
A Role for ER-Beta in the Effects of Low-Density Lipoprotein Cholesterol and 27-Hydroxycholesterol on Breast Cancer Progression: Involvement of the IGF Signalling Pathway?




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. SiRNA Transfection
2.3. Cell Viability
2.4. Western Blotting
2.5. Trans-Well Cell Migration/Invasion Assay
2.6. Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) and the Cancer Genome Atlas (TCGA) Dataset Analysis
2.7. Radioimmunoassay (RIA)
2.8. Statistical Analysis
3. Results
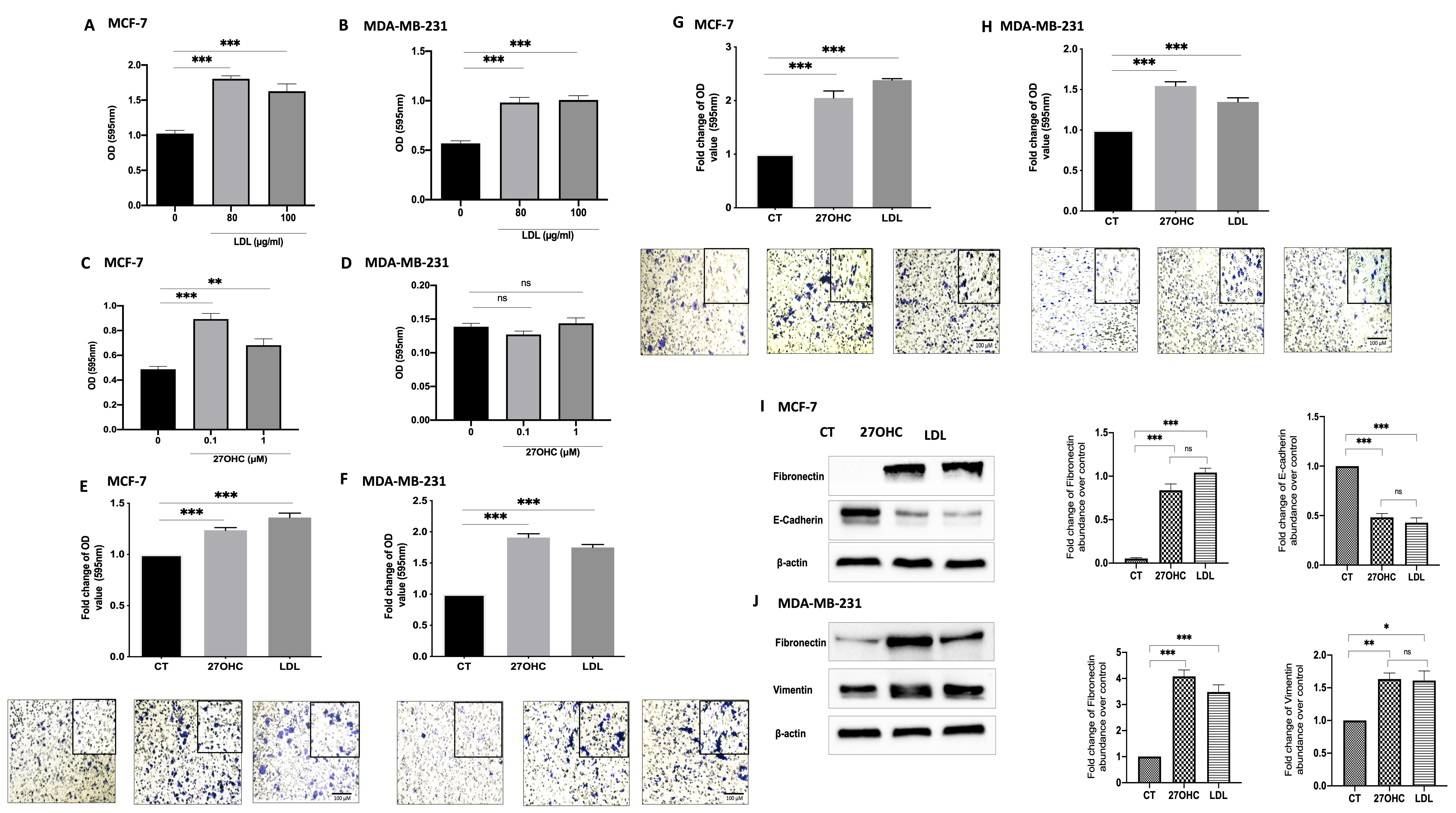
3.1. Cholesterol LDL and Its Metabolite 27OHC Increases Cells Proliferation, Migration, and Levels of EMT Markers in Breast Cancer Cells
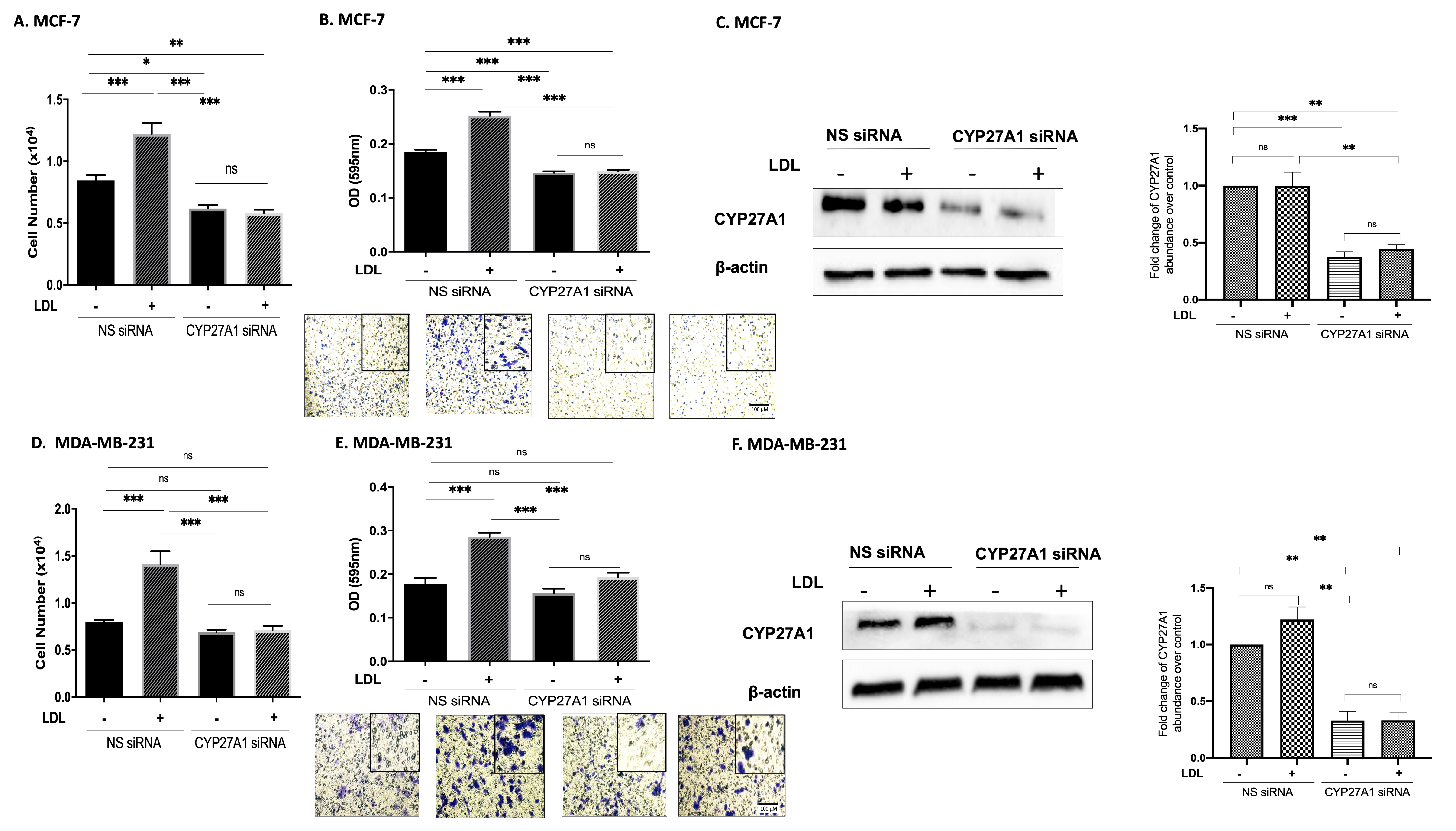
3.2. Cholesterol Increases Proliferation and Migration through 27OHC Production in Breast Cancer Cells
3.3. 27OHC Promotes Cell Proliferation, Migration, and Invasion of Breast Cancer Cells
3.4. 27OHC Increases Cell Migration and Invasion of MDA-MB-231 Breast Cancer Cells
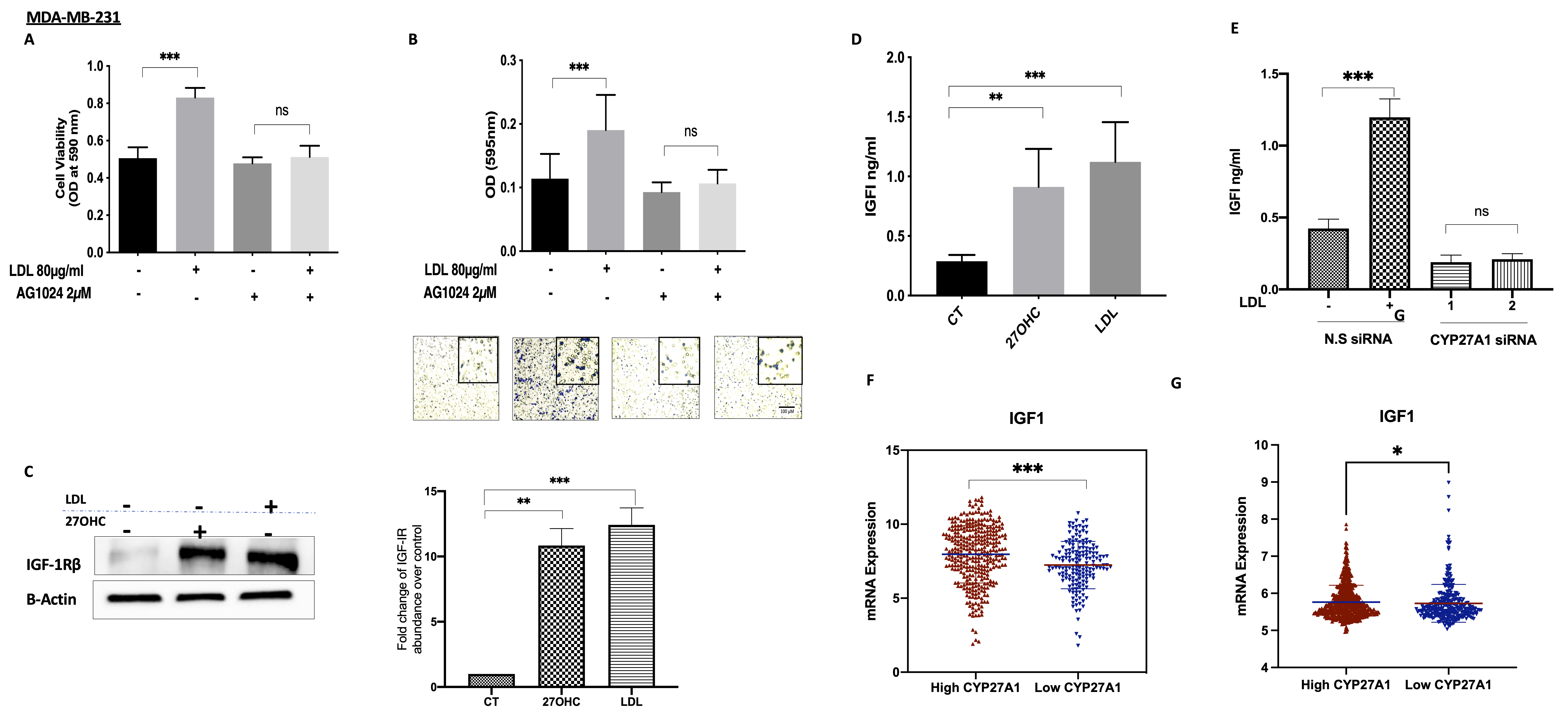
3.5. The Involvement of the IGF System in the Actions of Cholesterolin TNBC
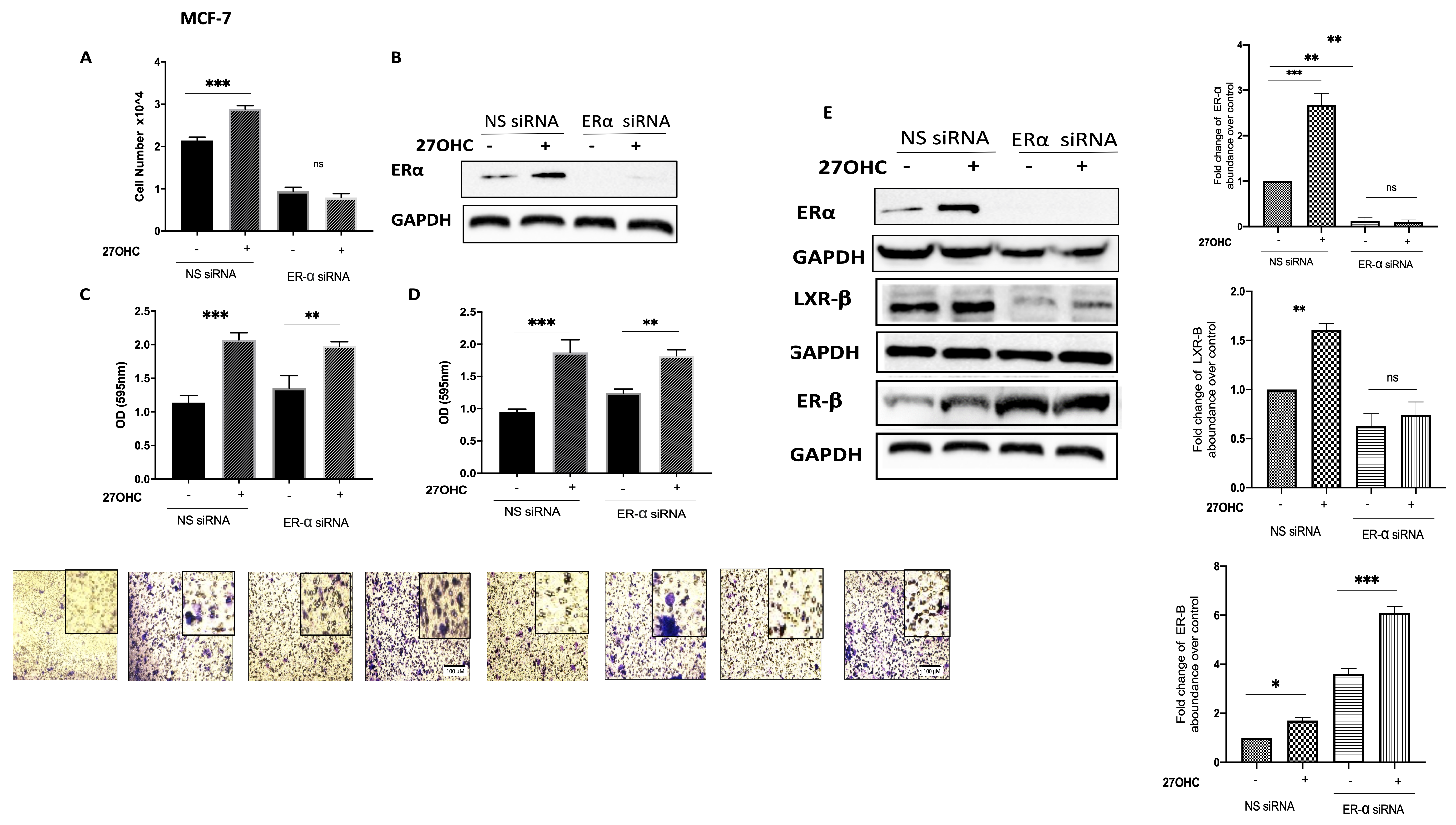
3.6. ER-β Regulates IGF-1 and EGF Receptors in TNBC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Mcdonnell, D.P.; Park, S.; Goulet, M.T.; Jasper, J.; Wardell, S.E.; Chang, C.-Y.; Norris, J.D.; Guyton, J.R.; Nelson, E.R. Obesity, Cholesterol Metabolism, and Breast Cancer Pathogenesis. Cancer Res. 2014, 74, 4976–4982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; Nelson, E.R. 27-Hydroxycholesterol, an Endogenous Selective Estrogen Receptor Modulator. Maturitas 2017, 104, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Omoto, Y.; Iwase, H.; Yamashita, H.; Toyama, T.; Coombes, R.C.; Filipovic, A.; Warner, M.; Gustafsson, J.A. Differential Expression of Estrogen Receptor α, Β1, and Β2 in Lobular and Ductal Breast Cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 1933–1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skliris, G.P.; Leygue, E.; Curtis-Snell, L.; Watson, P.H.; Murphy, L.C. Expression of Oestrogen Receptor-β in Oestrogen Receptor-α Negative Human Breast Tumours. Br. J. Cancer 2006, 95, 616–626. [Google Scholar] [CrossRef] [Green Version]
- Girgert, R.; Emons, G.; Gründker, C. Estrogen Signaling in ERα-Negative Breast Cancer: ERβ and GPER. Front. Endocrinol. 2019, 9, 781. [Google Scholar] [CrossRef] [Green Version]
- Haque, M.M.; Desai, K.V. Pathways to Endocrine Therapy Resistance in Breast Cancer. Front. Endocrinol. 2019, 10, 375. [Google Scholar] [CrossRef]
- Monaco, A.; Licitra, F.; Di Gisi, M.; Galasso, G.; Di Donato, M.; Giovannelli, P.; Migliaccio, A.; Castoria, G. ERβ in Triple-Negative Breast Cancer: Emerging Concepts and Therapeutic Possibilities. Endocrines 2021, 2, 33. [Google Scholar] [CrossRef]
- Minutolo, F.; Macchia, M.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A. Estrogen Receptor β Ligands: Recent Advances and Biomedical Applications. Med. Res. Rev. 2011, 31, 364–442. [Google Scholar] [CrossRef]
- Johnson, K.E.; Siewert, K.M.; Klarin, D.; Damrauer, S.M.; Chang, K.M.; Tsao, P.S.; Assimes, T.L.; Maxwell, K.N.; Voight, B.F. The Relationship between Circulating Lipids and Breast Cancer Risk: A Mendelian Randomization Study. PLoS Med. 2020, 17, e1003302. [Google Scholar] [CrossRef]
- Gallagher, E.J.; Zelenko, Z.; Neel, B.A.; Antoniou, I.M.; Rajan, L.; Kase, N.; LeRoith, D. Elevated Tumor LDLR Expression Accelerates LDL Cholesterol-Mediated Breast Cancer Growth in Mouse Models of Hyperlipidemia. Oncogene 2017, 36, 6462–6471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, D.W. Oxysterol Biosynthetic Enzymes. Biochim. Biophys. Acta 2000, 1529, 126–135. [Google Scholar] [CrossRef]
- Cruz, P.; Torres, C.; Ramírez, M.E.; Epuñán, M.J.; Valladares, L.E.; Sierralta, W.D. Proliferation of Human Mammary Cancer Cells Exposed to 27-Hydroxycholesterol. Exp. Ther. Med. 2010, 1, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Nelson, E.R.; Wardell, S.E.; Jasper, J.S.; Park, S.; Suchindran, S.; Howe, M.K.; Carver, N.J.; Pillai, R.V.; Sullivan, P.M.; Sondhi, V.; et al. 27-Hydroxycholesterol Links Hypercholesterolemia and Breast Cancer Pathophysiology. Science 2013, 342, 1094–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Ishikawa, T.; Sirianni, R.; Tang, H.; McDonald, J.G.; Yuhanna, I.S.; Thompson, B.; Girard, L.; Mineo, C.; Brekken, R.A.; et al. 27-Hydroxycholesterol Promotes Cell-Autonomous, ER-Positive Breast Cancer Growth. Cell Rep. 2013, 5, 637–645. [Google Scholar] [CrossRef] [Green Version]
- Dalenc, F.; Iuliano, L.; Filleron, T.; Zerbinati, C.; Voisin, M.; Arellano, C.; Chatelut, E.; Marquet, P.; Samadi, M.; Roché, H.; et al. Circulating Oxysterol Metabolites as Potential New Surrogate Markers in Patients with Hormone Receptor-Positive Breast Cancer: Results of the OXYTAM Study. J. Steroid Biochem. Mol. Biol. 2017, 169, 210–218. [Google Scholar] [CrossRef]
- Nguyen, V.T.M.; Barozzi, I.; Faronato, M.; Lombardo, Y.; Steel, J.H.; Patel, N.; Darbre, P.; Castellano, L.; Gyorffy, B.; Woodley, L.; et al. Differential Epigenetic Reprogramming in Response to Specific Endocrine Therapies Promotes Cholesterol Biosynthesis and Cellular Invasion. Nat. Commun. 2015, 6, 10044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiramitsu, S.; Ishikawa, T.; Lee, W.-R.; Khan, T.; Crumbley, C.; Khwaja, N.; Zamanian, F.; Asghari, A.; Sen, M.; Zhang, Y.; et al. Estrogen Receptor Beta-Mediated Modulation of Lung Cancer Cell Proliferation by 27-Hydroxycholesterol. Front. Endocrinol. 2018, 9, 470. [Google Scholar] [CrossRef] [PubMed]
- DuSell, C.D.; Umetani, M.; Shaul, P.W.; Mangelsdorf, D.J.; McDonnell, D.P. 27-Hydroxycholesterol Is an Endogenous Selective Estrogen Receptor Modulator. Mol. Endocrinol. 2008, 22, 65–77. [Google Scholar] [CrossRef] [Green Version]
- Starkey, N.J.E.; Li, Y.; Drenkhahn-Weinaug, S.K.; Liu, J.; Lubahn, D.B. 27-Hydroxycholesterol Is an Estrogen Receptor β -Selective Negative Allosteric Modifier of 17 β -Estradiol Binding. Endocrinology 2018, 159, 1972–1981. [Google Scholar] [CrossRef] [Green Version]
- Belardi, V.; Gallagher, E.J.; Novosyadlyy, R.; Leroith, D. Insulin and IGFs in Obesity-Related Breast Cancer. J. Mammary Gland Biol. Neoplasia 2013, 18, 277–289. [Google Scholar] [CrossRef]
- Sturtz, L.A.; Melley, J.; Mamula, K.; Shriver, C.D.; Ellsworth, R.E. Outcome Disparities in African American Women with Triple Negative Breast Cancer: A Comparison of Epidemiological and Molecular Factors between African American and Caucasian Women with Triple Negative Breast Cancer. BMC Cancer 2014, 14, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samani, A.A.; Yakar, S.; LeRoith, D.; Brodt, P. The Role of the IGF System in Cancer Growth and Metastasis: Overview and Recent Insights. Endo. Rev. 2007, 28, 20–47. [Google Scholar] [CrossRef]
- Kallirroi, V.; Aikaterini, B.; Maria, T.; Tzanakakis George, N.; Dragana, N. Insulin-like Growth Factor and Epidermal Growth Factor Signaling in Breast Cancer Cell Growth: Focus on Endocrine Resistant Disease. Anal. Cell Pathol. 2015, 2015, 975495. [Google Scholar] [CrossRef] [Green Version]
- Christopoulos, P.F.; Corthay, A.; Koutsilieris, M. Aiming for the Insulin-like Growth Factor-1 System in Breast Cancer Therapeutics. Cancer Treat. Rev. 2018, 63, 79–95. [Google Scholar] [CrossRef] [PubMed]
- Richardson, A.E.; Hamilton, N.; Davis, W.; Brito, C.; De León, D. Insulin-like Growth Factor-2 (IGF-2) Activates Estrogen Receptor-α and-β via the IGF-1 and the Insulin Receptors in Breast Cancer Cells. Growth Factors 2011, 29, 82–93. [Google Scholar] [CrossRef]
- Hamilton, N.; Márquez-Garbán, D.; Mah, V.; Fernando, G.; Elshimali, Y.; Garbán, H.; Elashoff, D.; Vadgama, J.; Goodglick, L.; Pietras, R. Biologic Roles of Estrogen Receptor- β and Insulin-like Growth Factor-2 in Triple-Negative Breast Cancer. Biomed Res. Int. 2015, 2015, 925703. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Biernacka, K.M.; Holly, J.M.P.; Jarrett, C.; Morrison, A.A.; Morgan, A.; Winters, Z.E.; Foulstone, E.J.; Shield, J.P.; Perks, C.M. Hyperglycaemia Confers Resistance to Chemotherapy on Breast Cancer Cells: The Role of Fatty Acid Synthase. Endocr. Relat. Cancer 2010, 17, 539–551. [Google Scholar] [CrossRef] [Green Version]
- Feoktistova, M.; Geserick, P.; Leverkus, M. Crystal Violet Assay for Determining Viability of Cultured Cells. Cold Spring Harb. Protoc. 2016, 2016, 343–346. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Davies, S.C.; Wass, J.A.H.; Ross, R.J.M.; Cotterill, A.M.; Buchanan, C.R.; Coulson, V.J.; Holly, J.M.P. The Induction of a Specific Protease for Insulin-like Growth Factor Binding Protein-3 in the Circulation during Severe Illness. J. Endocrinol. 1991, 130, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yu, D.D.; Yan, D.L.; Hu, Y.; Chen, D.; Liu, Y.; Zhang, H.D.; Yu, S.R.; Cao, H.X.; Feng, J.F. Liver X Receptor as a Drug Target for the Treatment of Breast Cancer. Anti-Cancer Drugs. 2016, 27, 373–382. [Google Scholar] [CrossRef]
- Beckwitt, C.H.; Brufsky, A.; Oltvai, Z.N.; Wells, A. Statin Drugs to Reduce Breast Cancer Recurrence and Mortality. Breast Cancer Res. 2018, 20, 144. [Google Scholar] [CrossRef] [PubMed]
- Lipovka, Y.; Konhilas, J.P. The Complex Nature of Oestrogen Signalling in Breast Cancer: Enemy or Ally? Biosci. Rep. 2016, 36, e00352. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, Y.; Rogers, M.A. Sterol Metabolism and Transport in Atherosclerosis and Cancer. Front. Endocrinol. 2018, 9, 509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Zhang, L.; Xian, G.; Lv, Y.; Lin, Y.; Wang, Y. 25-Hydroxycholesterol Promotes Migration and Invasion of Lung Adenocarcinoma Cells. Biochem. Biophys. Res. Commun. 2017, 484, 857–863. [Google Scholar] [CrossRef]
- Zeng, L.; Zielinska, H.A.; Arshad, A.; Shield, J.P.; Bahl, A.; Holly, J.M.P.; Perks, C.M. Hyperglycaemia-Induced Chemoresistance in Breast Cancer Cells: Role of the Estrogen Receptor. Endocr. Relat. Cancer 2016, 23, 125–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leygue, E.; Murphy, L.C. A Bi-Faceted Role of Estrogen Receptor β in Breast Cancer in: Endocrine-Related Cancer Volume 20 Issue 3. Endocr. Relat. Cancer 2013, 20, R127–R139. [Google Scholar] [CrossRef]
- Sun, Y.-S.; Zhao, Z.; Yang, Z.-N.; Xu, F.; Lu, H.-J.; Zhu, Z.-Y.; Shi, W.; Jiang, J.; Yao, P.-P.; Zhu, H.-P. Risk Factors and Preventions of Breast Cancer. Int. J. Biol. Sci. 2017, 13, 1387–1397. [Google Scholar] [CrossRef] [Green Version]
- Sellitto, A.; D’agostino, Y.; Alexandrova, E.; Lamberti, J.; Pecoraro, G.; Memoli, D.; Rocco, D.; Coviello, E.; Giurato, G.; Nassa, G.; et al. Insights into the Role of Estrogen Receptor β in Triple-Negative Breast Cancer. Cancers 2020, 12, 1477. [Google Scholar] [CrossRef]
- Song, I.S.; Jeong, Y.J.; Jeong, S.H.; Kim, J.E.; Han, J.; Kim, T.H.; Jang, S.W. Modulation of Mitochondrial ERβ Expression Inhibits Triple-Negative Breast Cancer Tumor Progression by Activating Mitochondrial Function. Cell. Physiol. Biochem. 2019, 52, 468–485. [Google Scholar] [CrossRef] [Green Version]
- Carder, P.J.; Murphy, C.E.; Dervan, P.; Kennedy, M.; McCann, A.; Saunders, P.T.K.; Shaaban, A.M.; Foster, C.S.; Witton, C.J.; Bartlett, J.M.S.; et al. A Multi-Centre Investigation towards Reaching a Consensus on the Immunohistochemical Detection of ERβ in Archival Formalin-Fixed Paraffin Embedded Human Breast Tissue. Breast Cancer Res. Treat. 2005, 92, 287–293. [Google Scholar] [CrossRef]
- Raza, S.; Meyer, M.; Goodyear, C.; Hammer, K.D.P.; Guo, B.; Ghribi, O. The Cholesterol Metabolite 27-Hydroxycholesterol Stimulates Cell Proliferation via ERβ in Prostate Cancer Cells. Cancer Cell Int. 2017, 17, 52. [Google Scholar] [CrossRef] [Green Version]
- Graham, N.A.; Minasyan, A.; Lomova, A.; Cass, A.; Balanis, N.G.; Friedman, M.; Chan, S.; Zhao, S.; Delgado, A.; Go, J.; et al. Recurrent Patterns of DNA Copy Number Alterations in Tumors Reflect Metabolic Selection Pressures. Mol. Syst. Biol. 2017, 13, 914. [Google Scholar] [CrossRef] [PubMed]
- Umetani, M. Re-Adopting Classical Nuclear Receptors by Cholesterol Metabolites. J. Steroid Biochem. Mol. Biol. 2016, 157, 20–26. [Google Scholar] [CrossRef] [Green Version]
- Di Donato, M.; Galasso, G.; Giovannelli, P.; Sinisi, A.A.; Migliaccio, A.; Castoria, G. Targeting the Nerve Growth Factor Signaling Impairs the Proliferative and Migratory Phenotype of Triple-Negative Breast Cancer Cells. Front. Cell Dev. Biol. 2021, 9, 676568. [Google Scholar] [CrossRef]
- Sekine, Y.; Furuya, Y.; Nishii, M.; Koike, H.; Matsui, H.; Suzuki, K. Simvastatin Inhibits the Proliferation of Human Prostate Cancer PC-3 Cells via down-Regulation of the Insulin-like Growth Factor 1 Receptor. Biochem. Biophys. Res. Commun. 2008, 372, 356–361. [Google Scholar] [CrossRef]
- Sekine, Y.; Nakayama, H.; Miyazawa, Y.; Kato, H.; Furuya, Y.; Arai, S.; Koike, H.; Matsui, H.; Shibata, Y.; Ito, K.; et al. Simvastatin in Combination with Meclofenamic Acid Inhibits the Proliferation and Migration of Human Prostate Cancer PC-3 Cells via an AKR1C3 Mechanism. Oncol. Lett. 2018, 15, 3167–3172. [Google Scholar] [CrossRef] [PubMed]
- De Laurentiis, A.; Donovan, L.; Arcaro, A. Lipid Rafts and Caveolae in Signaling by Growth Factor Receptors. Open Biochem. J. 2007, 1, 12–32. [Google Scholar] [CrossRef]
- Bin, L.; Jin, L.; Wen Wen, X.; Xin Yuan, G.; Yan Ru, Q.; Li Yi, Z.; Simon, L.; Sai Wah, T.; Annie, L.M.C. Suppression of Esophageal Tumor Growth and Chemoresistance by Directly Targeting the PI3K/AKT Pathway. Oncotarget 2014, 5, 11576–11587. [Google Scholar] [CrossRef] [Green Version]
- Bin, L.; Tsao, S.W.; Chan, K.W.; Ludwig, D.L.; Novosyadlyy, R.; Li, Y.Y.; He, Q.Y.; Cheun, A.L.M. Id1-Induced IGF-II and Its Autocrine/Endocrine Promotion of Esophageal Cancer Progression and Chemoresistance--Implications for IGF-II and IGF-IR-Targeted Therapy. Clin. Cancer Res. 2014, 20, 2651–2662. [Google Scholar] [CrossRef] [Green Version]
- Araya, Z.; Tang, W.; Wikvall, K. Hormonal Regulation of the Human Sterol 27-Hydroxylase Gene CYP27A1. Biochem. J. 2003, 372, 529–534. [Google Scholar] [CrossRef]
- Lee, J.; Hong, E.M.; Jang, J.A.; Park, S.W.; Koh, D.H.; Choi, M.H.; Jang, H.J.; Kae, S.H. Simvastatin Induces Apoptosis and Suppresses Insulin-like Growth Factor 1 Receptor in Bile Duct Cancer Cells. Gut Liver 2016, 10, 310–317. [Google Scholar] [CrossRef] [Green Version]
- Yee, D.; Lee, A.V. Crosstalk between the Insulin-like Growth Factors and Estrogens in Breast Cancer. J. Mammary Gland. Biol. Neoplasia 2000, 5, 107–115. [Google Scholar] [CrossRef]
- Ignatov, A.; Ignatov, T.; Roessner, A.; Costa, S.D.; Kalinski, T. Role of GPR30 in the Mechanisms of Tamoxifen Resistance in Breast Cancer MCF-7 Cells. Breast Cancer Res. Treat. 2010, 123, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Li, X.; Blanchard, A.; Bramwell, V.H.C.; Pritchard, K.I.; Tu, D.; Shepherd, L.; Myal, Y.; Penner, C.; Watson, P.H.; et al. Expression of Both Estrogen Receptor-Beta 1 (ER-Β1) and Its Co-Regulator Steroid Receptor RNA Activator Protein (SRAP) Are Predictive for Benefit from Tamoxifen Therapy in Patients with Estrogen Receptor-Alpha (ER-α)-Negative Early Breast Cancer (EBC). Ann. Oncol. 2013, 24, 1986–1993. [Google Scholar] [CrossRef]
- Hershberger, P.A.; Stabile, L.P.; Kanterewicz, B.; Rothstein, M.E.; Gubish, C.T.; Land, S.; Shuai, Y.; Siegfried, J.M.; Nichols, M. Estrogen Receptor Beta (ERβ) Subtype-Specific Ligands Increase Transcription, P44/P42 Mitogen Activated Protein Kinase (MAPK) Activation and Growth in Human Non-Small Cell Lung Cancer Cells. J. Steroid Biochem. Mol. Biol. 2009, 116, 102–109. [Google Scholar] [CrossRef] [Green Version]
- Bialesova, L.; Xu, L.; Gustafsson, J.Å.; Haldosen, L.A.; Zhao, C.; Dahlman-Wright, K. Estrogen Receptor SS2 Induces Proliferation and Invasiveness of Triple Negative Breast Cancer Cells; Association with Regulation of PHD3 and HIF-1α. Oncotarget 2017, 8, 76622–76633. [Google Scholar] [CrossRef] [Green Version]
- Jensen, E.V.; Cheng, G.; Palmieri, C.; Saji, S.; Mäkelä, S.; Van Noorden, S.; Wahlström, T.; Warner, M.; Coombes, R.C.; Gustafsson, J.Å. Estrogen Receptors and Proliferation Markers in Primary and Recurrent Breast Cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 15197–15202. [Google Scholar] [CrossRef] [Green Version]
- Austin, D.; Hamilton, N.; Elshimali, Y.; Pietras, R.; Wu, Y.; Vadgama, J. Estrogen Receptor-Beta Is a Potential Target for Triple Negative Breast Cancer Treatment. Oncotarget 2018, 9, 33912–33930. [Google Scholar] [CrossRef] [Green Version]
- Božović, A.; Mandušić, V.; Todorović, L.; Krajnović, M. Estrogen Receptor Beta: The Promising Biomarker and Potential Target in Metastases. Int. J. Mol. Sci. 2021, 22, 1656. [Google Scholar] [CrossRef]
- Piperigkou, Z.; Bouris, P.; Onisto, M.; Franchi, M.; Kletsas, D.; Theocharis, A.D.; Karamanos, N.K. Estrogen Receptor Beta Modulates Breast Cancer Cells Functional Properties, Signaling and Expression of Matrix Molecules. Matrix Biol. 2016, 56, 4–23. [Google Scholar] [CrossRef]
- Yan, S.; Dey, P.; Ziegler, Y.; Jiao, X.; Kim, S.H.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Contrasting Activities of Estrogen Receptor Beta Isoforms in Triple Negative Breast Cancer. Breast Cancer Res. Treat. 2020, 185, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Andersson, S.; Sundberg, M.; Pristovsek, N.; Ibrahim, A.; Jonsson, P.; Katona, B.; Clausson, C.M.; Zieba, A.; Ramström, M.; Söderberg, O.; et al. Insufficient Antibody Validation Challenges Oestrogen Receptor Beta Research. Nat. Commun. 2017, 8, 15840. [Google Scholar] [CrossRef]
- Novelli, F.; Milella, M.; Melucci, E.; Di Benedetto, A.; Sperduti, I.; Perrone-Donnorso, R.; Perracchio, L.; Venturo, I.; Nisticò, C.; Fabi, A.; et al. A Divergent Role for Estrogen Receptor-Beta in Node-Positive and Node-Negative Breast Cancer Classified According to Molecular Subtypes: An Observational Prospective Study. Breast Cancer Res. 2008, 10, R74. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mashat, R.M.; Zielinska, H.A.; Holly, J.M.P.; Perks, C.M. A Role for ER-Beta in the Effects of Low-Density Lipoprotein Cholesterol and 27-Hydroxycholesterol on Breast Cancer Progression: Involvement of the IGF Signalling Pathway? Cells 2022, 11, 94. https://doi.org/10.3390/cells11010094
Mashat RM, Zielinska HA, Holly JMP, Perks CM. A Role for ER-Beta in the Effects of Low-Density Lipoprotein Cholesterol and 27-Hydroxycholesterol on Breast Cancer Progression: Involvement of the IGF Signalling Pathway? Cells. 2022; 11(1):94. https://doi.org/10.3390/cells11010094
Chicago/Turabian StyleMashat, Reham M., Hanna A. Zielinska, Jeff M. P. Holly, and Claire M. Perks. 2022. "A Role for ER-Beta in the Effects of Low-Density Lipoprotein Cholesterol and 27-Hydroxycholesterol on Breast Cancer Progression: Involvement of the IGF Signalling Pathway?" Cells 11, no. 1: 94. https://doi.org/10.3390/cells11010094
APA StyleMashat, R. M., Zielinska, H. A., Holly, J. M. P., & Perks, C. M. (2022). A Role for ER-Beta in the Effects of Low-Density Lipoprotein Cholesterol and 27-Hydroxycholesterol on Breast Cancer Progression: Involvement of the IGF Signalling Pathway? Cells, 11(1), 94. https://doi.org/10.3390/cells11010094