
TRPC6 Inactivation Reduces Albuminuria Induced by Protein Overload in Sprague Dawley Rats

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
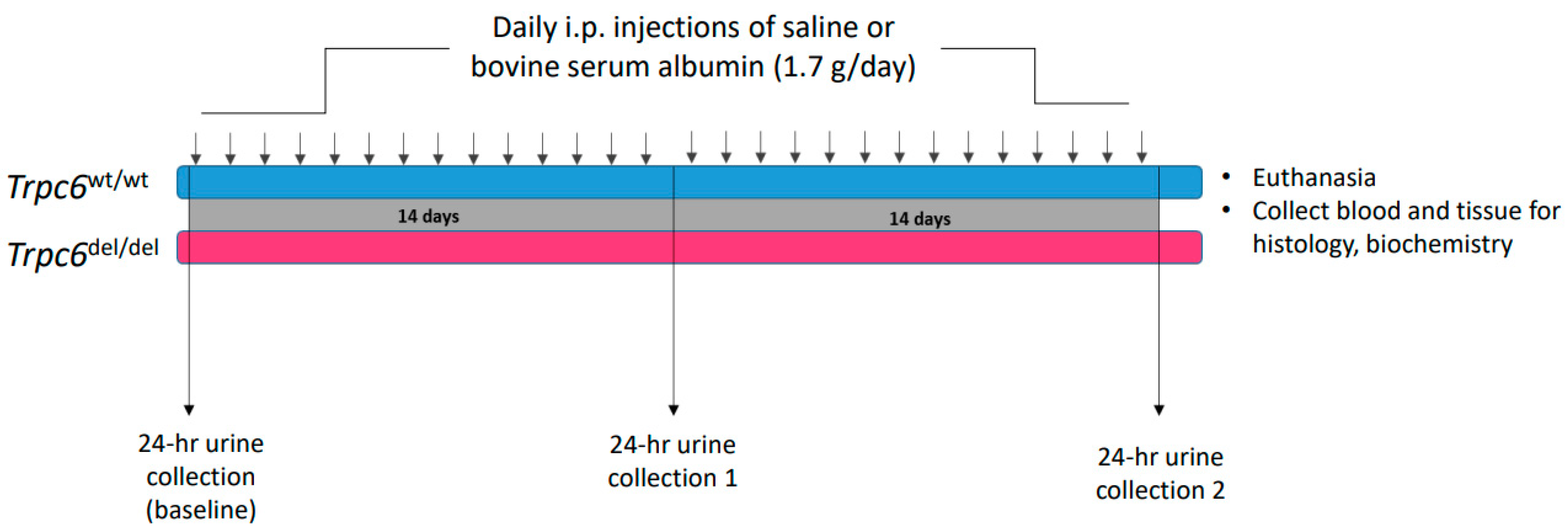
2. Materials and Methods
2.1. Animals
2.2. Albumin Overload and Histology
2.3. Immunoblot Analysis
2.4. Statistical Analyses
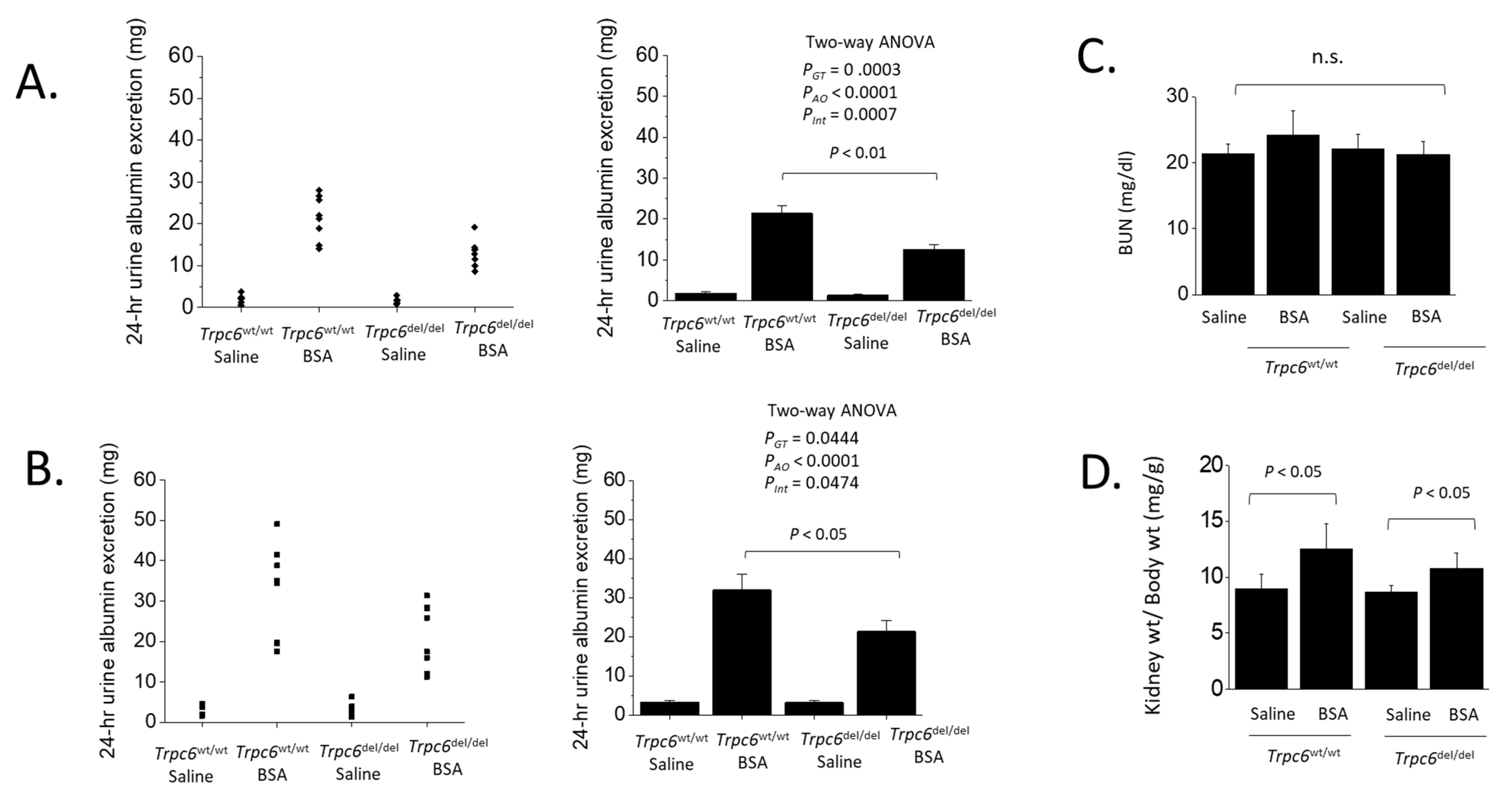
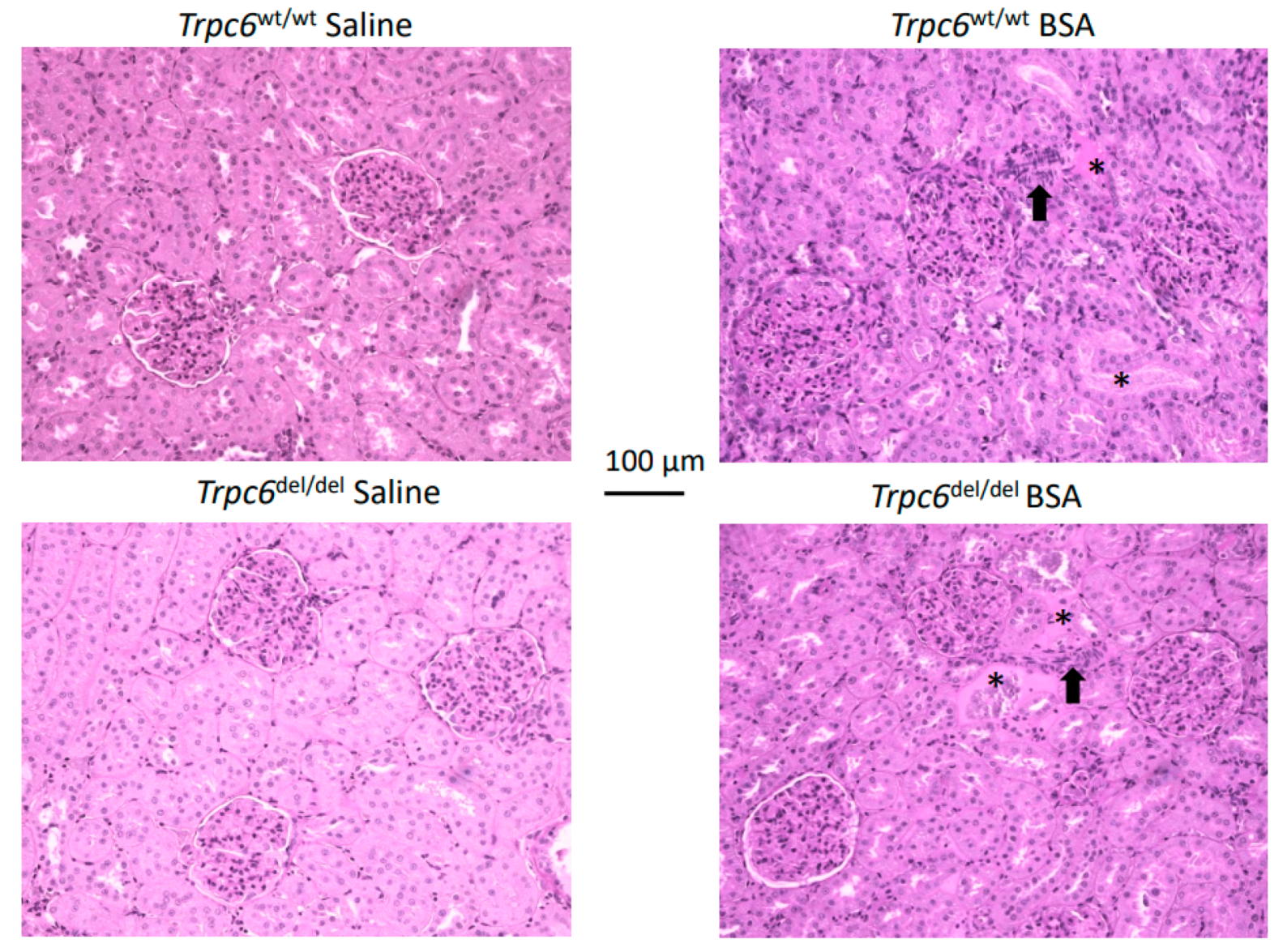
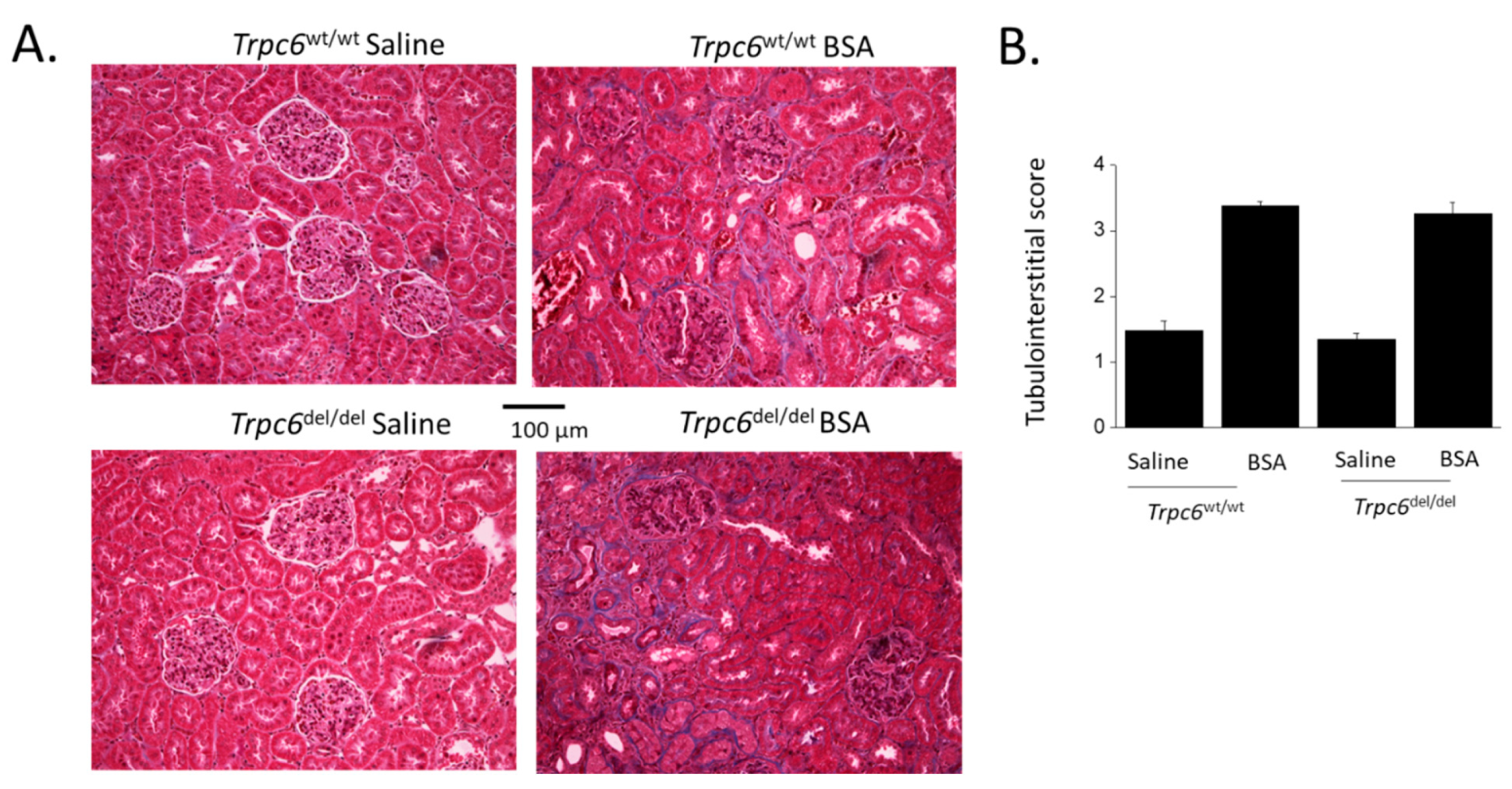
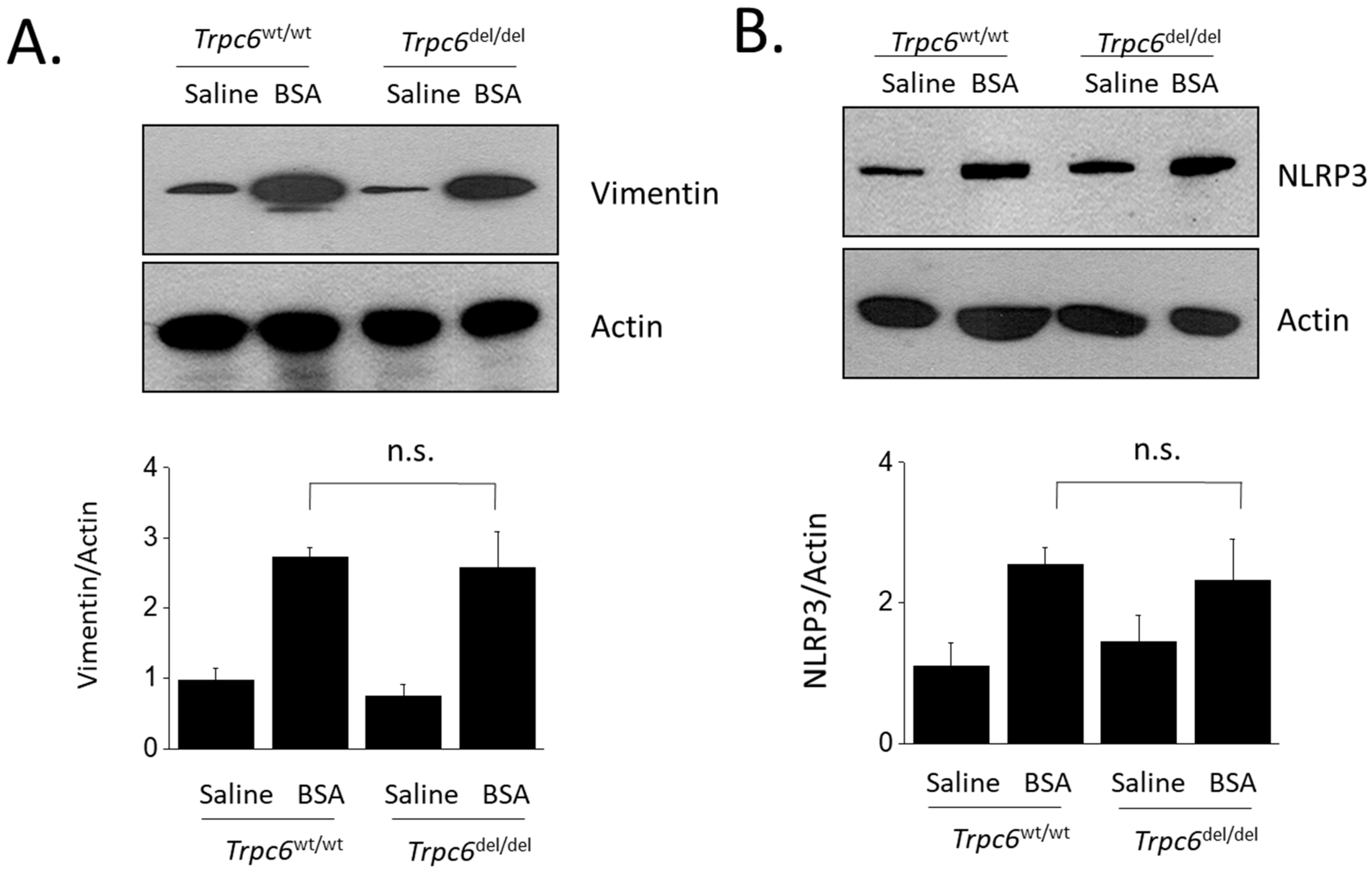
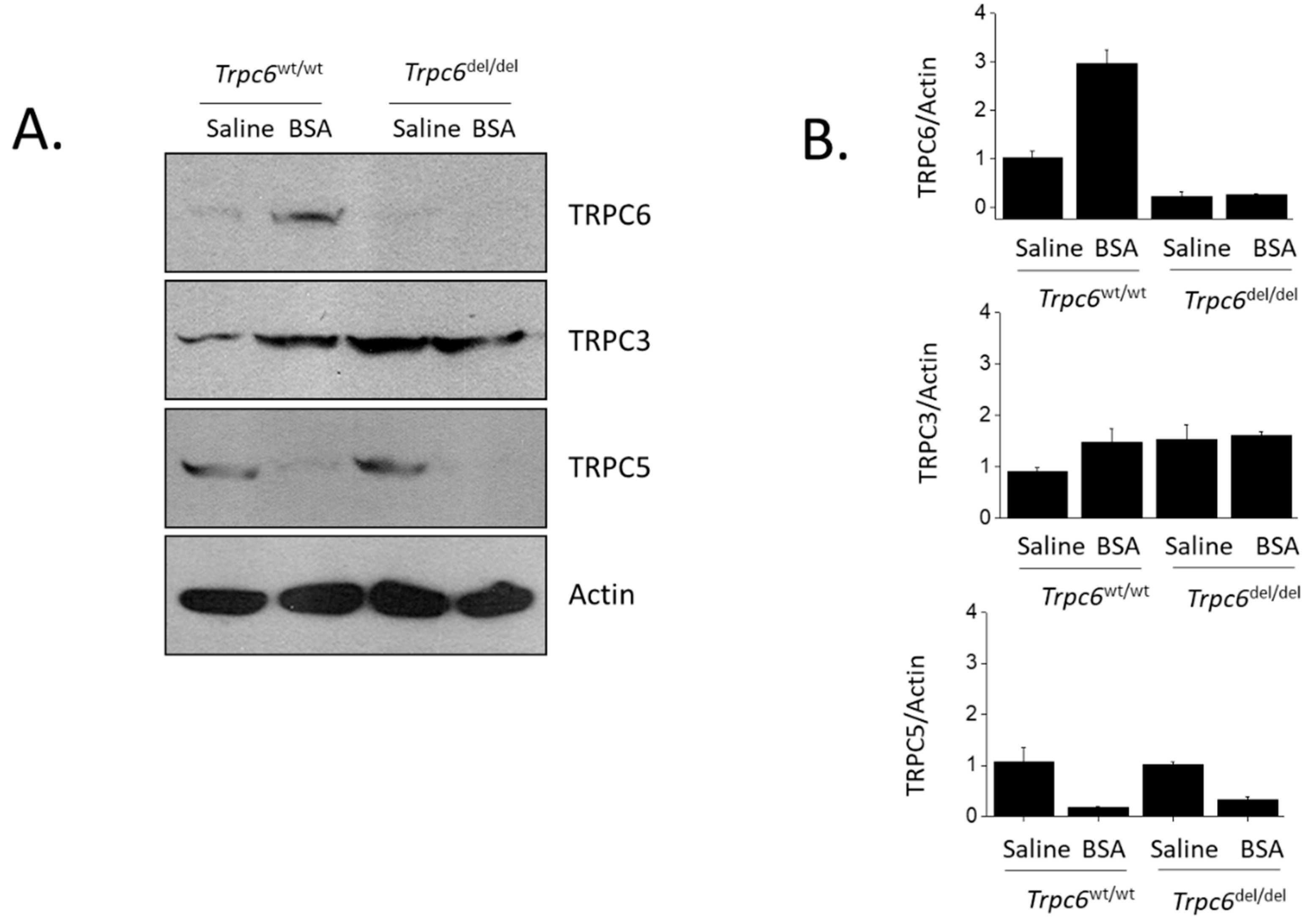
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Dryer, S.E.; Roshanravan, H.; Kim, E.Y. TRPC channels: Regulation, dysregulation and contributions to chronic kidney disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2019, 1865, 1041–1066. [Google Scholar] [CrossRef] [PubMed]
- Ilatovskaya, D.V.; Palygin, O.; Chubinskiy-Nadezhdin, V.; Negulyaev, Y.A.; Ma, R.; Birnbaumer, L.; Staruschenko, A. Angiotensin II has acute effects on TRPC6 channels in podocytes of freshly isolated glomeruli. Kidney Int. 2014, 86, 506–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, M.; Roshanravan, H.; Khine, J.; Dryer, S.E. Angiotensin II activation of TRPC6 channels in rat podocytes requires generation of reactive oxygen species. J. Cell. Physiol. 2014, 229, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Roshanravan, H.; Dryer, S.E. ATP acting through P2Y receptors causes activation of podocyte TRPC6 channels: Role of podocin and reactive oxygen species. Am. J. Physiol.-Ren. Physiol. 2014, 306, 1088–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, M.; Kim, E.Y.; Hagmann, H.; Benzing, T.; Dryer, S.E. Opposing effects of podocin on the gating of podocyte TRPC6 channels evoked by membrane stretch or diacylglycerol. Am. J. Physiol.-Cell Physiol. 2013, 305, C276–C289. [Google Scholar] [CrossRef]
- Gyarmati, G.; Toma, I.; Izuhara, A.; Burford, J.L.; Shroff, U.N.; Papadouri, S.; Deepak, S.; Peti-Peterdi, J. The role of TRPC6 calcium channels and P2 purinergic receptors in podocyte mechanical and metabolic sensing. Physiol. Int. 2022, 109, 31–45. [Google Scholar]
- Winn, M.P.; Conlon, P.J.; Lynn, K.L.; Farrington, M.K.; Creazzo, T.; Hawkins, A.F.; Daskalakis, N.; Kwan, S.Y.; Ebersviller, S.; Burchette, J.L.; et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 2005, 308, 1801–1804. [Google Scholar]
- Reiser, J.; Polu, K.R.; Möller, C.C.; Kenlan, P.; Altintas, M.M.; Wei, C.; Faul, C.; Herbert, S.; Villegas, I.; Avila-Casado, C.; et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat. Genet. 2005, 37, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Heeringa, S.F.; Möller, C.C.; Du, J.Y.; Yue, L.X.; Hinkes, B.; Chernin, G.; Vlangos, C.N.; Hoyer, P.F.; Reiser, J.; Hildebrandt, F. A novel TRPC6 mutation that causes childhood FSGS. PLoS ONE 2009, 4, e7771. [Google Scholar] [CrossRef]
- Santín, S.; Ars, E.; Rossetti, S.; Salido, E.; Silva, I.; García-Maset, R.; Giménez, I.; Ruíz, P.; Mendizábal, S.; Luciano Nieto, J.; et al. TRPC6 mutational analysis in a large cohort of patients with focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2009, 24, 3089–3096. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.; Chen, N.; Wang, Z.H.; Pan, X.X.; Ren, H.; Zhang, W.; Wang, W.M. Identification and functional analysis of a novel TRPC6 mutation associated with late onset familial focal segmental glomerulosclerosis in Chinese patients. Mutat. Res. 2009, 664, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Gigante, M.; Caridi, G.; Montemurno, E.; Soccio, M.; d’Apolito, M.; Cerullo, G.; Aucella, F.; Schirinzi, A.; Emma, F.; Massella, L.; et al. TRPC6 mutations in children with steroid-resistant nephrotic syndrome and atypical phenotype. Clin. J. Am. Soc. Nephrol. 2011, 6, 1626–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mir, S.; Yavascan, O.; Berdeli, A.; Sozeri, B. TRPC6 gene variants in Turkish children with steroid-resistant nephrotic syndrome. Nephrol. Dial. Transplant. 2012, 27, 205–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofstra, J.M.; Lainez, S.; van Kuijk, W.H.; Schoots, J.; Baltissen, M.P.; Hoefsloot, L.H.; Knoers, N.V.; Berden, J.H.; Bindels, R.J.; van der Vlag, J.; et al. New TRPC6 gain-of-function mutation in a non-consanguineous Dutch family with late-onset focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2013, 28, 1830–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Möller, C.C.; Wei, C.; Altintas, M.M.; Li, J.; Greka, A.; Ohse, T.; Pippin, J.W.; Rastaldi, M.P.; Wawersik, S.; Schiavi, S.; et al. Induction of TRPC6 channel in acquired forms of proteinuric kidney disease. J. Am. Soc. Nephrol. 2007, 18, 29–36. [Google Scholar] [CrossRef]
- Krall, P.; Canales, C.P.; Kairath, P.; Carmona-Mora, P.; Molina, J.; Carpio, J.D.; Ruiz, P.; Mezzano, S.A.; Li, J.; Wei, C.; et al. Podocyte-specific overexpression of wild type or mutant trpc6 in mice is sufficient to cause glomerular disease. PLoS ONE 2010, 5, e12859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canales, C.P.; Krall, P.; Kairath, P.; Perez, I.C.; Fragoso, M.A.; Carmona-Mora, P.; Ruiz, P.; Reiser, J.; Young, J.I.; Walz, K. Characterization of a Trpc6 transgenic mouse associated with early onset FSGS. Br. J. Med. Med. Res. 2015, 5, 1198–2012. [Google Scholar] [CrossRef]
- Kim, E.Y.; Yazdizadeh Shotorbani, P.; Dryer, S.E. Trpc6 inactivation confers protection in a model of severe nephrosis in rats. J. Mol. Med. 2018, 96, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.Y.; Roshanravan, H.; Dryer, S.E. Changes in podocyte TRPC channels evoked by plasma and sera from patients with recurrent FSGS and by putative glomerular permeability factors. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 2342–2354. [Google Scholar] [CrossRef]
- Kim, E.Y.; Hassanzadeh Khayyat, N.; Dryer, S.E. Mechanisms underlying modulation of podocyte TRPC6 channels by suPAR: Role of NADPH oxidases and Src family tyrosine kinases. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1864, 3527–3536. [Google Scholar] [CrossRef]
- Kim, E.Y.; Shotorbani, P.Y.; Dryer, S.E. TRPC6 inactivation does not affect loss of renal function in nephrotoxic serum glomerulonephritis in rats, but reduces severity of glomerular lesions. Biochem. Biophys. Rep. 2019, 17, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Dryer, S.E. Effects of TRPC6 inactivation on glomerulosclerosis and renal fibrosis in aging rats. Cells 2021, 10, 856. [Google Scholar] [CrossRef] [PubMed]
- Spires, D.; Ilatovskaya, D.V.; Levchenko, V.; North, P.E.; Geurts, A.M.; Palygin, O.; Staruschenko, A. Protective role of Trpc6 knockout in the progression of diabetic kidney disease. Am. J. Physiol.-Ren. Physiol. 2018, 315, F1091–F1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassanzadeh Khayyat, N.; Kim, E.Y.; Dryer, S.E. TRPC6 inactivation does not protect against diabetic kidney disease in streptozotocin (STZ)-treated Sprague-Dawley rats. FASEB BioAdvances 2019, 1, 773–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, B.L.; Matera, D.; Doerner, J.F.; Zheng, N.; Del Camino, D.; Mishra, S.; Bian, H.; Zeveleva, S.; Zhen, X.; Blair, N.T.; et al. In vivo selective inhibition of TRPC6 by antagonist BI 749327 ameliorates fibrosis and dysfunction in cardiac and renal disease. Proc. Natl. Acad. Sci. USA 2019, 116, 10156–10161. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.L.; Xie, J.; An, S.W.; Oliver, N.; Barrezueta, N.X.; Lin, M.H.; Birnbaumer, L.; Huang, C.L. Inhibition of TRPC6 channels ameliorates renal fibrosis and contributes to renal protection by soluble klotho. Kidney Int. 2017, 91, 830–841. [Google Scholar] [CrossRef] [Green Version]
- Kong, W.; Haschler, T.N.; Nürnberg, B.; Krämer, S.; Gollasch, M.; Markó, L. Renal Fibrosis, immune cell infiltration and changes of TRPC channel expression after unilateral ureteral obstruction in Trpc6-/- mice. Cell. Physiol. Biochem. 2019, 52, 1484–1502. [Google Scholar]
- Ashworth, C.T.; James, J.A. Glomerular excretion of macromolecular substances. Electron microscopic study of rat kidney after administration of human serum albumin. Am. J. Pathol. 1961, 39, 307–316. [Google Scholar]
- Lawrence, G.M.; Brewer, D.B. Effect of strain and sex on the induction of hyperalbuminaemic proteinuria in the rat. Clin. Sci. 1981, 61, 751–756. [Google Scholar] [CrossRef]
- Eddy, A.A. Interstitial nephritis induced by protein-overload proteinuria. Am. J. Pathol. 1989, 135, 719–733. [Google Scholar]
- Eddy, A.A.; Kim, H.; López-Guisa, J.; Oda, T.; Soloway, P.D. Interstitial fibrosis in mice with overload proteinuria: Deficiency of TIMP-1 is not protective. Kidney Int. 2000, 58, 618–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoja, C.; Benigni, A.; Remuzzi, G. Cellular responses to protein overload: Key event in renal disease progression. Curr. Opin. Nephrol. Hypertens. 2004, 13, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Long, K.R.; Rbaibi, Y.; Gliozzi, M.L.; Ren, Q.; Weisz, O.A. Differential kidney proximal tubule cell responses to protein overload by albumin and its ligands. Am. J. Physiol.-Ren. Physiol. 2020, 318, F851–F859. [Google Scholar] [CrossRef] [PubMed]
- Eyre, J.; Ioannou, K.; Grubb, B.D.; Saleem, M.A.; Mathieson, P.W.; Brunskill, N.J.; Christensen, E.I.; Topham, P.S. Statin-sensitive endocytosis of albumin by glomerular podocytes. Am. J. Physiol.-Ren. Physiol. 2007, 292, 674–681. [Google Scholar] [CrossRef] [Green Version]
- Abbate, M.; Zoja, C.; Morigi, M.; Rottoli, D.; Angioletti, S.; Tomasoni, S.; Zanchi, C.; Longaretti, L.; Donadelli, R.; Remuzzi, G. Transforming growth factor-beta1 is up-regulated by podocytes in response to excess intraglomerular passage of proteins: A central pathway in progressive glomerulosclerosis. Am. J. Pathol. 2002, 161, 2179–2193. [Google Scholar] [CrossRef]
- Yoshida, S.; Nagase, M.; Shibata, S.; Fujita, T. Podocyte injury induced by albumin overload in vivo and in vitro: Involvement of TGF-beta and p38 MAPK. Nephron Exp. Nephrol. 2008, 108, 57–68. [Google Scholar] [CrossRef]
- Yanagida-Asanuma, E.; Asanuma, K.; Kim, K.; Donnelly, M.; Young Choi, H.; Hyung Chang, J.; Suetsugu, S.; Tomino, Y.; Takenawa, T.; Faul, C.; et al. Synaptopodin protects against proteinuria by disrupting Cdc42:IRSp53:Mena signaling complexes in kidney podocytes. Am. J. Pathol. 2007, 171, 415–427. [Google Scholar] [CrossRef] [Green Version]
- He, F.; Chen, S.; Wang, H.; Shao, N.; Tian, X.; Jiang, H.; Liu, J.; Zhu, Z.; Meng, X.; Zhang, C. Regulation of CD2-associated protein influences podocyte endoplasmic reticulum stress-mediated apoptosis induced by albumin overload. Gene 2011, 484, 18–25. [Google Scholar] [CrossRef]
- Chen, S.; He, F.F.; Wang, H.; Fang, Z.; Shao, N.; Tian, X.J.; Liu, J.S.; Zhu, Z.H.; Wang, Y.M.; Wang, S.; et al. Calcium entry via TRPC6 mediates albumin overload-induced endoplasmic reticulum stress and apoptosis in podocytes. Cell Calcium 2011, 50, 523–529. [Google Scholar] [CrossRef]
- Liu, Y.; Echtermeyer, F.; Thilo, F.; Theilmeier, G.; Schmidt, A.; Schülein, R.; Jensen, B.L.; Loddenkempe, C.; Jankowski, V.; Marcussen, N.; et al. The proteoglycan syndecan 4 regulates transient receptor potential canonical 6 channels via RhoA/Rho-associated protein kinase signaling. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 378–385. [Google Scholar] [CrossRef] [Green Version]
- Thongprayoon, C.; Cheungpasitporn, W.; Mao, M.A.; Sakhuha, A.; Kashani, K. U-shaped association of serum albumin level and acute kidney injury risk in hospitalized patients. PLoS ONE 2018, 13, e0199153. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Guess, A.J.; Chanley, M.A.; Smoyer, W.E. Albumin-induced podocyte injury and protection are associated with regulation of COX-2. Kidney Int. 2014, 86, 1150–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamura, L.; Dummer, P.; Kopp, J.; Qiu, L.; Levi, M.; Faubel, S.; Blaine, J. Endocytosis of albumin by podocytes elicits an inflammatory response and induces apoptotic cell death. PLoS ONE 2013, 8, e54817. [Google Scholar]
- Martínez-Klimova, E.; Aparicio-Trejo, O.E.; Tapia, E.; Pedraza-Chaverri, J. Unilateral ureteral obstruction as a model to investigate fibrosis-attenuating treatments. Biomolecules 2019, 9, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaldecker, T.; Kim, S.; Tarabanis, C.; Tian, D.; Hakroush, S.; Castonguay, P.; Ahn, W.; Wallentin, H.; Heid, H.; Hopkins, C.R.; et al. Inhibition of the TRPC5 ion channel protects the kidney filter. J. Clin. Investig. 2013, 123, 5298–5309. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Castonguay, P.; Sidhom, E.H.; Clark, A.R.; Dvela-Levitt, M.; Kim, S.; Sieber, J.; Wieder, N.; Jung, J.Y.; Andreeva, S.; et al. A small-molecule inhibitor of TRPC5 ion channels suppresses progressive kidney disease in animal models. Science 2017, 358, 1332–1336. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Kim, C.; Pablo, J.L.B.; Zhang, F.; Jung, J.Y.; Xiao, L.; Bazua-Valenti, S.; Emani, M.; Hopkins, C.R.; Weins, A.; et al. TRPC5 Channel inhibition protects podocytes in puromycin-aminonucleoside induced nephrosis models. Front. Med. 2021, 8, 721865. [Google Scholar] [CrossRef]
- Wang, X.; Dande, R.R.; Yu, H.; Samelko, B.; Miller, R.E.; Altintas, M.M.; Reiser, J. TRPC5 does not cause or aggravate glomerular disease. J. Am. Soc. Nephrol. 2018, 29, 409–415. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, E.Y.; Dryer, S.E. TRPC6 Inactivation Reduces Albuminuria Induced by Protein Overload in Sprague Dawley Rats. Cells 2022, 11, 1985. https://doi.org/10.3390/cells11131985
Kim EY, Dryer SE. TRPC6 Inactivation Reduces Albuminuria Induced by Protein Overload in Sprague Dawley Rats. Cells. 2022; 11(13):1985. https://doi.org/10.3390/cells11131985
Chicago/Turabian StyleKim, Eun Young, and Stuart E. Dryer. 2022. "TRPC6 Inactivation Reduces Albuminuria Induced by Protein Overload in Sprague Dawley Rats" Cells 11, no. 13: 1985. https://doi.org/10.3390/cells11131985
APA StyleKim, E. Y., & Dryer, S. E. (2022). TRPC6 Inactivation Reduces Albuminuria Induced by Protein Overload in Sprague Dawley Rats. Cells, 11(13), 1985. https://doi.org/10.3390/cells11131985