
Endocannabinoid and Nitric Oxide-Dependent IGF-I-Mediated Synaptic Plasticity at Mice Barrel Cortex


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement and Animals
2.2. Slice Preparation
2.3. Electrophysiological Recordings
2.4. Data Analysis
3. Results
3.1. IGF-I Modulates Synaptic Transmission
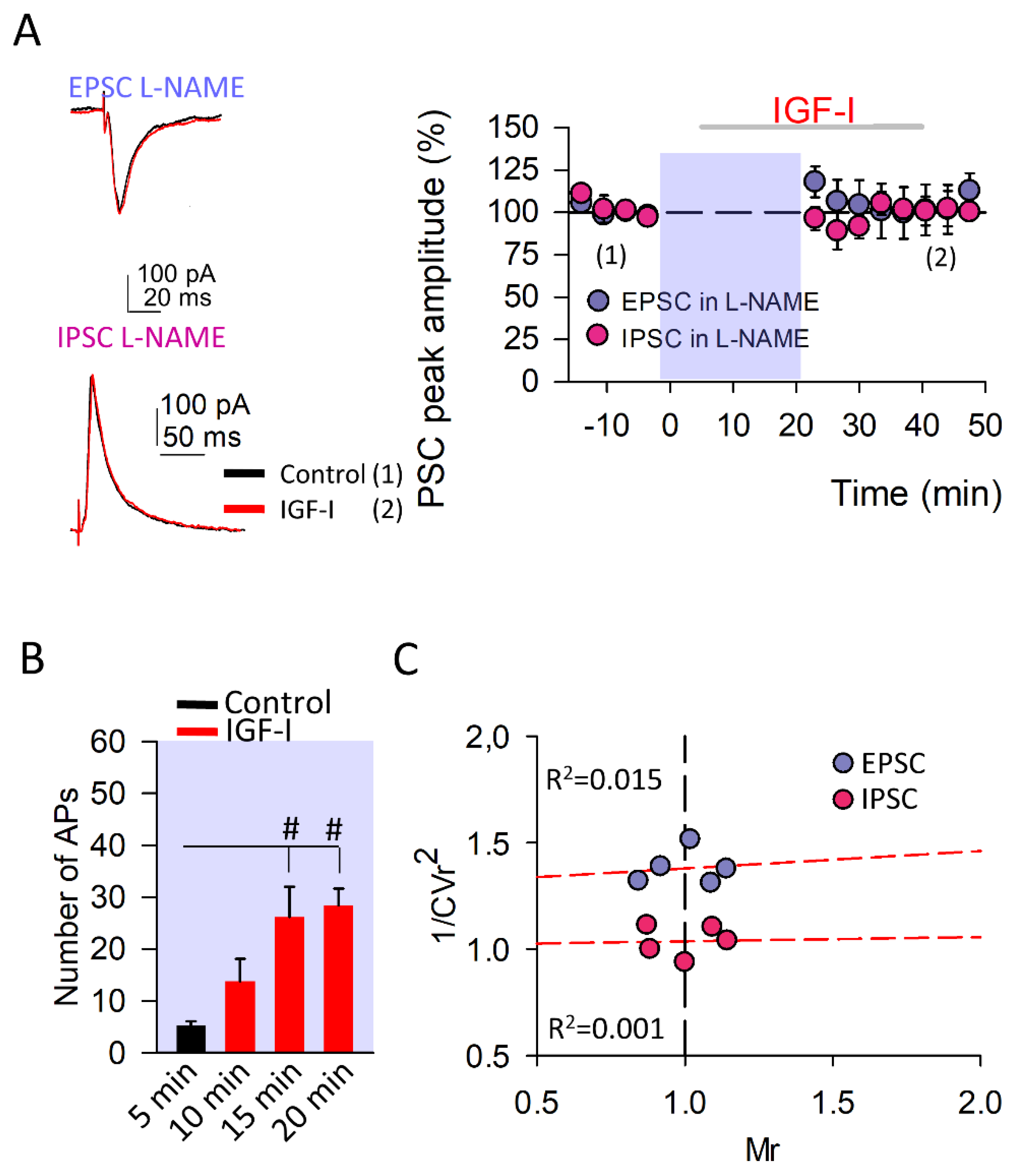
3.2. The Induction of IGF-I-Mediated Synaptic Plasticity at Both Excitatory and Inhibitory Synapses Requires the Nitric Oxide Synthesis
3.3. Endocannabinoids Determine the Sign of the IGF-I-Mediated Synaptic Plasticity at Excitatory Synapses
3.4. IGF-I-Mediated Facilitation of the t-LTP Requires CB1R Activation
4. Discussion
Functional Role of the Modulation of Synaptic Transmission by IGF-I
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, Z.; Chen, Z.; Shang, C.; Yan, F.; Shi, Y.; Zhang, J.; Qu, B.; Han, H.; Wang, Y.; Li, D.; et al. IGF1-Dependent Synaptic Plasticity of Mitral Cells in Olfactory Memory during Social Learning. Neuron 2017, 95, 106–122.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aleman, A.; Torres-Alemán, I. Circulating Insulin-like Growth Factor I and Cognitive Function: Neuromodulation throughout the Lifespan. Prog. Neurobiol. 2009, 89, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Trejo, J.I.; Piriz, J.; Llorens-Martin, M.V.; Fernandez, A.M.; Bolós, M.; LeRoith, D.; Nuñez, A.; Torres-Aleman, I. Central Actions of Liver-Derived Insulin-like Growth Factor I Underlying Its pro-Cognitive Effects. Mol. Psychiatry 2007, 12, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Pristerà, A.; Blomeley, C.; Lopes, E.; Threlfell, S.; Merlini, E.; Burdakov, D.; Cragg, S.; Guillemot, F.; Ang, S.-L.; Palmiter, R.D. Dopamine Neuron-Derived IGF-1 Controls Dopamine Neuron Firing, Skill Learning, and Exploration. Proc. Natl. Acad. Sci. USA 2019, 116, 3817–3826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto, M.; Cai, W.; Konishi, M.; Kahn, C.R. Insulin Signaling in the Hippocampus and Amygdala Regulates Metabolism and Neurobehavior. Proc. Natl. Acad. Sci. USA 2019, 116, 6379–6384. [Google Scholar] [CrossRef] [Green Version]
- Nuñez, A.; Carro, E.; Torres-Alemán, I. Insulin-like Growth Factor I Modifies Electrophysiological Properties of Rat Brain Stem Neurons. J. Neurophysiol. 2003, 89, 3008–3017. [Google Scholar] [CrossRef] [Green Version]
- Gazit, N.; Vertkin, I.; Shapira, I.; Helm, M.; Slomowitz, E.; Sheiba, M.; Mor, Y.; Rizzoli, S.; Slutsky, I. IGF-1 Receptor Differentially Regulates Spontaneous and Evoked Transmission via Mitochondria at Hippocampal Synapses. Neuron 2016, 89, 583–597. [Google Scholar] [CrossRef] [Green Version]
- Maglio, L.E.; Noriega-Prieto, J.A.; Maroto, I.B.; Martin-Cortecero, J.; Muñ oz-Callejas, A.; Callejo-Mó stoles, M.; Ferná ndez de Sevilla, D. IGF-1 Facilitates Extinction of Conditioned Fear. eLife 2021, 10, e67267. [Google Scholar] [CrossRef]
- Araujo, D.M.; Lapchak, P.A.; Collier, B.; Chabot, J.G.; Quirion, R. Insulin-like Growth Factor-1 (Somatomedin-C) Receptors in the Rat Brain: Distribution and Interaction with the Hippocampal Cholinergic System. Brain Res. 1989, 484, 130–138. [Google Scholar] [CrossRef]
- Seto, D.; Zheng, W.H.; McNicoll, A.; Collier, B.; Quirion, R.; Kar, S. Insulin-like Growth Factor-I Inhibits Endogenous Acetylcholine Release from the Rat Hippocampal Formation: Possible Involvement of GABA in Mediating the Effects. Neuroscience 2002, 115, 603–612. [Google Scholar] [CrossRef]
- Castro-alamancos, M.A.; Torres-aleman, I. Long-Term Depression of Glutamate-Induced y-Aminobutyric Acid Release in Cerebellum by Insulin-like Growth Factor I. Proc. Natl. Acad. Sci. USA 1993, 90, 7386–7390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maya-Vetencourt, J.F.; Baroncelli, L.; Viegi, A.; Tiraboschi, E.; Castren, E.; Cattaneo, A.; Maffei, L. IGF-1 Restores Visual Cortex Plasticity in Adult Life by Reducing Local GABA Levels. Neural Plast. 2012, 2012, 250421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvell, G.E.; Simons, D.J. Biometric Analyses of Vibrissal Tactile Discrimination in the Rat. J. Neurosci. 1990, 10, 2638–2648. [Google Scholar] [CrossRef] [PubMed]
- Mysoet, J.; Dupont, E.; Bastide, B.; Canu, M.H. Role of IGF-1 in Cortical Plasticity and Functional Deficit Induced by Sensorimotor Restriction. Behav. Brain Res. 2015, 290, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Lines, J.; Martin, E.D.; Kofuji, P.; Aguilar, J.; Araque, A. Astrocytes Modulate Sensory-Evoked Neuronal Network Activity. Nat. Commun. 2020, 11, 3689. [Google Scholar] [CrossRef]
- Noriega-Prieto, J.A.; Maglio, L.E.; Zegarra-Valdivia, J.A.; Pignatelli, J.; Fernandez, A.M.; Martinez-Rachadell, L.; Fernandes, J.; Núñez, Á.; Araque, A.; Torres-Alemán, I.; et al. Astrocytic IGF-IRs Induce Adenosine-Mediated Inhibitory Downregulation and Improve Sensory Discrimination. J. Neurosci. 2021, 41, 4768–4781. [Google Scholar] [CrossRef]
- Ahn, K.; McKinney, M.K.; Cravatt, B.F. Enzymatic Pathways That Regulate Endocannabinoid Signaling in the Nervous System. Chem. Rev. 2008, 108, 1687–1707. [Google Scholar] [CrossRef] [Green Version]
- Piomelli, D. The Molecular Logic of Endocannabinoid Signalling. Nat. Rev. Neurosci. 2003, 4, 873–884. [Google Scholar] [CrossRef] [Green Version]
- di Marzo, V. The Endocannabinoid System: Its General Strategy of Action, Tools for Its Pharmacological Manipulation and Potential Therapeutic Exploitation. Pharmacol. Res. 2009, 60, 77–84. [Google Scholar] [CrossRef]
- Castillo, P.E.; Younts, T.J.; Chávez, A.E.; Hashimotodani, Y. Endocannabinoid Signaling and Synaptic Function. Neuron 2012, 76, 70–81. [Google Scholar] [CrossRef] [Green Version]
- Martin-Fernandez, M.; Jamison, S.; Robin, L.M.; Zhao, Z.; Martin, E.D.; Aguilar, J.; Benneyworth, M.A.; Marsicano, G.; Araque, A. Synapse-Specific Astrocyte Gating of Amygdala-Related Behavior. Nat. Neurosci. 2017, 20, 1540–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maglio, L.E.; Noriega-Prieto, J.A.; Maraver, M.J.; Fernández de Sevilla, D. Endocannabinoid-Dependent Long-Term Potentiation of Synaptic Transmission at Rat Barrel Cortex. Cereb. Cortex 2018, 28, 1568–1581. [Google Scholar] [CrossRef] [PubMed]
- Covelo, A.; Araque, A. Neuronal Activity Determines Distinct Gliotransmitter Release from a Single Astrocyte. eLife 2018, 7, e32237. [Google Scholar] [CrossRef] [PubMed]
- Noriega-Prieto, J.A.; Araque, A. Sensing and Regulating Synaptic Activity by Astrocytes at Tripartite Synapse. Neurochem. Res. 2021, 46, 2580–2585. [Google Scholar] [CrossRef]
- Stella, N.; Piomelli, D. Receptor-Dependent Formation of Endogenous Cannabinoids in Cortical Neurons. Eur. J. Pharmacol. 2001, 425, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Taqatqeh, F.; Mergia, E.; Neitz, A.; Eysel, U.T.; Koesling, D.; Mittmann, T. More than a Retrograde Messenger: Nitric Oxide Needs Two CGMP Pathways to Induce Hippocampal Long-Term Potentiation. J. Neurosci. 2009, 29, 9344–9350. [Google Scholar] [CrossRef]
- Kakazu, Y.; Uchida, S.; Nakagawa, T.; Akaike, N.; Nabekura, J. Reversibility and Cation Selectivity of the K+-Cl− Cotransport in Rat Central Neurons. J. Neurophysiol. 2000, 84, 281–288. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.R.; Payne, J.A. Cation transport by the neuronal K+-Cl− cotransporter KCC2: Thermodynamics and kinetics of alternate transport modes. Am. J. Physiol. Physiol. 2004, 287, C919–C931. [Google Scholar] [CrossRef] [Green Version]
- Creager, R.; Dunwiddie, T.; Lynch, G. Paired-pulse and Frequency Facilitation in the CA1 Region of the in Vitro Rat Hippocampus. J. Physiol. 1980, 299, 409–424. [Google Scholar] [CrossRef]
- Clark, K.A.; Randall, A.D.; Collingridge, G.L. A Comparison of Paired-Pulse Facilitation of AMPA and NMDA Receptor-Mediated Excitatory Postsynaptic Currents in the Hippocampus. Exp. Brain Res. 1994, 101, 272–278. [Google Scholar] [CrossRef]
- Kuhnt, U.; Voronin, L.L. Interaction between Paired-Pulse Facilitation and Long-Term Potentiation in Area Ca1 of Guinea-Pig Hippocampal Slices: Application of Quantal Analysis. Neuroscience 1994, 101, 391–397. [Google Scholar] [CrossRef]
- Noriega-Prieto, J.A.; Maglio, L.E.; Gallero-Salas, Y.; Fernández de Sevilla, D. Nitric Oxide-Dependent LTD at Infralimbic Cortex. Neuroscience 2019, 418, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Robello, M.; Amico, C.; Bucossi, G.; Cupello, A.; Rapallino, M.V.; Thellung, S. Nitric oxide and GABA A Receptor Function the Rat Cerebral Cortex and Cerebellar Granule Cells. Neuroscience 1996, 74, 99–105. [Google Scholar] [CrossRef]
- Volgushev, M.; Balaban, P.; Chistiakova, M.; Eysel, U.T. Retrograde Signalling with Nitric Oxide at Neocortical Synapses. Eur. J. Neurosci. 2000, 12, 4255–4267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardingham, N.; Fox, K. The Role of Nitric Oxide and GluR1 in Presynaptic and Postsynaptic Components of Neocortical Potentiation. J. Neurosci. 2006, 26, 7395–7404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sjöström, P.J.; Turrigiano, G.G.; Nelson, S.B. Multiple Forms of Long-Term Plasticity at Unitary Neocortical Layer 5 Synapses. Neuropharmacology 2007, 52, 176–184. [Google Scholar] [CrossRef]
- Haul, S.; Gödecke, A.; Schrader, J.; Haas, H.L.; Luhmann, H.J. Impairment of Neocortical Long-Term Potentiation in Mice Deficient of Endothelial Nitric Oxide Synthase. J. Neurophysiol. 1999, 81, 494–497. [Google Scholar] [CrossRef] [Green Version]
- Heifets, B.D.; Castillo, P.E. Endocannabinoid Signaling and Long-Term Synaptic Plasticity. Annu. Rev. Physiol. 2009, 71, 283–306. [Google Scholar] [CrossRef] [Green Version]
- Bodor, Á.L.; Katona, I.; Nyíri, G.; Mackie, K.; Ledent, C.; Hájos, N.; Freund, T.F. Endocannabinoid Signaling in Rat Somatosensory Cortex: Laminar Differences and Involvement of Specific Interneuron Types. J. Neurosci. 2005, 25, 6845–6856. [Google Scholar] [CrossRef] [Green Version]
- Fortin, D.A.; Levine, E.S. Differential Effects of Endocannabinoids on Glutamatergic and GABAergic Inputs to Layer 5 Pyramidal Neurons. Cereb. Cortex 2006, 17, 163–174. [Google Scholar] [CrossRef] [Green Version]
- Araque, A.; Carmignoto, G.; Haydon, P.G.; Oliet, S.H.R.; Robitaille, R.; Volterra, A. Gliotransmitters Travel in Time and Space. Neuron 2014, 81, 728–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarrete, M.; Araque, A. Endocannabinoids Potentiate Synaptic Transmission through Stimulation of Astrocytes. Neuron 2010, 68, 113–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Gonzalo, M.; Navarrete, M.; Perea, G.; Covelo, A.; Martín-Fernández, M.; Shigemoto, R.; Luján, R.; Araque, A. Endocannabinoids Induce Lateral Long-Term Potentiation of Transmitter Release by Stimulation of Gliotransmission. Cereb. Cortex 2015, 25, 3699–3712. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noriega-Prieto, J.A.; Maglio, L.E.; Ibáñez-Santana, S.; de Sevilla, D.F. Endocannabinoid and Nitric Oxide-Dependent IGF-I-Mediated Synaptic Plasticity at Mice Barrel Cortex. Cells 2022, 11, 1641. https://doi.org/10.3390/cells11101641
Noriega-Prieto JA, Maglio LE, Ibáñez-Santana S, de Sevilla DF. Endocannabinoid and Nitric Oxide-Dependent IGF-I-Mediated Synaptic Plasticity at Mice Barrel Cortex. Cells. 2022; 11(10):1641. https://doi.org/10.3390/cells11101641
Chicago/Turabian StyleNoriega-Prieto, José Antonio, Laura Eva Maglio, Sara Ibáñez-Santana, and David Fernández de Sevilla. 2022. "Endocannabinoid and Nitric Oxide-Dependent IGF-I-Mediated Synaptic Plasticity at Mice Barrel Cortex" Cells 11, no. 10: 1641. https://doi.org/10.3390/cells11101641
APA StyleNoriega-Prieto, J. A., Maglio, L. E., Ibáñez-Santana, S., & de Sevilla, D. F. (2022). Endocannabinoid and Nitric Oxide-Dependent IGF-I-Mediated Synaptic Plasticity at Mice Barrel Cortex. Cells, 11(10), 1641. https://doi.org/10.3390/cells11101641