
Uncovering Functional Contributions of PMAT (Slc29a4) to Monoamine Clearance Using Pharmacobehavioral Tools

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Genotyping
2.3. Drugs
2.4. Behavior Tests
2.4.1. Tail Suspension Test (TST)
2.4.2. Post-TST Locomotor Testing
2.4.3. Psychostimulant-Induced Locomotor Sensitization—Common Methods
2.4.3.1. Cocaine-Induced Locomotor Sensitization
2.4.3.2. D-Amphetamine Induced Locomotor Sensitization
2.5. Statistical Analyses
3. Results
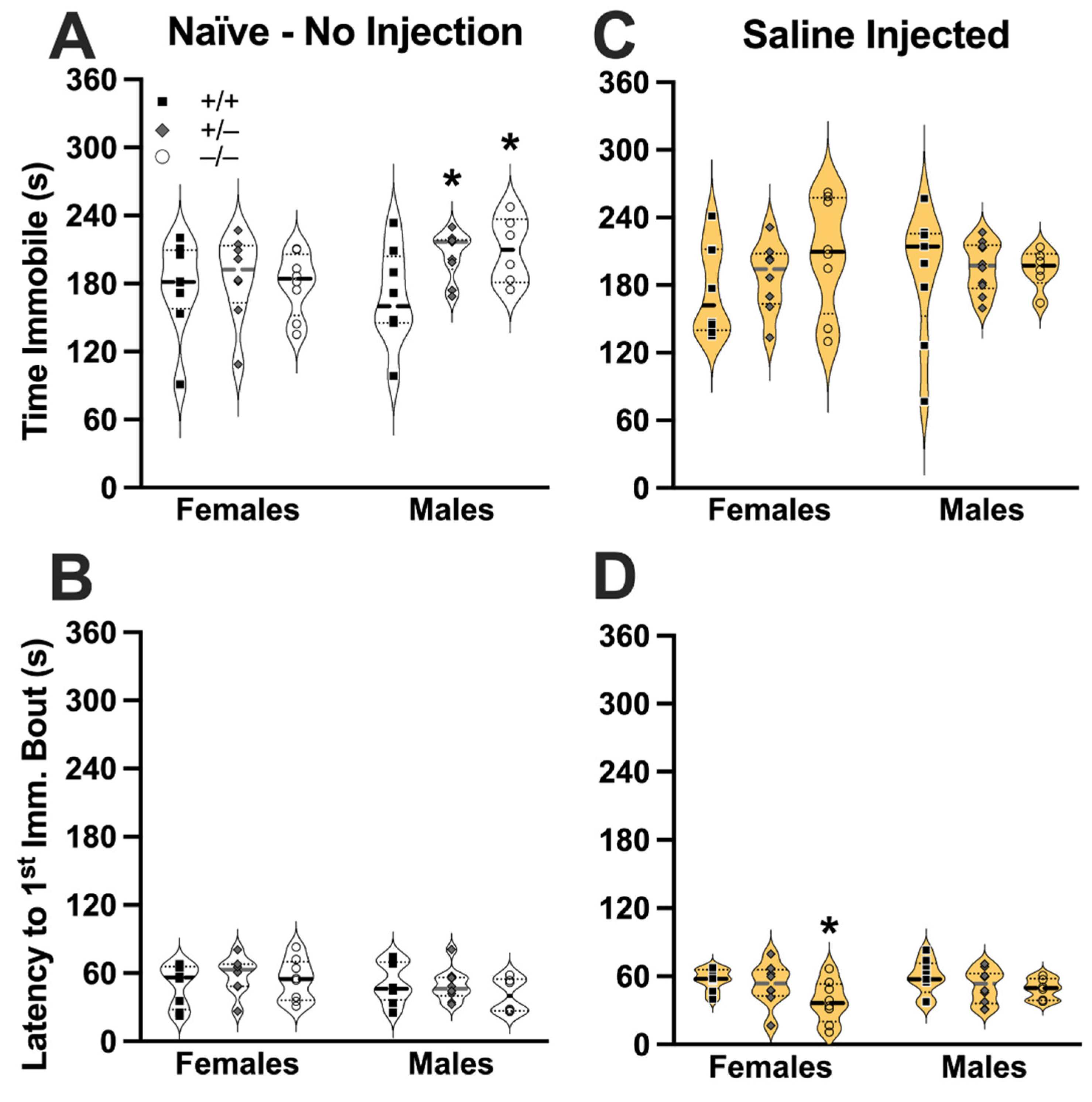
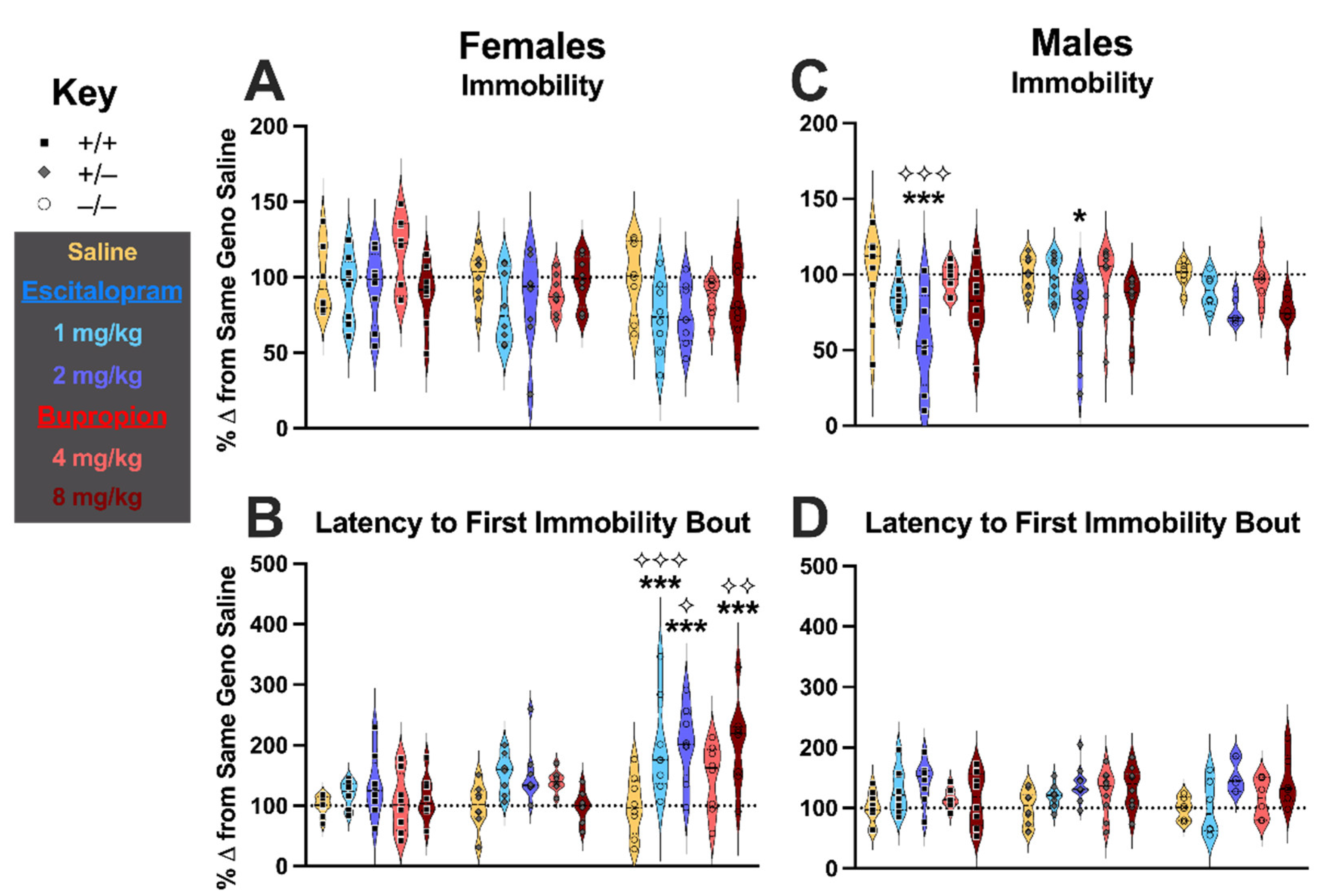
3.1. TST Behavior
3.2. Post-TST Locomotor Behavior
3.3. Cocaine-Induced Locomotor Sensitization
3.4. Amphetamine-Induced Locomotor Sensitization
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aggarwal, S.; Mortensen, O.V. Overview of Monoamine Transporters. Curr. Protoc. Pharm. 2017, 79, 12.16.1–12.16.17. [Google Scholar] [CrossRef] [PubMed]
- Daws, L.C. Unfaithful neurotransmitter transporters: Focus on serotonin uptake and implications for antidepressant efficacy. Pharm. Ther. 2009, 121, 89–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bönisch, H. Organic Cation Transporters in the Central Nervous System. Handb. Exp. Pharm. 2021, 266, 119–167. [Google Scholar] [CrossRef]
- Maier, J.; Niello, M.; Rudin, D.; Daws, L.C.; Sitte, H.H. The Interaction of Organic Cation Transporters 1-3 and PMAT with Psychoactive Substances. Handb. Exp. Pharmacol. 2021, 266, 199–214. [Google Scholar] [CrossRef]
- Gu, H.; Wall, S.C.; Rudnick, G. Stable expression of biogenic amine transporters reveals differences in inhibitor sensitivity, kinetics, and ion dependence. J. Biol. Chem. 1994, 269, 7124–7130. [Google Scholar] [CrossRef]
- Wang, J. The plasma membrane monoamine transporter (PMAT): Structure, function, and role in organic cation disposition. Clin. Pharm. Ther. 2016, 100, 489–499. [Google Scholar] [CrossRef] [Green Version]
- Shirasaka, Y.; Lee, N.; Duan, H.; Ho, H.; Pak, J.; Wang, J. Interspecies comparison of the functional characteristics of plasma membrane monoamine transporter (PMAT) between human, rat and mouse. J. Chem. Neuroanat. 2017, 83, 99–106. [Google Scholar] [CrossRef]
- Mayer, F.P.; Schmid, D.; Owens, W.A.; Gould, G.G.; Apuschkin, M.; Kudlacek, O.; Salzer, I.; Boehm, S.; Chiba, P.; Williams, P.H.; et al. An unsuspected role for organic cation transporter 3 in the actions of amphetamine. Neuropsychopharmacology 2018, 43, 2408–2417. [Google Scholar] [CrossRef]
- Dahlin, A.; Xia, L.; Kong, W.; Hevner, R.; Wang, J. Expression and immunolocalization of the plasma membrane monoamine transporter in the brain. Neuroscience 2007, 146, 1193–1211. [Google Scholar] [CrossRef] [Green Version]
- Duan, H.; Wang, J. Selective Transport of Monoamine Neurotransmitters by Human Plasma Membrane Monoamine Transporter and Organic Cation Transporter 3. J. Pharm. Exp. Ther. 2010, 335, 743–753. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Jørgensen, T.N.; Loland, C.J.; Newman, A.H. A rhodamine-labeled citalopram analogue as a high-affinity fluorescent probe for the serotonin transporter. Bioorg. Med. Chem. Lett. 2013, 23, 323–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verrico, C.D.; Miller, G.M.; Madras, B.K. MDMA (Ecstasy) and human dopamine, norepinephrine, and serotonin transporters: Implications for MDMA-induced neurotoxicity and treatment. Psychopharmacology 2007, 189, 489–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angenoorth, T.J.; Stankovic, S.; Niello, M.; Holy, M.; Brandt, S.D.; Sitte, H.H.; Maier, J. Interaction Profiles of Central Nervous System Active Drugs at Human Organic Cation Transporters 1–3 and Human Plasma Membrane Monoamine Transporter. Int. J. Mol. Sci. 2021, 22, 12995. [Google Scholar] [CrossRef] [PubMed]
- Clauss, N.J.; Koek, W.; Daws, L.C. Role of Organic Cation Transporter 3 and Plasma Membrane Monoamine Transporter in the Rewarding Properties and Locomotor Sensitizing Effects of Amphetamine in Male and Female Mice. Int. J. Mol. Sci. 2021, 22, 13420. [Google Scholar] [CrossRef] [PubMed]
- Elliot, E.E. Cocaine sensitization in the mouse using a cumulative dosing regime. Behav. Pharm. 2002, 13, 407–415. [Google Scholar] [CrossRef]
- Zakharova, E.; Wade, D.; Izenwasser, S. Sensitivity to cocaine conditioned reward depends on sex and age. Pharm. Biochem. Behav. 2009, 92, 131–134. [Google Scholar] [CrossRef] [Green Version]
- Milesi-Hallé, A.; McMillan, D.E.; Laurenzana, E.M.; Byrnes-Blake, K.A.; Owens, S.M. Sex differences in (+)-amphetamine- and (+)-methamphetamine-induced behavioral response in male and female Sprague-Dawley rats. Pharm. Biochem. Behav. 2007, 86, 140–149. [Google Scholar] [CrossRef] [Green Version]
- Becker, J.B.; McClellan, M.L.; Reed, B.G. Sex differences, gender and addiction: Sex, Gender, and Addiction. J. Neurosci. Res. 2016, 95, 136–147. [Google Scholar] [CrossRef] [Green Version]
- Wei, R.; Gust, S.L.; Tandio, D.; Maheux, A.; Nguyen, K.H.; Wang, J.; Bourque, S.; Plane, F.; Hammond, J.R. Deletion of murine slc29a4 modifies vascular responses to adenosine and 5-hydroxytryptamine in a sexually dimorphic manner. Physiol. Rep. 2020, 8, e14395. [Google Scholar] [CrossRef] [Green Version]
- Gilman, T.L.; George, C.M.; Vitela, M.; Herrera-Rosales, M.; Basiouny, M.S.; Koek, W.; Daws, L.C. Constitutive plasma membrane monoamine transporter (PMAT, Slc29a4) deficiency subtly affects anxiety-like and coping behaviours. Eur. J. Neurosci. 2018, 48, 1706–1716. [Google Scholar] [CrossRef]
- Duan, H.; Wang, J. Impaired Monoamine and Organic Cation Uptake in Choroid Plexus in Mice with Targeted Disruption of the Plasma Membrane Monoamine Transporter (Slc29a4) Gene. J. Biol. Chem. 2013, 288, 3535–3544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Research Council. Guide for the Care and Use of Laboratory Animals, 8th ed.; National Academies Press: Washington, DC, USA, 2011. [Google Scholar] [CrossRef]
- Ripoll, N.; David, D.J.P.; Dailly, E.; Hascoët, M.; Bourin, M. Antidepressant-like effects in various mice strains in the tail suspension test. Behav. Brain Res. 2003, 143, 193–200. [Google Scholar] [CrossRef]
- Mitchell, N.C.; Gould, G.G.; Koek, W.; Daws, L.C. Ontogeny of SERT Expression and Antidepressant-like Response to Escitalopram in Wild-Type and SERT Mutant Mice. J. Pharm. Exp. Ther. 2016, 358, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, W.M.S.; Burch, R.L. The Principles of Humane Experimental Technique. Nature 1959, 184, 1675–1676. [Google Scholar] [CrossRef]
- Robinson, T.E.; Becker, J.B. Enduring changes in brain and behavior produced by chronic amphetamine administration: A review and evaluation of animal models of amphetamine psychosis. Brain Res. Rev. 1986, 11, 157–198. [Google Scholar] [CrossRef]
- Yates, J.W.; Meij, J.T.A.; Sullivan, J.R.; Richtand, N.M.; Yu, L. Bimodal effect of amphetamine on motor behaviors in C57BL/6 mice. Neurosci. Lett. 2007, 427, 66–70. [Google Scholar] [CrossRef] [Green Version]
- El-Ghundi, M.B.; Fan, T.; Karasinska, J.M.; Yeung, J.; Zhou, M.; O’Dowd, B.F.; George, S.R. Restoration of amphetamine-induced locomotor sensitization in dopamine D1 receptor-deficient mice. Psychopharmacology 2010, 207, 599–618. [Google Scholar] [CrossRef] [Green Version]
- Han, D.D.; Gu, H.H. Comparison of the monoamine transporters from human and mouse in their sensitivities to psychostimulant drugs. BMC Pharm. 2006, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Haenisch, B.; Bönisch, H. Interaction of the human plasma membrane monoamine transporter (hPMAT) with antidepressants and antipsychotics. Naunyn-Schmiedeberg’s Arch. Pharm. 2010, 381, 33–39. [Google Scholar] [CrossRef]
- Kirsch, I.; Huedo-Medina, T.B.; Pigott, H.E.; Johnson, B.T. Do Outcomes of Clinical Trials Resemble Those “Real World” Patients? A Reanalysis of the STAR*D Antidepressant Data Set. Psychol. Conscious Theory Res. Pract. 2018, 5, 339–345. [Google Scholar] [CrossRef]
- Chekroud, A.M.; Gueorguieva, R.; Krumholz, H.M.; Trivedi, M.H.; Krystal, J.H.; McCarthy, G. Reevaluating the Efficacy and Predictability of Antidepressant Treatments: A Symptom Clustering Approach. JAMA Psychiatry 2017, 74, 370. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Allen, S.; Haque, M.N.; Angelescu, I.; Baumeister, D.; Tracy, D.K. Bupropion: A systematic review and meta-analysis of effectiveness as an antidepressant. Ther. Adv. Psychopharmacol. 2016, 6, 99–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, G.P.; McGowan, O.O.; Dalton, C.F. Pharmacogenomics in psychiatry: The relevance of receptor and transporter polymorphisms. Br. J. Clin. Pharm. 2014, 77, 654–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porcelli, S.; Fabbri, C.; Serretti, A. Meta-analysis of serotonin transporter gene promoter polymorphism (5-HTTLPR) association with antidepressant efficacy. Eur. Neuropsychopharm. 2012, 22, 239–258. [Google Scholar] [CrossRef]
- Andre, K.; Kampman, O.; Illi, A.; Viikki, M.; Setälä-Soikkeli, E.; Mononen, N.; Lehtimäki, T.; Haraldsson, S.; Koivisto, P.A.; Leinonen, E. SERT and NET polymorphisms, temperament and antidepressant response. Nord. J. Psychiatry 2015, 69, 531–538. [Google Scholar] [CrossRef]
- Christensen, M.M.; Brasch-Andersen, C.; Green, H.; Nielsen, F.; Damkier, P.; Beck-Nielsen, H.; Brosen, K. The pharmacogenetics of metformin and its impact on plasma metformin steady-state levels and glycosylated hemoglobin A1c. Pharm. Genom. 2011, 21, 837–850. [Google Scholar] [CrossRef]
- Moeez, S.; Khalid, S.; Shaeen, S.; Khalid, M.; Zia, A.; Gul, A.; Niazi, R.; Khalid, Z. Clinically significant findings of high-risk mutations in human SLC29A4 gene associated with diabetes mellitus type 2 in Pakistani population. J. Biomol. Struct. Dyn. 2021, 1–14. [Google Scholar] [CrossRef]
- Dawed, A.Y.; Zhou, K.; van Leeuwen, N.; Mahajan, A.; Robertson, N.; Koivula, R.; Elders, P.J.; Rauh, S.P.; Jones, A.G.; Holl, R.W.; et al. Variation in the Plasma Membrane Monoamine Transporter (PMAT, Encoded in SLC29A4) and Organic Cation Transporter 1 (OCT1, Encoded in SLC22A1) and Gastrointestinal Intolerance to Metformin in Type 2 Diabetes: An IMI DIRECT Study. Diabetes Care 2019, 42, dc182182. [Google Scholar] [CrossRef] [Green Version]
- Duncan, L.E.; Keller, M.C. A Critical Review of the First 10 Years of Candidate Gene-by-Environment Interaction Research in Psychiatry. Am. J. Psychiatry 2011, 168, 1041–1049. [Google Scholar] [CrossRef]
- Schinka, J.A.; Letsch, E.A.; Crawford, F.C. DRD4 and novelty seeking: Results of meta-analyses. Am. J. Med. Genet. 2002, 114, 643–648. [Google Scholar] [CrossRef]
- Gizer, I.R.; Ficks, C.; Waldman, I.D. Candidate gene studies of ADHD: A meta-analytic review. Hum. Genet. 2009, 126, 51–90. [Google Scholar] [CrossRef] [PubMed]
- Commons, K.G.; Cholanians, A.B.; Babb, J.A.; Ehlinger, D.G. The Rodent Forced Swim Test Measures Stress-Coping Strategy, Not Depression-Like Behavior. ACS Chem. Neurosci. 2017, 8, 955–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandler, R.; Keay, K.A.; Floyd, N.; Price, J. Central circuits mediating patterned autonomic activity during active vs. passive emotional coping. Brain Res. Bull. 2000, 53, 95–104. [Google Scholar] [CrossRef]
- De Kloet, E.R.; Molendijk, M.L. Coping with the Forced Swim Stressor: Towards Understanding an Adaptive Mechanism. Neural Plast. 2016, 2016, 6503162. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beaver, J.N.; Weber, B.L.; Ford, M.T.; Anello, A.E.; Kassis, S.K.; Gilman, T.L. Uncovering Functional Contributions of PMAT (Slc29a4) to Monoamine Clearance Using Pharmacobehavioral Tools. Cells 2022, 11, 1874. https://doi.org/10.3390/cells11121874
Beaver JN, Weber BL, Ford MT, Anello AE, Kassis SK, Gilman TL. Uncovering Functional Contributions of PMAT (Slc29a4) to Monoamine Clearance Using Pharmacobehavioral Tools. Cells. 2022; 11(12):1874. https://doi.org/10.3390/cells11121874
Chicago/Turabian StyleBeaver, Jasmin N., Brady L. Weber, Matthew T. Ford, Anna E. Anello, Sarah K. Kassis, and T. Lee Gilman. 2022. "Uncovering Functional Contributions of PMAT (Slc29a4) to Monoamine Clearance Using Pharmacobehavioral Tools" Cells 11, no. 12: 1874. https://doi.org/10.3390/cells11121874
APA StyleBeaver, J. N., Weber, B. L., Ford, M. T., Anello, A. E., Kassis, S. K., & Gilman, T. L. (2022). Uncovering Functional Contributions of PMAT (Slc29a4) to Monoamine Clearance Using Pharmacobehavioral Tools. Cells, 11(12), 1874. https://doi.org/10.3390/cells11121874