; Teh, M.Y.; Crosse, K.M.; Monson, E.A.; Smith, M.; Tran, E.N.; Standish, A.J.; Morona, R.; Beard, M.R. The interferon stimulated gene viperin, restricts Shigella. flexneri in vitro. Sci. Rep. 2019, 9, 15598. [Google Scholar] [CrossRef] [Green Version]
- Dorrington, M.G.; Bradfield, C.J.; Lack, J.B.; Lin, B.; Liang, J.J.; Starr, T.; Ernst, O.; Gross, J.L.; Sun, J.; Miller, A.H.; et al. Type I IFNs facilitate innate immune control of the opportunistic bacteria Burkholderia cenocepacia in the macrophage cytosol. PLoS Pathog. 2021, 17, e1009395. [Google Scholar] [CrossRef]
- Qiu, H.; Fan, Y.; Joyee, A.G.; Wang, S.; Han, X.; Bai, H.; Jiao, L.; Van Rooijen, N.; Yang, X. Type I IFNs enhance susceptibility to Chlamydia muridarum lung infection by enhancing apoptosis of local macrophages. J. Immunol. 2008, 181, 2092–2102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagarajan, U.M.; Prantner, D.; Sikes, J.D.; Andrews, C.W., Jr.; Goodwin, A.M.; Nagarajan, S.; Darville, T. Type I interferon signaling exacerbates Chlamydia muridarum genital infection in a murine model. Infect. Immun. 2008, 76, 4642–4648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auerbuch, V.; Brockstedt, D.G.; Meyer-Morse, N.; O’Riordan, M.; Portnoy, D.A. Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J. Exp. Med. 2004, 200, 527–533. [Google Scholar] [CrossRef] [Green Version]
- Carrero, J.A.; Calderon, B.; Unanue, E.R. Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J. Exp. Med. 2004, 200, 535–540. [Google Scholar] [CrossRef]
- Jia, T.; Leiner, I.; Dorothee, G.; Brandl, K.; Pamer, E.G. MyD88 and Type I interferon receptor-mediated chemokine induction and monocyte recruitment during Listeria monocytogenes infection. J. Immunol. 2009, 183, 1271–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connell, R.M.; Saha, S.K.; Vaidya, S.A.; Bruhn, K.W.; Miranda, G.A.; Zarnegar, B.; Perry, A.K.; Nguyen, B.O.; Lane, T.F.; Taniguchi, T.; et al. Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J. Exp. Med. 2004, 200, 437–445. [Google Scholar] [CrossRef] [Green Version]
- Carrero, J.A.; Calderon, B.; Unanue, E.R. Lymphocytes are detrimental during the early innate immune response against Listeria monocytogenes. J. Exp. Med. 2006, 203, 933–940. [Google Scholar] [CrossRef]
- Rayamajhi, M.; Humann, J.; Penheiter, K.; Andreasen, K.; Lenz, L.L. Induction of IFN-alphabeta enables Listeria monocytogenes to suppress macrophage activation by IFN-gamma. J. Exp. Med. 2010, 207, 327–337. [Google Scholar] [CrossRef] [Green Version]
- Brzoza-Lewis, K.L.; Hoth, J.J.; Hiltbold, E.M. Type I interferon signaling regulates the composition of inflammatory infiltrates upon infection with Listeria monocytogenes. Cell. Immunol. 2012, 273, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Henry, T.; Kirimanjeswara, G.S.; Ruby, T.; Jones, J.W.; Peng, K.; Perret, M.; Ho, L.; Sauer, J.D.; Iwakura, Y.; Metzger, D.W.; et al. Type I IFN signaling constrains IL-17A/F secretion by gammadelta T cells during bacterial infections. J. Immunol. 2010, 184, 3755–3767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, K.A.; Durack, J.; Portnoy, D.A. STING-dependent type I IFN production inhibits cell-mediated immunity to Listeria monocytogenes. PLoS Pathog. 2014, 10, e1003861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborne, S.E.; Sit, B.; Shaker, A.; Currie, E.; Tan, J.M.; van Rijn, J.; Higgins, D.E.; Brumell, J.H. Type I interferon promotes cell-to-cell spread of Listeria monocytogenes. Cell. Microbiol. 2017, 19, e12660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, J.M.J.; Garner, M.E.; Regeimbal, J.M.; Greene, C.J.; Marquez, J.D.R.; Ammendolia, D.A.; McCluggage, A.R.R.; Li, T.; Wu, K.J.; Cemma, M.; et al. Listeria exploits IFITM3 to suppress antibacterial activity in phagocytes. Nat. Commun. 2021, 12, 4999. [Google Scholar] [CrossRef]
- Frantz, R.; Teubner, L.; Schultze, T.; La Pietra, L.; Muller, C.; Gwozdzinski, K.; Pillich, H.; Hain, T.; Weber-Gerlach, M.; Panagiotidis, G.D.; et al. The secRNome of Listeria monocytogenes Harbors Small Noncoding RNAs That Are Potent Inducers of Beta Interferon. mBio 2019, 10, e01223-19. [Google Scholar] [CrossRef] [Green Version]
- Pagliuso, A.; Tham, T.N.; Allemand, E.; Robertin, S.; Dupuy, B.; Bertrand, Q.; Becavin, C.; Koutero, M.; Najburg, V.; Nahori, M.A.; et al. An RNA-Binding Protein Secreted by a Bacterial Pathogen Modulates RIG-I Signaling. Cell Host Microbe 2019, 26, 823–835.e811. [Google Scholar] [CrossRef] [Green Version]
- Malakhova, O.A.; Kim, K.I.; Luo, J.K.; Zou, W.; Kumar, K.G.; Fuchs, S.Y.; Shuai, K.; Zhang, D.E. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 2006, 25, 2358–2367. [Google Scholar] [CrossRef]
- Shaabani, N.; Honke, N.; Nguyen, N.; Huang, Z.; Arimoto, K.I.; Lazar, D.; Loe, T.K.; Lang, K.S.; Prinz, M.; Knobeloch, K.P.; et al. The probacterial effect of type I interferon signaling requires its own negative regulator USP18. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Kernbauer, E.; Maier, V.; Rauch, I.; Muller, M.; Decker, T. Route of Infection Determines the Impact of Type I Interferons on Innate Immunity to Listeria monocytogenes. PLoS ONE 2013, 8, e65007. [Google Scholar] [CrossRef] [Green Version]
- Pitts, M.G.; Myers-Morales, T.; D’Orazio, S.E. Type I IFN Does Not Promote Susceptibility to Foodborne Listeria monocytogenes. J. Immunol. 2016, 196, 3109–3116. [Google Scholar] [CrossRef] [Green Version]
- Henry, T.; Brotcke, A.; Weiss, D.S.; Thompson, L.J.; Monack, D.M. Type I interferon signaling is required for activation of the inflammasome during Francisella infection. J. Exp. Med. 2007, 204, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Roark, C.L.; Simonian, P.L.; Fontenot, A.P.; Born, W.K.; O’Brien, R.L. Gammadelta T cells: An important source of IL-17. Curr. Opin. Immunol. 2008, 20, 353–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storek, K.M.; Gertsvolf, N.A.; Ohlson, M.B.; Monack, D.M. cGAS and Ifi204 cooperate to produce type I IFNs in response to Francisella infection. J. Immunol. 2015, 194, 3236–3245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, J.W.; Kayagaki, N.; Broz, P.; Henry, T.; Newton, K.; O’Rourke, K.; Chan, S.; Dong, J.; Qu, Y.; Roose-Girma, M.; et al. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc. Natl. Acad. Sci. USA 2010, 107, 9771–9776. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Man, S.M.; Karki, R.; Malireddi, R.K.S.; Kanneganti, T.D. Detrimental Type I Interferon Signaling Dominates Protective AIM2 Inflammasome Responses during Francisella novicida Infection. Cell Rep. 2018, 22, 3168–3174. [Google Scholar] [CrossRef] [Green Version]
- Perkins, D.J.; Rajaiah, R.; Tennant, S.M.; Ramachandran, G.; Higginson, E.E.; Dyson, T.N.; Vogel, S.N. Salmonella Typhimurium Co-Opts the Host Type I IFN System to Restrict Macrophage Innate Immune Transcriptional Responses Selectively. J. Immunol. 2015, 195, 2461–2471. [Google Scholar] [CrossRef] [Green Version]
- Robinson, N.; McComb, S.; Mulligan, R.; Dudani, R.; Krishnan, L.; Sad, S. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat. Immunol. 2012, 13, 954–962. [Google Scholar] [CrossRef]
- Dhariwala, M.O.; Olson, R.M.; Anderson, D.M. Induction of Type I Interferon through a Noncanonical Toll-Like Receptor 7 Pathway during Yersinia pestis Infection. Infect. Immun. 2017, 85, e00570-17. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.A.; Lee-Lewis, H.; Hughes-Hanks, J.; Lewis, C.A.; Anderson, D.M. Opposing roles for interferon regulatory factor-3 (IRF-3) and type I interferon signaling during plague. PLoS Pathog. 2012, 8, e1002817. [Google Scholar] [CrossRef]
- De Almeida, L.A.; Carvalho, N.B.; Oliveira, F.S.; Lacerda, T.L.; Vasconcelos, A.C.; Nogueira, L.; Bafica, A.; Silva, A.M.; Oliveira, S.C. MyD88 and STING signaling pathways are required for IRF3-mediated IFN-beta induction in response to Brucella abortus infection. PLoS ONE 2011, 6, e23135. [Google Scholar] [CrossRef] [Green Version]
- Donovan, M.L.; Schultz, T.E.; Duke, T.J.; Blumenthal, A. Type I Interferons in the Pathogenesis of Tuberculosis: Molecular Drivers and Immunological Consequences. Front. Immunol. 2017, 8, 1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, M.P.; Graham, C.M.; McNab, F.W.; Xu, Z.; Bloch, S.A.; Oni, T.; Wilkinson, K.A.; Banchereau, R.; Skinner, J.; Wilkinson, R.J.; et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 2010, 466, 973–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maertzdorf, J.; Repsilber, D.; Parida, S.K.; Stanley, K.; Roberts, T.; Black, G.; Walzl, G.; Kaufmann, S.H. Human gene expression profiles of susceptibility and resistance in tuberculosis. Genes Immun. 2011, 12, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Zak, D.E.; Penn-Nicholson, A.; Scriba, T.J.; Thompson, E.; Suliman, S.; Amon, L.M.; Mahomed, H.; Erasmus, M.; Whatney, W.; Hussey, G.D.; et al. A blood RNA signature for tuberculosis disease risk: A prospective cohort study. Lancet 2016, 387, 2312–2322. [Google Scholar] [CrossRef] [Green Version]
- Manca, C.; Tsenova, L.; Bergtold, A.; Freeman, S.; Tovey, M.; Musser, J.M.; Barry, C.E., 3rd; Freedman, V.H.; Kaplan, G. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-alpha/beta. Proc. Natl. Acad. Sci. USA 2001, 98, 5752–5757. [Google Scholar] [CrossRef] [Green Version]
- Manca, C.; Tsenova, L.; Freeman, S.; Barczak, A.K.; Tovey, M.; Murray, P.J.; Barry, C.; Kaplan, G. Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J. Interf. Cytokine Res. 2005, 25, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Ordway, D.; Henao-Tamayo, M.; Harton, M.; Palanisamy, G.; Troudt, J.; Shanley, C.; Basaraba, R.J.; Orme, I.M. The hypervirulent Mycobacterium tuberculosis strain HN878 induces a potent TH1 response followed by rapid down-regulation. J. Immunol. 2007, 179, 522–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanley, S.A.; Johndrow, J.E.; Manzanillo, P.; Cox, J.S. The Type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J. Immunol. 2007, 178, 3143–3152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorhoi, A.; Yeremeev, V.; Nouailles, G.; Weiner, J., 3rd; Jorg, S.; Heinemann, E.; Oberbeck-Muller, D.; Knaul, J.K.; Vogelzang, A.; Reece, S.T.; et al. Type I IFN signaling triggers immunopathology in tuberculosis-susceptible mice by modulating lung phagocyte dynamics. Eur. J. Immunol. 2014, 44, 2380–2393. [Google Scholar] [CrossRef]
- Antonelli, L.R.; Gigliotti Rothfuchs, A.; Goncalves, R.; Roffe, E.; Cheever, A.W.; Bafica, A.; Salazar, A.M.; Feng, C.G.; Sher, A. Intranasal Poly-IC treatment exacerbates tuberculosis in mice through the pulmonary recruitment of a pathogen-permissive monocyte/macrophage population. J. Clin. Investig. 2010, 120, 1674–1682. [Google Scholar] [CrossRef]
- Dauphinee, S.M.; Richer, E.; Eva, M.M.; McIntosh, F.; Paquet, M.; Dangoor, D.; Burkart, C.; Zhang, D.E.; Gruenheid, S.; Gros, P.; et al. Contribution of increased ISG15, ISGylation and deregulated type I IFN signaling in Usp18 mutant mice during the course of bacterial infections. Genes Immun. 2014, 15, 282–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNab, F.W.; Ewbank, J.; Rajsbaum, R.; Stavropoulos, E.; Martirosyan, A.; Redford, P.S.; Wu, X.; Graham, C.M.; Saraiva, M.; Tsichlis, P.; et al. TPL-2-ERK1/2 signaling promotes host resistance against intracellular bacterial infection by negative regulation of type I IFN production. J. Immunol. 2013, 191, 1732–1743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer-Barber, K.D.; Andrade, B.B.; Oland, S.D.; Amaral, E.P.; Barber, D.L.; Gonzales, J.; Derrick, S.C.; Shi, R.; Kumar, N.P.; Wei, W.; et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature 2014, 511, 99–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novikov, A.; Cardone, M.; Thompson, R.; Shenderov, K.; Kirschman, K.D.; Mayer-Barber, K.D.; Myers, T.G.; Rabin, R.L.; Trinchieri, G.; Sher, A.; et al. Mycobacterium tuberculosis triggers host type I IFN signaling to regulate IL-1beta production in human macrophages. J. Immunol. 2011, 187, 2540–2547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, D.X.; Yamashiro, L.H.; Chen, K.J.; Mukaida, N.; Kramnik, I.; Darwin, K.H.; Vance, R.E. Type I interferon-driven susceptibility to Mycobacterium tuberculosis is mediated by IL-1Ra. Nat. Microbiol. 2019, 4, 2128–2135. [Google Scholar] [CrossRef] [PubMed]
- Ji, D.X.; Witt, K.C.; Kotov, D.I.; Margolis, S.R.; Louie, A.; Chevee, V.; Chen, K.J.; Gaidt, M.M.; Dhaliwal, H.S.; Lee, A.Y.; et al. Role of the transcriptional regulator SP140 in resistance to bacterial infections via repression of type I interferons. Elife 2021, 10, e67290. [Google Scholar] [CrossRef] [PubMed]
- Desvignes, L.; Wolf, A.J.; Ernst, J.D. Dynamic roles of type I and type II IFNs in early infection with Mycobacterium tuberculosis. J. Immunol. 2012, 188, 6205–6215. [Google Scholar] [CrossRef] [Green Version]
- Moreira-Teixeira, L.; Sousa, J.; McNab, F.W.; Torrado, E.; Cardoso, F.; Machado, H.; Castro, F.; Cardoso, V.; Gaifem, J.; Wu, X.; et al. Type I IFN Inhibits Alternative Macrophage Activation during Mycobacterium tuberculosis Infection and Leads to Enhanced Protection in the Absence of IFN-gamma Signaling. J. Immunol. 2016, 197, 4714–4726. [Google Scholar] [CrossRef] [Green Version]
- Kuchtey, J.; Fulton, S.A.; Reba, S.M.; Harding, C.V.; Boom, W.H. Interferon-alphabeta mediates partial control of early pulmonary Mycobacterium bovis bacillus Calmette-Guerin infection. Immunology 2006, 118, 39–49. [Google Scholar] [CrossRef]
- Wang, J.; Hussain, T.; Zhang, K.; Liao, Y.; Yao, J.; Song, Y.; Sabir, N.; Cheng, G.; Dong, H.; Li, M.; et al. Inhibition of type I interferon signaling abrogates early Mycobacterium bovis infection. BMC Infect. Dis. 2019, 19, 1031. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, G.; Midiri, A.; Biondo, C.; Beninati, C.; Zummo, S.; Galbo, R.; Tomasello, F.; Gambuzza, M.; Macri, G.; Ruggeri, A.; et al. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J. Immunol. 2007, 178, 3126–3133. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Ruby, T.; Belhocine, K.; Bouley, D.M.; Kayagaki, N.; Dixit, V.M.; Monack, D.M. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 2012, 490, 288–291. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Pathogen | Is STING Protective? | References |
|---|---|---|
| Brucella spp. | Yes | [49,55,56,74] |
| Burkholderia spp. | No | [67] |
| Chlamydia spp. | Inconclusive | [70] |
| Legionella pneumophila | Yes | [62,63,64] |
| Listeria monocytogenes | Yes (partially) | [65,71] |
| M. avium ssp. paratuberculosis | Inconclusive | [79] |
| Mycobacterium smegmatis | Inconclusive | [79] |
| Mycobacterium tuberculosis | No | [75] |
| Salmonella typhimurium | Yes (partially) | [72] |
| Yersinia pestis | Yes | [66] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guimarães, E.S.; Marinho, F.V.; de Queiroz, N.M.G.P.; Antunes, M.M.; Oliveira, S.C. Impact of STING Inflammatory Signaling during Intracellular Bacterial Infections. Cells 2022, 11, 74. https://doi.org/10.3390/cells11010074
Guimarães ES, Marinho FV, de Queiroz NMGP, Antunes MM, Oliveira SC. Impact of STING Inflammatory Signaling during Intracellular Bacterial Infections. Cells. 2022; 11(1):74. https://doi.org/10.3390/cells11010074
Chicago/Turabian StyleGuimarães, Erika S., Fabio V. Marinho, Nina M. G. P. de Queiroz, Maísa M. Antunes, and Sergio C. Oliveira. 2022. "Impact of STING Inflammatory Signaling during Intracellular Bacterial Infections" Cells 11, no. 1: 74. https://doi.org/10.3390/cells11010074
APA StyleGuimarães, E. S., Marinho, F. V., de Queiroz, N. M. G. P., Antunes, M. M., & Oliveira, S. C. (2022). Impact of STING Inflammatory Signaling during Intracellular Bacterial Infections. Cells, 11(1), 74. https://doi.org/10.3390/cells11010074