
Detecting DNA: An Overview of DNA Recognition by Inflammasomes and Protection against Bacterial Respiratory Infections


Abstract
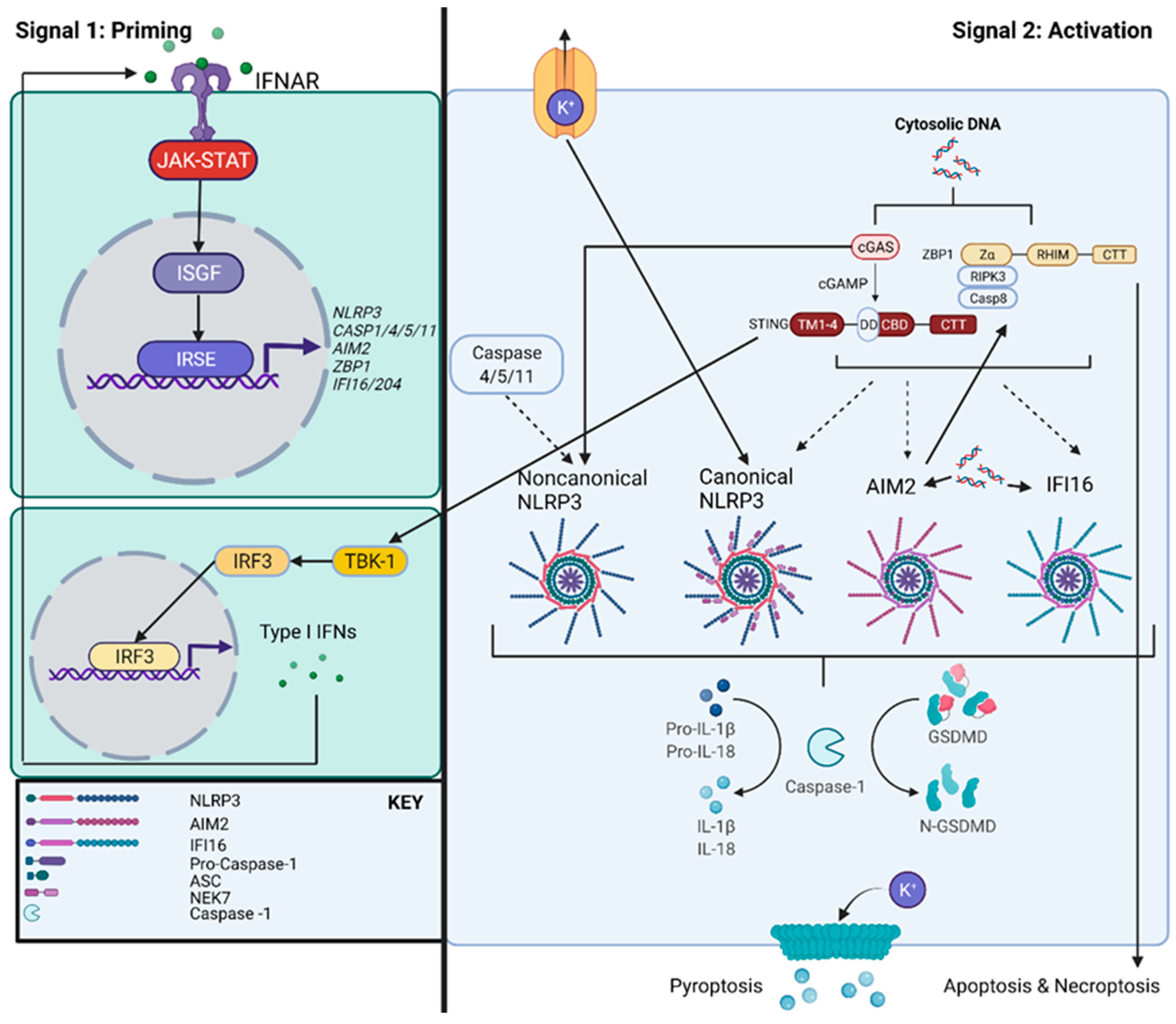
:1. Introduction
1.1. Inflammasome Sensing of Bacteria
1.2. Nucleic Acid Sensing by Inflammasomes
2. Bacterial DNA Sensing Effectors Associated with Inflammasome Activation
2.1. Stimulator of Interferon Genes (STING)
2.2. Z-DNA Binding Protein 1 (ZBP1)
3. Bacterial Respiratory Diseases Implicated with DNA-Induced Inflammasome Activation
3.1. Brucella spp.
3.2. Burkholderia spp.
3.3. Francisella tularensis/novicida
3.4. Legionella pneumophila
3.5. Mycobacterium tuberculosis
3.6. Staphylococcus aureus
3.7. Streptococcus pneumoniae
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coutermarsh-Ott, S.; Eden, K.; Allen, I.C. Beyond the inflammasome: Regulatory NOD-like receptor modulation of the host immune response following virus exposure. J. Gen. Virol. 2016, 97, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Muñoz-Planillo, R.; Núñez, G. Sensing and reacting to microbes through the inflammasomes. Nat. Immunol. 2012, 13, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Paludan, S.R.; Bowie, A.G. Immune sensing of DNA. Immunity 2013, 38, 870–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guimarães, E.S.; Marinho, F.V.; de Queiroz, N.M.G.P.; Antunes, M.M.; Oliveira, S.C. Impact of STING Inflammatory Signaling during Intracellular Bacterial Infections. Cells 2022, 11, 74. [Google Scholar] [CrossRef]
- Malireddi, R.K.S.; Kanneganti, T.-D. Role of type I interferons in inflammasome activation, cell death, and disease during microbial infection. Front. Cell Infect. Microbiol. 2013, 3, 77. [Google Scholar] [CrossRef] [Green Version]
- Briard, B.; Place, D.E.; Kanneganti, T.D. DNA Sensing in the Innate Immune Response. Physiology 2020, 35, 112–124. [Google Scholar] [CrossRef]
- Kuriakose, T.; Kanneganti, T.-D. ZBP1: Innate Sensor Regulating Cell Death and Inflammation. Trends Immunol. 2018, 39, 123–134. [Google Scholar] [CrossRef]
- Kaiser, W.J.; Upton, J.W.; Mocarski, E.S. Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa B activation via the DNA-dependent activator of IFN regulatory factors. J. Immunol. 2008, 181, 6427–6434. [Google Scholar] [CrossRef] [Green Version]
- Malireddi, R.K.S.; Kesavardhana, S.; Kanneganti, T.-D. ZBP1 and TAK1: Master Regulators of NLRP3 Inflammasome/Pyroptosis, Apoptosis, and Necroptosis (PAN-optosis). Front. Cell Infect. Microbiol. 2019, 9, 406. [Google Scholar] [CrossRef] [Green Version]
- Swanson, K.V.; Deng, M.; Ting, J.P.Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Yi, Y.-S. Caspase-11 non-canonical inflammasome: A critical sensor of intracellular lipopolysaccharide in macrophage-mediated inflammatory responses. Immunology 2017, 152, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, K.-P.; Hornung, V. Molecular mechanisms and cellular functions of cGAS–STING signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Alnemri, T.; Yu, J.W.; Datta, P.; Wu, J.; Alnemri, E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef] [Green Version]
- Tupik, J.D.; Nagai-Singer, M.A.; Allen, I.C. To protect or adversely affect? The dichotomous role of the NLRP1 inflammasome in human disease. Mol. Asp. Med. 2020, 76, 100858. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.-Z.; Sun, H.-S.; Lv, J.-C.; Guo, L.; Yang, Q.-R. Expression and clinical significance of the NEK7-NLRP3 inflammasome signaling pathway in patients with systemic lupus erythematosus. J. Inflamm. 2018, 15, 16. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- He, W.-t.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.-H.; Zhong, C.-Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Booty, L.M.; Bryant, C.E. Gasdermin D and Beyond—Gasdermin-mediated Pyroptosis in Bacterial Infections. J. Mol. Biol. 2022, 434, 167409. [Google Scholar] [CrossRef]
- Wang, J.; Deobald, K.; Re, F. Gasdermin D Protects from Melioidosis through Pyroptosis and Direct Killing of Bacteria. J. Immunol. 2019, 202, 3468–3473. [Google Scholar] [CrossRef]
- Liu, Z.-Z.; Yang, Y.-J.; Zhou, F.-H.; Ma, K.; Lin, X.-Q.; Yan, S.-Q.; Gao, Y.; Chen, W. GSDMD contributes to host defence against Staphylococcus aureus skin infection by suppressing the Cxcl1–Cxcr2 axis. Vet. Res. 2021, 52, 71. [Google Scholar] [CrossRef]
- Chen, K.W.; Monteleone, M.; Boucher, D.; Sollberger, G.; Ramnath, D.; Condon, N.D.; Pein, J.B.v.; Broz, P.; Sweet, M.J.; Schroder, K. Noncanonical inflammasome signaling elicits gasdermin D dependent neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Lucrezia, R.D.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B.; et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koizumi, Y.; Toma, C.; Higa, N.; Nohara, T.; Nakasone, N.; Suzuki, T. Inflammasome activation via intracellular NLRs triggered by bacterial infection. Cell Microbiol. 2012, 14, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, I.; Jha, S. Comprehensive review of ASC structure and function in immune homeostasis and disease. Mol. Biol. Rep. 2020, 47, 3077–3096. [Google Scholar] [CrossRef] [PubMed]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS activates caspase-11: Implications in TLR4-independent endotoxic shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Walle, L.V.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef]
- Kerur, N.; Veettil, M.V.; Sharma-Walia, N.; Bottero, V.; Sadagopan, S.; Otageri, P.; Chandran, B. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe 2011, 9, 363–375. [Google Scholar] [CrossRef] [Green Version]
- Albrecht, M.; Choubey, D.; Lengauer, T. The HIN domain of IFI-200 proteins consists of two OB folds. Biochem. Biophys. Res. Commun. 2005, 327, 679–687. [Google Scholar] [CrossRef]
- Atianand, M.K.; Fitzgerald, K.A. Molecular basis of DNA recognition in the immune system. J. Immunol. 2013, 190, 1911–1918. [Google Scholar] [CrossRef]
- Yan, H.; Dalal, K.; Hon, B.K.; Youkharibache, P.; Lau, D.; Pio, F. RPA nucleic acid-binding properties of IFI16-HIN200. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2008, 1784, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Diner, B.A.; Chen, J.; Cristea, I.M. Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc. Natl. Acad. Sci. USA 2012, 109, 10558–10563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, T.S. The nucleic acid-sensing inflammasomes. Immunol. Rev. 2015, 265, 103–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, A.; Li, Y.; Yin, Q.; Ruan, J.; Yu, X.; Egelman, E.; Wu, H. Plasticity in PYD assembly revealed by cryo-EM structure of the PYD filament of AIM2. Cell Discov. 2015, 1, 15013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamkanfi, M.; Kanneganti, T.-D. Nlrp3: An immune sensor of cellular stress and infection. Int. J. Biochem. Cell Biol. 2010, 42, 792–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Zheng, M.; Kanneganti, T.D. The regulation of the ZBP1-NLRP3 inflammasome and its implications in pyroptosis, apoptosis, and necroptosis (PANoptosis). Immunol. Rev. 2020, 297, 26–38. [Google Scholar] [CrossRef]
- Wang, W.; Hu, D.; Wu, C.; Feng, Y.; Li, A.; Liu, W.; Wang, Y.; Chen, K.; Tian, M.; Xiao, F.; et al. STING promotes NLRP3 localization in ER and facilitates NLRP3 deubiquitination to activate the inflammasome upon HSV-1 infection. PLoS Pathog. 2020, 16, e1008335. [Google Scholar] [CrossRef] [Green Version]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS–STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Ramshorn, K.; Pinci, F.; Zuber, S.; O’Duill, F.; Schmid-Burgk, J.L.; Hoss, F.; Buhmann, R.; et al. The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell 2017, 171, 1110–1124.E18. [Google Scholar] [CrossRef]
- Mishra, S.; Raj, A.S.; Kumar, A.; Rajeevan, A.; Kumari, P.; Kumar, H. Innate immune sensing of influenza A viral RNA through IFI16 promotes pyroptotic cell death. iScience 2021, 25, 103714. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, R.; Guo, W.; Xie, L.; Qiao, Z.; Chen, S.; Zhu, J.; Huang, C.; Huang, J.; Chen, B.; et al. STING-Mediated IFI16 Degradation Negatively Controls Type I Interferon Production. Cell Rep. 2019, 29, 1249–1260.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, S.J.; Brode, S.; Ellis, L.; Fitzmaurice, T.J.; Elder, M.J.; Gekara, N.O.; Tourlomousis, P.; Bryant, C.; Clare, S.; Chee, R.; et al. Detection of a microbial metabolite by STING regulates inflammasome activation in response to Chlamydia trachomatis infection. PLoS Pathog. 2017, 13, e1006383. [Google Scholar] [CrossRef] [PubMed]
- Lee-Kirsch, M.A.; Gong, M.; Chowdhury, D.; Senenko, L.; Engel, K.; Lee, Y.A.; de Silva, U.; Bailey, S.L.; Witte, T.; Vyse, T.J.; et al. Mutations in the gene encoding the 3’-5’ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat. Genet. 2007, 39, 1065–1067. [Google Scholar] [CrossRef]
- Kato, Y.; Park, J.; Takamatsu, H.; Konaka, H.; Aoki, W.; Aburaya, S.; Ueda, M.; Nishide, M.; Koyama, S.; Hayama, Y.; et al. Apoptosis-derived membrane vesicles drive the cGAS-STING pathway and enhance type I IFN production in systemic lupus erythematosus. Ann. Rheum. Dis. 2018, 77, 1507–1515. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Xie, L.; Qiao, Z.; Mai, S.; Zhu, J.; Zhang, F.; Chen, S.; Li, L.; Shen, F.; Qin, Y.; et al. STING-mediated degradation of IFI16 negatively regulates apoptosis by inhibiting p53 phosphorylation at serine 392. J. Biol. Chem. 2021, 297, 100930. [Google Scholar] [CrossRef]
- Corrales, L.; Woo, S.-R.; Williams, J.B.; McWhirter, S.M.; Dubensky, T.W., Jr.; Gajewski, T.F. Antagonism of the STING Pathway via Activation of the AIM2 Inflammasome by Intracellular DNA. J. Immunol. 2016, 196, 3191–3198. [Google Scholar] [CrossRef]
- Kuriakose, T.; Zheng, M.; Neale, G.; Kanneganti, T.D. IRF1 Is a Transcriptional Regulator of ZBP1 Promoting NLRP3 Inflammasome Activation and Cell Death during Influenza Virus Infection. J. Immunol. 2018, 200, 1489–1495. [Google Scholar] [CrossRef] [Green Version]
- Banoth, B.; Tuladhar, S.; Karki, R.; Sharma, B.R.; Briard, B.; Kesavardhana, S.; Burton, A.; Kanneganti, T.D. ZBP1 promotes fungi-induced inflammasome activation and pyroptosis, apoptosis, and necroptosis (PANoptosis). J. Biol. Chem. 2020, 295, 18276–18283. [Google Scholar] [CrossRef]
- Muendlein, H.I.; Connolly, W.M.; Magri, Z.; Smirnova, I.; Ilyukha, V.; Gautam, A.; Degterev, A.; Poltorak, A. ZBP1 promotes LPS-induced cell death and IL-1β release via RHIM-mediated interactions with RIPK1. Nat. Commun. 2021, 12, 86. [Google Scholar] [CrossRef]
- Kuriakose, T.; Man, S.M.; Malireddi, R.K.S.; Karki, R.; Kesavardhana, S.; Place, D.E.; Neale, G.; Vogel, P.; Kanneganti, T.-D. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 2016, 1, aag2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Karki, R.; Wang, Y.; Nguyen, L.N.; Kalathur, R.C.; Kanneganti, T.-D. AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence. Nature 2021, 597, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, K.; Eryilmazlar, D.; Triantafilou, M. Herpes simplex virus 2-induced activation in vaginal cells involves Toll-like receptors 2 and 9 and DNA sensors DAI and IFI16. Am. J. Obstet. Gynecol. 2014, 210, 122.e1–122.e10. [Google Scholar] [CrossRef]
- Kesavardhana, S.; Kuriakose, T.; Guy, C.S.; Samir, P.; Malireddi, R.K.S.; Mishra, A.; Kanneganti, T.-D. ZBP1/DAI ubiquitination and sensing of influenza vRNPs activate programmed cell death. J. Exp. Med. 2017, 214, 2217–2229. [Google Scholar] [CrossRef] [PubMed]
- Thapa, R.J.; Ingram, J.P.; Ragan, K.B.; Nogusa, S.; Boyd, D.F.; Benitez, A.A.; Sridharan, H.; Kosoff, R.; Shubina, M.; Landsteiner, V.J.; et al. DAI Senses Influenza A Virus Genomic RNA and Activates RIPK3-Dependent Cell Death. Cell Host Microbe 2016, 20, 674–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, T.H.; Kwon, K.M.; Kim, Y.-E.; Kim, K.K.; Ahn, J.-H. DNA sensing-independent inhibition of herpes simplex virus 1 replication by DAI/ZBP1. J. Virol. 2013, 87, 3076–3086. [Google Scholar] [CrossRef] [Green Version]
- Gomes, M.T.; Campos, P.C.; Oliveira, F.S.; Corsetti, P.P.; Bortoluci, K.R.; Cunha, L.D.; Zamboni, D.S.; Oliveira, S.C. Critical role of ASC inflammasomes and bacterial type IV secretion system in caspase-1 activation and host innate resistance to Brucella abortus infection. J. Immunol. 2013, 190, 3629–3638. [Google Scholar] [CrossRef] [Green Version]
- Hielpos, M.S.; Fernández, A.G.; Falivene, J.; Alonso Paiva, I.M.; Muñoz González, F.; Ferrero, M.C.; Campos, P.C.; Vieira, A.T.; Oliveira, S.C.; Baldi, P.C. IL-1R and Inflammasomes Mediate Early Pulmonary Protective Mechanisms in Respiratory Brucella Abortus Infection. Front. Cell Infect. Microbiol. 2018, 8, 391. [Google Scholar] [CrossRef]
- Costa Franco, M.M.; Marim, F.; Guimarães, E.S.; Assis, N.R.G.; Cerqueira, D.M.; Alves-Silva, J.; Harms, J.; Splitter, G.; Smith, J.; Kanneganti, T.D.; et al. Brucella abortus Triggers a cGAS-Independent STING Pathway to Induce Host Protection That Involves Guanylate-Binding Proteins and Inflammasome Activation. J. Immunol. 2018, 200, 607–622. [Google Scholar] [CrossRef] [Green Version]
- Cerqueira, D.M.; Gomes, M.T.R.; Silva, A.L.N.; Rungue, M.; Assis, N.R.G.; Guimarães, E.S.; Morais, S.B.; Broz, P.; Zamboni, D.S.; Oliveira, S.C. Guanylate-binding protein 5 licenses caspase-11 for Gasdermin-D mediated host resistance to Brucella abortus infection. PLoS Pathog. 2018, 14, e1007519. [Google Scholar] [CrossRef]
- Costa Franco, M.M.S.; Marim, F.M.; Alves-Silva, J.; Cerqueira, D.; Rungue, M.; Tavares, I.P.; Oliveira, S.C. AIM2 senses Brucella abortus DNA in dendritic cells to induce IL-1β secretion, pyroptosis and resistance to bacterial infection in mice. Microbes Infect. 2019, 21, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Tupik, J.D.; Coutermarsh-Ott, S.L.; Benton, A.H.; King, K.A.; Kiryluk, H.D.; Caswell, C.C.; Allen, I.C. ASC-Mediated Inflammation and Pyroptosis Attenuates Brucella abortus Pathogenesis Following the Recognition of gDNA. Pathogens 2020, 9, 1008. [Google Scholar] [CrossRef] [PubMed]
- Lacey, C.A.; Mitchell, W.J.; Dadelahi, A.S.; Skyberg, J.A. Caspase-1 and Caspase-11 Mediate Pyroptosis, Inflammation, and Control of Brucella Joint Infection. Infect. Immun. 2018, 86, e00361-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahoo, M.; Lantier, L.; Re, F. Role of Canonical and Non-canonical Inflammasomes During Burkholderia Infection. Curr. Top. Microbiol. Immunol. 2016, 397, 199–214. [Google Scholar] [CrossRef]
- Ceballos-Olvera, I.; Sahoo, M.; Miller, M.A.; Del Barrio, L.; Re, F. Inflammasome-dependent pyroptosis and IL-18 protect against Burkholderia pseudomallei lung infection while IL-1β is deleterious. PLoS Pathog. 2011, 7, e1002452. [Google Scholar] [CrossRef]
- Lichtenegger, S.; Stiehler, J.; Saiger, S.; Zauner, A.; Kleinhappl, B.; Bernecker, C.; Schlenke, P.; Wagner, G.E.; Krause, K.; Gastager, M.; et al. Burkholderia pseudomallei triggers canonical inflammasome activation in a human primary macrophage-based infection model. PLoS Negl. Trop. Dis. 2020, 14, e0008840. [Google Scholar] [CrossRef]
- Wang, J.; Sahoo, M.; Lantier, L.; Warawa, J.; Cordero, H.; Deobald, K.; Re, F. Caspase-11-dependent pyroptosis of lung epithelial cells protects from melioidosis while caspase-1 mediates macrophage pyroptosis and production of IL-18. PLoS Pathog. 2018, 14, e1007105. [Google Scholar] [CrossRef]
- Bast, A.; Krause, K.; Schmidt, I.H.; Pudla, M.; Brakopp, S.; Hopf, V.; Breitbach, K.; Steinmetz, I. Caspase-1-dependent and -independent cell death pathways in Burkholderia pseudomallei infection of macrophages. PLoS Pathog. 2014, 10, e1003986. [Google Scholar] [CrossRef] [Green Version]
- Miao, E.A.; Leaf, I.A.; Treuting, P.M.; Mao, D.P.; Dors, M.; Sarkar, A.; Warren, S.E.; Wewers, M.D.; Aderem, A. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 2010, 11, 1136–1142. [Google Scholar] [CrossRef]
- Place, D.E.; Christgen, S.; Tuladhar, S.; Vogel, P.; Malireddi, R.K.S.; Kanneganti, T.D. Hierarchical Cell Death Program Disrupts the Intracellular Niche Required for Burkholderia thailandensis Pathogenesis. mBio 2021, 12, e0105921. [Google Scholar] [CrossRef]
- Ku, J.W.K.; Chen, Y.; Lim, B.J.W.; Gasser, S.; Crasta, K.C.; Gan, Y.H. Bacterial-induced cell fusion is a danger signal triggering cGAS-STING pathway via micronuclei formation. Proc. Natl. Acad. Sci. USA 2020, 117, 15923–15934. [Google Scholar] [CrossRef] [PubMed]
- Dilucca, M.; Ramos, S.; Shkarina, K.; Santos, J.C.; Broz, P. Guanylate-Binding Protein-Dependent Noncanonical Inflammasome Activation Prevents Burkholderia thailandensis-Induced Multinucleated Giant Cell Formation. mBio 2021, 12, e0205421. [Google Scholar] [CrossRef] [PubMed]
- Gavrilin, M.A.; Abdelaziz, D.H.; Mostafa, M.; Abdulrahman, B.A.; Grandhi, J.; Akhter, A.; Abu Khweek, A.; Aubert, D.F.; Valvano, M.A.; Wewers, M.D.; et al. Activation of the pyrin inflammasome by intracellular Burkholderia cenocepacia. J. Immunol. 2012, 188, 3469–3477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallet, P.; Lagrange, B.; Henry, T. Francisella Inflammasomes: Integrated Responses to a Cytosolic Stealth Bacterium. In Inflammasome Signaling and Bacterial Infections; Backert, S., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 229–256. [Google Scholar] [CrossRef]
- Atianand, M.K.; Duffy, E.B.; Shah, A.; Kar, S.; Malik, M.; Harton, J.A. Francisella tularensis reveals a disparity between human and mouse NLRP3 inflammasome activation. J. Biol. Chem. 2011, 286, 39033–39042. [Google Scholar] [CrossRef] [Green Version]
- Periasamy, S.; Le, H.T.; Duffy, E.B.; Chin, H.; Harton, J.A. Inflammasome-Independent NLRP3 Restriction of a Protective Early Neutrophil Response to Pulmonary Tularemia. PLoS Pathog. 2016, 12, e1006059. [Google Scholar] [CrossRef] [Green Version]
- Belhocine, K.; Monack, D.M. Francisella infection triggers activation of the AIM2 inflammasome in murine dendritic cells. Cell. Microbiol. 2012, 14, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Fernandes-Alnemri, T.; Yu, J.-W.; Juliana, C.; Solorzano, L.; Kang, S.; Wu, J.; Datta, P.; McCormick, M.; Huang, L.; McDermott, E.; et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 2010, 11, 385–393. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.W.; Kayagaki, N.; Broz, P.; Henry, T.; Newton, K.; O’Rourke, K.; Chan, S.; Dong, J.; Qu, Y.; Roose-Girma, M.; et al. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc. Natl. Acad. Sci. USA 2010, 107, 9771–9776. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Zheng, M.; Balakrishnan, A.; Karki, R.; Kanneganti, T.D. Gasdermin D Promotes AIM2 Inflammasome Activation and Is Required for Host Protection against Francisella novicida. J. Immunol. 2018, 201, 3662–3668. [Google Scholar] [CrossRef] [Green Version]
- Man, S.M.; Karki, R.; Malireddi, R.K.S.; Neale, G.; Vogel, P.; Yamamoto, M.; Lamkanfi, M.; Kanneganti, T.-D. The transcription factor IRF1 and guanylate-binding proteins target activation of the AIM2 inflammasome by Francisella infection. Nat. Immunol. 2015, 16, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.W.; Broz, P.; Monack, D.M. Innate immune recognition of francisella tularensis: Activation of type-I interferons and the inflammasome. Front. Microbiol. 2011, 2, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meunier, E.; Wallet, P.; Dreier, R.F.; Costanzo, S.; Anton, L.; Rühl, S.; Dussurgey, S.; Dick, M.S.; Kistner, A.; Rigard, M.; et al. Guanylate-binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nat. Immunol. 2015, 16, 476–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storek, K.M.; Gertsvolf, N.A.; Ohlson, M.B.; Monack, D.M. cGAS and Ifi204 cooperate to produce type I IFNs in response to Francisella infection. J. Immunol. 2015, 194, 3236–3245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.; Man, S.M.; Karki, R.; Malireddi, R.K.S.; Kanneganti, T.D. Detrimental Type I Interferon Signaling Dominates Protective AIM2 Inflammasome Responses during Francisella novicida Infection. Cell Rep. 2018, 22, 3168–3174. [Google Scholar] [CrossRef] [Green Version]
- Pierini, R.; Juruj, C.; Perret, M.; Jones, C.L.; Mangeot, P.; Weiss, D.S.; Henry, T. AIM2/ASC triggers caspase-8-dependent apoptosis in Francisella-infected caspase-1-deficient macrophages. Cell Death Differ. 2012, 19, 1709–1721. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, I.; Behl, B.; Mendonca, M.; Shrivastava, G.; Russo, A.J.; Menoret, A.; Ghosh, A.; Vella, A.T.; Vanaja, S.K.; Sarkar, S.N.; et al. Gasdermin D Restrains Type I Interferon Response to Cytosolic DNA by Disrupting Ionic Homeostasis. Immunity 2018, 49, 413–426.e5. [Google Scholar] [CrossRef] [Green Version]
- Gunn, J.S.; Ernst, R.K. The structure and function of Francisella lipopolysaccharide. Ann. N. Y. Acad. Sci. 2007, 1105, 202–218. [Google Scholar] [CrossRef]
- Dotson, R.J.; Rabadi, S.M.; Westcott, E.L.; Bradley, S.; Catlett, S.V.; Banik, S.; Harton, J.A.; Bakshi, C.S.; Malik, M. Repression of inflammasome by Francisella tularensis during early stages of infection. J. Biol. Chem. 2013, 288, 23844–23857. [Google Scholar] [CrossRef] [Green Version]
- Ulland, T.K.; Buchan, B.W.; Ketterer, M.R.; Fernandes-Alnemri, T.; Meyerholz, D.K.; Apicella, M.A.; Alnemri, E.S.; Jones, B.D.; Nauseef, W.M.; Sutterwala, F.S. Cutting edge: Mutation of Francisella tularensis mviN leads to increased macrophage absent in melanoma 2 inflammasome activation and a loss of virulence. J. Immunol. 2010, 185, 2670–2674. [Google Scholar] [CrossRef] [Green Version]
- Peng, K.; Broz, P.; Jones, J.; Joubert, L.M.; Monack, D. Elevated AIM2-mediated pyroptosis triggered by hypercytotoxic Francisella mutant strains is attributed to increased intracellular bacteriolysis. Cell Microbiol. 2011, 13, 1586–1600. [Google Scholar] [CrossRef] [Green Version]
- Cunha, L.D.; Zamboni, D.S. Recognition of Legionella pneumophila nucleic acids by innate immune receptors. Microbes Infect. 2014, 16, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Case, C.L.; Shin, S.; Roy, C.R. Asc and Ipaf Inflammasomes direct distinct pathways for caspase-1 activation in response to Legionella pneumophila. Infect. Immun. 2009, 77, 1981–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katagiri, N.; Shobuike, T.; Chang, B.; Kukita, A.; Miyamoto, H. The human apoptosis inhibitor NAIP induces pyroptosis in macrophages infected with Legionella pneumophila. Microbes Infect. 2012, 14, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Case, C.L.; Kohler, L.J.; Lima, J.B.; Strowig, T.; de Zoete, M.R.; Flavell, R.A.; Zamboni, D.S.; Roy, C.R. Caspase-11 stimulates rapid flagellin-independent pyroptosis in response to Legionella pneumophila. Proc. Natl. Acad. Sci. USA 2013, 110, 1851–1856. [Google Scholar] [CrossRef] [Green Version]
- Cunha, L.D.; Silva, A.L.N.; Ribeiro, J.M.; Mascarenhas, D.P.A.; Quirino, G.F.S.; Santos, L.L.; Flavell, R.A.; Zamboni, D.S. AIM2 Engages Active but Unprocessed Caspase-1 to Induce Noncanonical Activation of the NLRP3 Inflammasome. Cell Rep. 2017, 20, 794–805. [Google Scholar] [CrossRef]
- Lippmann, J.; Müller, H.C.; Naujoks, J.; Tabeling, C.; Shin, S.; Witzenrath, M.; Hellwig, K.; Kirschning, C.J.; Taylor, G.A.; Barchet, W.; et al. Dissection of a type I interferon pathway in controlling bacterial intracellular infection in mice. Cell Microbiol. 2011, 13, 1668–1682. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.C.; Sarhan, J.; Panda, A.; Muendlein, H.I.; Ilyukha, V.; Coers, J.; Yamamoto, M.; Isberg, R.R.; Poltorak, A. Constitutive Interferon Maintains GBP Expression Required for Release of Bacterial Components Upstream of Pyroptosis and Anti-DNA Responses. Cell Rep. 2018, 24, 155–168.e5. [Google Scholar] [CrossRef] [Green Version]
- Pilla, D.M.; Hagar, J.A.; Haldar, A.K.; Mason, A.K.; Degrandi, D.; Pfeffer, K.; Ernst, R.K.; Yamamoto, M.; Miao, E.A.; Coers, J. Guanylate binding proteins promote caspase-11-dependent pyroptosis in response to cytoplasmic LPS. Proc. Natl. Acad. Sci. USA 2014, 111, 6046–6051. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Moreno, J.S.; Hamann, L.; Shah, J.A.; Verbon, A.; Mockenhaupt, F.P.; Puzianowska-Kuznicka, M.; Naujoks, J.; Sander, L.E.; Witzenrath, M.; Cambier, J.C.; et al. The common HAQ STING variant impairs cGAS-dependent antibacterial responses and is associated with susceptibility to Legionnaires’ disease in humans. PLoS Pathog. 2018, 14, e1006829. [Google Scholar] [CrossRef]
- Ge, J.; Gong, Y.N.; Xu, Y.; Shao, F. Preventing bacterial DNA release and absent in melanoma 2 inflammasome activation by a Legionella effector functioning in membrane trafficking. Proc. Natl. Acad. Sci. USA 2012, 109, 6193–6198. [Google Scholar] [CrossRef] [Green Version]
- Beckwith, K.S.; Beckwith, M.S.; Ullmann, S.; Sætra, R.S.; Kim, H.; Marstad, A.; Åsberg, S.E.; Strand, T.A.; Haug, M.; Niederweis, M.; et al. Plasma membrane damage causes NLRP3 activation and pyroptosis during Mycobacterium tuberculosis infection. Nat. Commun. 2020, 11, 2270. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Zhou, J.; Zhou, Y.; Xie, Y.; Jiang, Y.; Wu, J.; Luo, Z.; Liu, G.; Yin, L.; Zhang, X.L. Mycobacterial EST12 activates a RACK1-NLRP3-gasdermin D pyroptosis-IL-1β immune pathway. Sci. Adv. 2020, 6, eaba4733. [Google Scholar] [CrossRef] [PubMed]
- Stutz, M.D.; Ojaimi, S.; Allison, C.; Preston, S.; Arandjelovic, P.; Hildebrand, J.M.; Sandow, J.J.; Webb, A.I.; Silke, J.; Alexander, W.S.; et al. Necroptotic signaling is primed in Mycobacterium tuberculosis-infected macrophages, but its pathophysiological consequence in disease is restricted. Cell Death Differ. 2018, 25, 951–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdalla, H.; Srinivasan, L.; Shah, S.; Mayer-Barber, K.D.; Sher, A.; Sutterwala, F.S.; Briken, V. Mycobacterium tuberculosis infection of dendritic cells leads to partially caspase-1/11-independent IL-1β and IL-18 secretion but not to pyroptosis. PLoS ONE 2012, 7, e40722. [Google Scholar] [CrossRef]
- Rastogi, S.; Ellinwood, S.; Augenstreich, J.; Mayer-Barber, K.D.; Briken, V. Mycobacterium tuberculosis inhibits the NLRP3 inflammasome activation via its phosphokinase PknF. PLoS Pathog. 2021, 17, e1009712. [Google Scholar] [CrossRef]
- Saiga, H.; Kitada, S.; Shimada, Y.; Kamiyama, N.; Okuyama, M.; Makino, M.; Yamamoto, M.; Takeda, K. Critical role of AIM2 in Mycobacterium tuberculosis infection. Int. Immunol. 2012, 24, 637–644. [Google Scholar] [CrossRef] [Green Version]
- Figueira, M.B.A.; de Lima, D.S.; Boechat, A.L.; Filho, M.; Antunes, I.A.; Matsuda, J.D.S.; Ribeiro, T.R.A.; Felix, L.S.; Gonçalves, A.S.F.; da Costa, A.G.; et al. Single-Nucleotide Variants in the AIM2—Absent in Melanoma 2 Gene (rs1103577) Associated With Protection for Tuberculosis. Front. Immunol. 2021, 12, 604975. [Google Scholar] [CrossRef]
- Yan, S.; Shen, H.; Lian, Q.; Jin, W.; Zhang, R.; Lin, X.; Gu, W.; Sun, X.; Meng, G.; Tian, Z.; et al. Deficiency of the AIM2-ASC Signal Uncovers the STING-Driven Overreactive Response of Type I IFN and Reciprocal Depression of Protective IFN-γ Immunity in Mycobacterial Infection. J. Immunol. 2018, 200, 1016–1026. [Google Scholar] [CrossRef]
- Manzanillo, P.S.; Shiloh, M.U.; Portnoy, D.A.; Cox, J.S. Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell Host Microbe 2012, 11, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Watson, R.O.; Bell, S.L.; MacDuff, D.A.; Kimmey, J.M.; Diner, E.J.; Olivas, J.; Vance, R.E.; Stallings, C.L.; Virgin, H.W.; Cox, J.S. The Cytosolic Sensor cGAS Detects Mycobacterium tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host Microbe 2015, 17, 811–819. [Google Scholar] [CrossRef] [Green Version]
- Collins, A.C.; Cai, H.; Li, T.; Franco, L.H.; Li, X.D.; Nair, V.R.; Scharn, C.R.; Stamm, C.E.; Levine, B.; Chen, Z.J.; et al. Cyclic GMP-AMP Synthase Is an Innate Immune DNA Sensor for Mycobacterium tuberculosis. Cell Host Microbe 2015, 17, 820–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marinho, F.V.; Benmerzoug, S.; Rose, S.; Campos, P.C.; Marques, J.T.; Báfica, A.; Barber, G.; Ryffel, B.; Oliveira, S.C.; Quesniaux, V.F.J. The cGAS/STING Pathway Is Important for Dendritic Cell Activation but Is Not Essential to Induce Protective Immunity against Mycobacterium tuberculosis Infection. J. Innate Immun. 2018, 10, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Bohsali, A.; Ahlbrand, S.E.; Srinivasan, L.; Rathinam, V.A.; Vogel, S.N.; Fitzgerald, K.A.; Sutterwala, F.S.; Briken, V. Cutting edge: Mycobacterium tuberculosis but not nonvirulent mycobacteria inhibits IFN-β and AIM2 inflammasome-dependent IL-1β production via its ESX-1 secretion system. J. Immunol. 2013, 191, 3514–3518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, G.; Idosa, B.A.; Bäckman, A.; Monecke, S.; Strålin, K.; Särndahl, E.; Söderquist, B. Caspase-1 inflammasome activity in patients with Staphylococcus aureus bacteremia. Microbiol. Immunol. 2019, 63, 487–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, K.M.; Ramanan, K.; Clay, M.E.; McHugh, K.J.; Pilewski, M.J.; Nickolich, K.L.; Corey, C.; Shiva, S.; Wang, J.; Muzumdar, R.; et al. The inflammasome potentiates influenza/Staphylococcus aureus superinfection in mice. JCI Insight 2018, 3, e97470. [Google Scholar] [CrossRef] [Green Version]
- Kitur, K.; Wachtel, S.; Brown, A.; Wickersham, M.; Paulino, F.; Peñaloza, H.F.; Soong, G.; Bueno, S.; Parker, D.; Prince, A. Necroptosis Promotes Staphylococcus aureus Clearance by Inhibiting Excessive Inflammatory Signaling. Cell Rep. 2016, 16, 2219–2230. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Liu, M.; Geng, N.; Du, Y.; Li, Z.; Gao, X.; Han, B.; Liu, J.; Liu, Y. Staphylococcus aureus mediates pyroptosis in bovine mammary epithelial cell via activation of NLRP3 inflammasome. Vet. Res. 2022, 53, 10. [Google Scholar] [CrossRef]
- Accarias, S.; Lugo-Villarino, G.; Foucras, G.; Neyrolles, O.; Boullier, S.; Tabouret, G. Pyroptosis of resident macrophages differentially orchestrates inflammatory responses to Staphylococcus aureus in resistant and susceptible mice. Eur. J. Immunol. 2015, 45, 794–806. [Google Scholar] [CrossRef]
- Kebaier, C.; Chamberland, R.R.; Allen, I.C.; Gao, X.; Broglie, P.M.; Hall, J.D.; Jania, C.; Doerschuk, C.M.; Tilley, S.L.; Duncan, J.A. Staphylococcus aureus α-hemolysin mediates virulence in a murine model of severe pneumonia through activation of the NLRP3 inflammasome. J. Infect. Dis. 2012, 205, 807–817. [Google Scholar] [CrossRef]
- Craven, R.R.; Gao, X.; Allen, I.C.; Gris, D.; Bubeck Wardenburg, J.; McElvania-Tekippe, E.; Ting, J.P.; Duncan, J.A. Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS ONE 2009, 4, e7446. [Google Scholar] [CrossRef]
- Kitur, K.; Parker, D.; Nieto, P.; Ahn, D.S.; Cohen, T.S.; Chung, S.; Wachtel, S.; Bueno, S.; Prince, A. Toxin-induced necroptosis is a major mechanism of Staphylococcus aureus lung damage. PLoS Pathog. 2015, 11, e1004820. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhao, S.; Gao, X.; Jiang, S.; Ma, J.; Wang, R.; Li, Q.; Qin, L.; Tong, Z.; Wu, J.; et al. Staphylococcus aureus Induces IFN-β Production via a CARMA3-Independent Mechanism. Pathogens 2021, 10, 300. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.Z.; Yang, Y.J.; Zhou, C.K.; Yan, S.Q.; Ma, K.; Gao, Y.; Chen, W. STING Contributes to Host Defense Against Staphylococcus aureus Pneumonia Through Suppressing Necroptosis. Front. Immunol. 2021, 12, 636861. [Google Scholar] [CrossRef] [PubMed]
- Gries, C.M.; Bruger, E.L.; Moormeier, D.E.; Scherr, T.D.; Waters, C.M.; Kielian, T. Cyclic di-AMP Released from Staphylococcus aureus Biofilm Induces a Macrophage Type I Interferon Response. Infect. Immun. 2016, 84, 3564–3574. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Yu, S.X.; Zhou, F.H.; Zhang, X.J.; Gao, W.Y.; Li, K.Y.; Liu, Z.Z.; Han, W.Y.; Yang, Y.J. DNA Sensor IFI204 Contributes to Host Defense Against Staphylococcus aureus Infection in Mice. Front. Immunol. 2019, 10, 474. [Google Scholar] [CrossRef]
- Rabes, A.; Suttorp, N.; Opitz, B. Inflammasomes in Pneumococcal Infection: Innate Immune Sensing and Bacterial Evasion Strategies. In Inflammasome Signaling and Bacterial Infections; Backert, S., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 215–227. [Google Scholar] [CrossRef]
- Fang, R.; Tsuchiya, K.; Kawamura, I.; Shen, Y.; Hara, H.; Sakai, S.; Yamamoto, T.; Fernandes-Alnemri, T.; Yang, R.; Hernandez-Cuellar, E.; et al. Critical roles of ASC inflammasomes in caspase-1 activation and host innate resistance to Streptococcus pneumoniae infection. J. Immunol. 2011, 187, 4890–4899. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.; Chen, T.; Lei, G.; Hou, F.; Jiang, J.; Huang, Q.; Peng, Y.; Ye, C.; Hu, D.-L.; Fang, R. Absent in melanoma 2 inflammasome is required for host defence against Streptococcus pneumoniae infection. Innate Immun. 2019, 25, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Witzenrath, M.; Pache, F.; Lorenz, D.; Koppe, U.; Gutbier, B.; Tabeling, C.; Reppe, K.; Meixenberger, K.; Dorhoi, A.; Ma, J.; et al. The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia. J. Immunol. 2011, 187, 434–440. [Google Scholar] [CrossRef]
- Kim, J.Y.; Paton, J.C.; Briles, D.E.; Rhee, D.K.; Pyo, S. Streptococcus pneumoniae induces pyroptosis through the regulation of autophagy in murine microglia. Oncotarget 2015, 6, 44161–44178. [Google Scholar] [CrossRef] [Green Version]
- Karmakar, M.; Katsnelson, M.; Malak, H.A.; Greene, N.G.; Howell, S.J.; Hise, A.G.; Camilli, A.; Kadioglu, A.; Dubyak, G.R.; Pearlman, E. Neutrophil IL-1β processing induced by pneumolysin is mediated by the NLRP3/ASC inflammasome and caspase-1 activation and is dependent on K+ efflux. J. Immunol. 2015, 194, 1763–1775. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Du, H.; Feng, S.; Wu, R.; Chen, T.; Jiang, J.; Peng, Y.; Ye, C.; Fang, R. NLRP3/ASC/Caspase-1 axis and serine protease activity are involved in neutrophil IL-1β processing during Streptococcus pneumoniae infection. Biochem. Biophys. Res. Commun. 2019, 513, 675–680. [Google Scholar] [CrossRef] [PubMed]
- van Lieshout, M.H.; Scicluna, B.P.; Florquin, S.; van der Poll, T. NLRP3 and ASC differentially affect the lung transcriptome during pneumococcal pneumonia. Am. J. Respir. Cell Mol. Biol. 2014, 50, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.; Hara, H.; Sakai, S.; Hernandez-Cuellar, E.; Mitsuyama, M.; Kawamura, I.; Tsuchiya, K. Type I interferon signaling regulates activation of the absent in melanoma 2 inflammasome during Streptococcus pneumoniae infection. Infect. Immun. 2014, 82, 2310–2317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, D.; Martin, F.J.; Soong, G.; Harfenist, B.S.; Aguilar, J.L.; Ratner, A.J.; Fitzgerald, K.A.; Schindler, C.; Prince, A. Streptococcus pneumoniae DNA initiates type I interferon signaling in the respiratory tract. mBio 2011, 2, e00016-11. [Google Scholar] [CrossRef] [Green Version]
- Koppe, U.; Högner, K.; Doehn, J.M.; Müller, H.C.; Witzenrath, M.; Gutbier, B.; Bauer, S.; Pribyl, T.; Hammerschmidt, S.; Lohmeyer, J.; et al. Streptococcus pneumoniae stimulates a STING- and IFN regulatory factor 3-dependent type I IFN production in macrophages, which regulates RANTES production in macrophages, cocultured alveolar epithelial cells, and mouse lungs. J. Immunol. 2012, 188, 811–817. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.R.; Cho, S.J.; Harris, R.M.; Yang, J.; Bermejo, S.; Sharma, L.; Dela Cruz, C.S.; Xu, J.F.; Stout-Delgado, H.W. RIPK3 Activates MLKL-mediated Necroptosis and Inflammasome Signaling during Streptococcus Infection. Am. J. Respir. Cell Mol. Biol. 2021, 64, 579–591. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Bacteria | Mechanisms of DNA-Mediated Inflammasome Activation | Function in Disease | References |
|---|---|---|---|
| Brucella spp. | STING-dependent but cGAS independent recognition of DNA; DNA release is mediated by GBPs and regulates caspase-11 activation; DNA promotes activation of the AIM2/ASC inflammasome and induces IL-1β secretion and pyroptosis in dendritic cells and macrophages; some implications of NLRP3 activation but not in the induction of pyroptosis | Protective against bacterial burden; pulmonary protection in alveolar macrophages | [57,58,59,60,61,62,63] |
| Burkholderia spp. | cGAS–STING recognition triggering autophagic cell death; GBP induction of non-canonical inflammasome activation and IFN stimulation; Caspase-1 meditation of pyroptosis in macrophages; Caspase-11 inflammasome mediated cleavage of Gasdermin-D pyroptosis and NLRP3 activation; PAN-optosis initiation of pyroptosis and apoptosis | Protection against bacterial replication and spread in murine lungs and human epithelial cells through pyroptosis and cell death | [26,64,65,66,67,68,69,70,71,72,73] |
| Francisella tularensis/novicida | GBP/STING/Ifi204 initiation of IFN signaling; IFN regulation of AIM2 inflammasome components; AIM2/ASC initiation of cytokine release and pyroptosis/apoptosis; GSDMD regulation of IFN signaling | Protective against bacterial load in lungs and mortality in murine models | [52,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91] |
| Legionella pneumophila | GBP/cGAS-STING initiation of IFN signaling and caspase-11 initiation of pyroptosis; AIM2/caspase-11 regulation of NLRP3 inflammasome activation | Protective against higher bacterial load and frequency of severe Legionnaire’s disease in humans | [92,93,94,95,96,97,98,99,100,101] |
| Mycobacterium tuberculosis | cGAS-STING activation of dendritic cells; ZBP1-mediated necroptosis; K+ efflux/ROS triggering NLRP3/ASC cytokine maturation and pyroptosis; IFN stimulation of AIM2 initiation of pyroptosis; AIM2 regulation of cGAS-STING overactivation of IFN signaling; caspase-11 initiation of pyroptosis | Protection against intratracheal infection through cytokine secretion; harmful when pyroptosis releases bacteria from phagosome | [102,103,104,105,106,107,108,109,110,111,112,113,114] |
| Staphylococcus aureus | STING-mediated suppression of necroptosis and IFN expression; Ifi204 initiation of IFN signaling and bacteria clearance; NLRP3/ASC activation with cytokine secretion and pyroptosis/necrosis; RIPK3 necroptosis initiation of bacteria clearance | Inflammasome protection against bacteria clearance but harmful initiation of excessive inflammation and mortality; DNA sensing may control excessive inflammation | [115,116,117,118,119,120,121,122,123,124,125,126] |
| Streptococcus pneumoniae | STING/ZBP1 mediation of IFN signaling and subsequent AIM2 component activation; AIM2/ASC and NLRP3 cytokine secretion and pyroptosis; RIPK3 initiation of NLRP3 activation and cell death | Reduced bacterial counts in nasal lavage fluid and reduced lung inflammation | [127,128,129,130,131,132,133,134,135,136,137,138] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tupik, J.D.; Markov Madanick, J.W.; Ivester, H.M.; Allen, I.C. Detecting DNA: An Overview of DNA Recognition by Inflammasomes and Protection against Bacterial Respiratory Infections. Cells 2022, 11, 1681. https://doi.org/10.3390/cells11101681
Tupik JD, Markov Madanick JW, Ivester HM, Allen IC. Detecting DNA: An Overview of DNA Recognition by Inflammasomes and Protection against Bacterial Respiratory Infections. Cells. 2022; 11(10):1681. https://doi.org/10.3390/cells11101681
Chicago/Turabian StyleTupik, Juselyn D., Justin W. Markov Madanick, Hannah M. Ivester, and Irving C. Allen. 2022. "Detecting DNA: An Overview of DNA Recognition by Inflammasomes and Protection against Bacterial Respiratory Infections" Cells 11, no. 10: 1681. https://doi.org/10.3390/cells11101681
APA StyleTupik, J. D., Markov Madanick, J. W., Ivester, H. M., & Allen, I. C. (2022). Detecting DNA: An Overview of DNA Recognition by Inflammasomes and Protection against Bacterial Respiratory Infections. Cells, 11(10), 1681. https://doi.org/10.3390/cells11101681