
Explaining Redundancy in CDK-Mediated Control of the Cell Cycle: Unifying the Continuum and Quantitative Models


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. How Many Separate Phases Are There in the Cell Cycle?
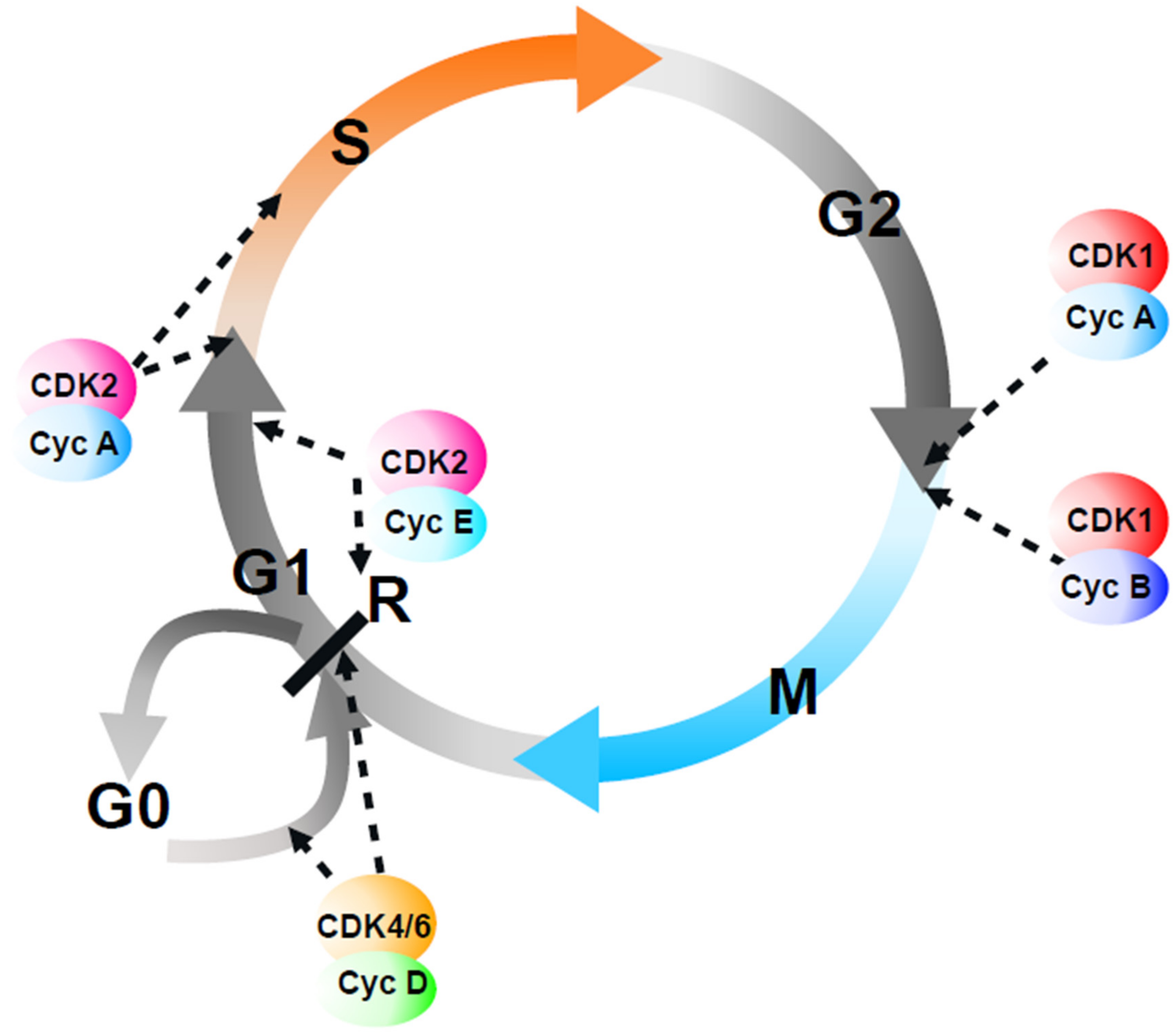
2. Building the Multiple CDK Model of Cell Cycle Control
3. “G1” Cyclin–CDK Complexes May Control Growth Rather Than S-Phase Onset
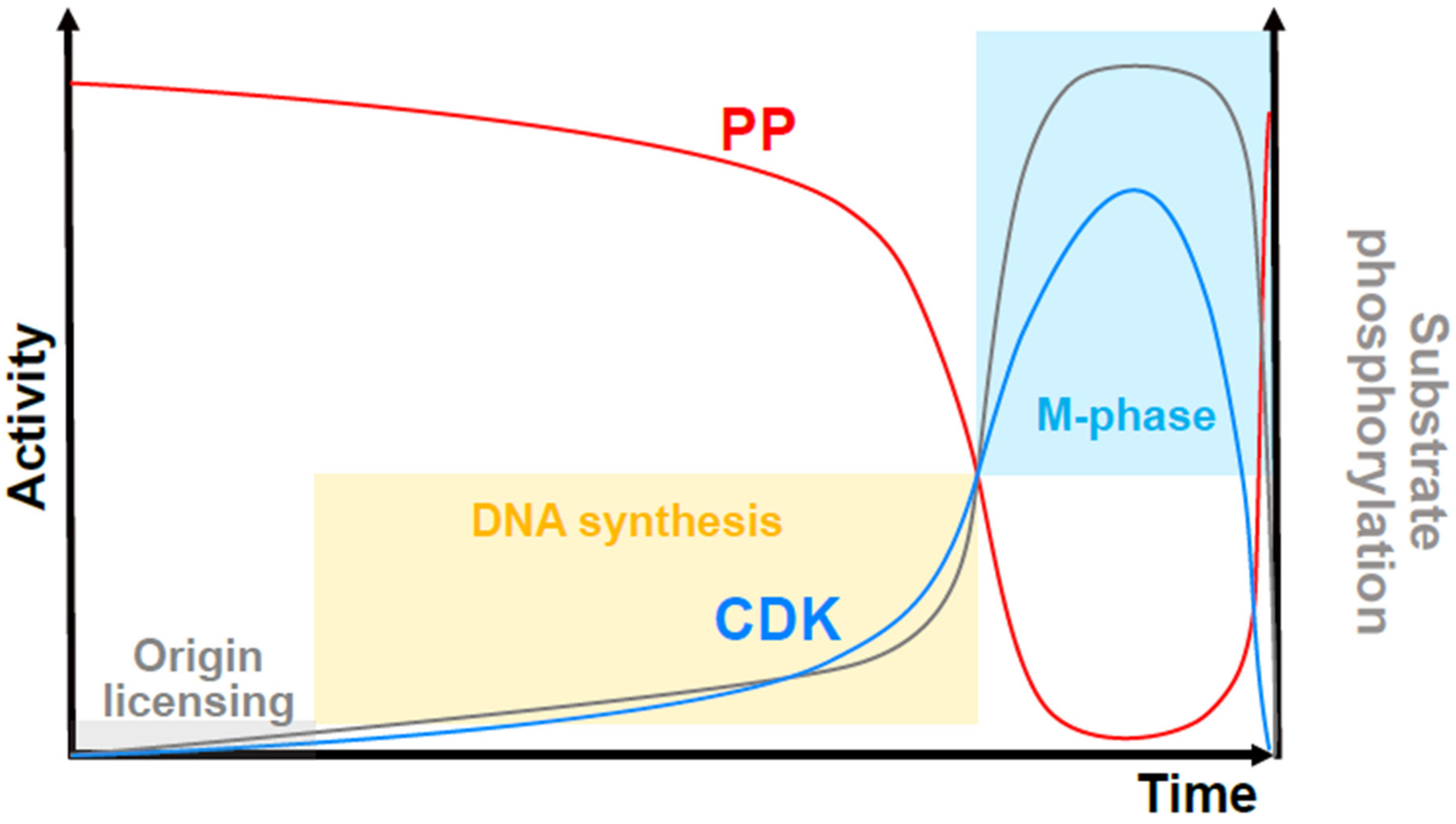
4. Specific Cyclins and CDKs Modulate Kinetics of Overall CDK Activity
5. Redundant CDK-Mediated Control of S-Phase Onset
6. Specific CDK–Cyclin Complexes Are Not Essential for Entry into Mitosis
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Uzbekov, R.; Prigent, C. A journey through time on the discovery of cell cycle regulation. Cells 2022, 11, 704. [Google Scholar] [CrossRef] [PubMed]
- Howard, A.; Pelc, S.R. Nuclear Incorporation of P32 as Demonstrated by Autoradiographs. Exp. Cell Res. 1951, 2, 178–187. [Google Scholar] [CrossRef]
- Quastler, H.; Sherman, F.G. Cell Population Kinetics in the Intestinal Epithelium of the Mouse. Exp. Cell Res. 1959, 17, 420–438. [Google Scholar] [CrossRef]
- Mendelsohn, M.L. Autoradiographic Analysis of Cell Proliferation in Spontaneous Breast Cancer of C3H Mouse. III. The Growth Fraction. J. Natl. Cancer Inst. 1962, 28, 1015–1029. [Google Scholar]
- Post, J.; Hoffman, J. Changes in the replication times and patterns of the liver cell during the life of the rat. Exp. Cell Res. 1964, 36, 111–123. [Google Scholar] [CrossRef]
- Wolfsberg, M.F. Cell population kinetics in the epithelium of the forestomach of the mouse. Exp. Cell Res. 1964, 35, 119–131. [Google Scholar] [CrossRef]
- Abercrombie, M.; Heaysman, J.E. Observations on the Social Behaviour of Cells in Tissue Culture. I. Speed of Movement of Chick Heart Fibroblasts in Relation to Their Mutual Contacts. Exp. Cell Res. 1953, 5, 111–131. [Google Scholar] [CrossRef]
- Stoker, M.G.; Rubin, H. Density Dependent Inhibition of Cell Growth in Culture. Nature 1967, 215, 171–172. [Google Scholar] [CrossRef]
- Tobey, R.A.; Ley, K.D. Regulation of Initiation of DNA Synthesis in Chinese Hamster Cells. I. Production of Stable, Reversible G1-Arrested Populations in Suspension Culture. J. Cell Biol. 1970, 46, 151–157. [Google Scholar] [CrossRef] [Green Version]
- Ley, K.D.; Tobey, R.A. Regulation of initiation of DNA synthesis in Chinese hamster cells: II. Induction of DNA Synthesis and Cell Division by Isoleucine and Glutamine in G(1)-Arrested Cells in Suspension Culture. J. Cell Biol. 1970, 47, 453–459. [Google Scholar] [CrossRef]
- Bürk, R.R. Growth Inhibitor of Hamster Fibroblast Cells. Nature 1966, 212, 1261–1262. [Google Scholar] [CrossRef]
- Kram, R.; Mamont, P.; Tomkins, G.M. Pleiotypic Control by Adenosine 3′:5′-Cyclic Monophosphate: A Model for Growth Control in Animal Cells. Proc. Natl. Acad. Sci. USA 1973, 70, 1432–1436. [Google Scholar] [CrossRef] [Green Version]
- Pardee, A.B. A Restriction Point for Control of Normal Animal Cell Proliferation. Proc. Natl. Acad. Sci. USA 1974, 71, 1286–1290. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, C.; Johnson, A.; Kõivomägi, M.; Zatulovskiy, E.; Kravitz, C.J.; Doncic, A.; Skotheim, J.M. A Precise Cdk Activity Threshold Determines Passage through the Restriction Point. Mol. Cell 2018, 69, 253–264.e5. [Google Scholar] [CrossRef]
- Cooper, S. Reappraisal of Serum Starvation, the Restriction Point, G0, and G1 Phase Arrest Points. FASEB J. 2003, 17, 333–340. [Google Scholar] [CrossRef]
- Cooper, S. The Anti-G0 Manifesto: Should a Problematic Construct (G0) with No Biological Reality Be Removed from the Cell Cycle? Yes! Bioessays 2021, 43, e2000270. [Google Scholar] [CrossRef]
- Brown, J.M. Long G1 or G0 State: A Method of Resolving the Dilemma for the Cell Cycle of an in Vivo Population. Exp. Cell Res. 1968, 52, 565–570. [Google Scholar] [CrossRef]
- Killander, D.; Zetterberg, A. A Quantitative Cytochemical Investigation of the Relationship between Cell Mass and Initiation of DNA Synthesis in Mouse Fibroblasts in Vitro. Exp. Cell Res. 1965, 40, 12–20. [Google Scholar] [CrossRef]
- Bürk, R.R. One-Step Growth Cycle for BHK21/13 Hamster Fibroblasts. Exp. Cell Res. 1970, 63, 309–316. [Google Scholar] [CrossRef]
- Cooper, S. On G0 and Cell Cycle Controls. Bioessays 1987, 7, 220–223. [Google Scholar] [CrossRef] [Green Version]
- Shackney, S.E.; Ford, S.S. Correlations between the DNA Content Distribution and Tritiated Thymidine Studies in Relation to Population Size in Sarcoma 180 in Vitro. Cancer Res. 1974, 34, 1401–1407. [Google Scholar]
- Shackney, S.E. On the Discreteness of the Phases of the Cell Cycle. J. Natl. Cancer Inst. 1975, 55, 827–829. [Google Scholar] [CrossRef]
- Magiera, M.M.; Gueydon, E.; Schwob, E. DNA Replication and Spindle Checkpoints Cooperate during S Phase to Delay Mitosis and Preserve Genome Integrity. J. Cell Biol. 2014, 204, 165–175. [Google Scholar] [CrossRef]
- Torres-Rosell, J.; De Piccoli, G.; Cordon-Preciado, V.; Farmer, S.; Jarmuz, A.; Machin, F.; Pasero, P.; Lisby, M.; Haber, J.E.; Aragón, L. Anaphase Onset before Complete DNA Replication with Intact Checkpoint Responses. Science 2007, 315, 1411–1415. [Google Scholar] [CrossRef]
- Ivanova, T.; Maier, M.; Missarova, A.; Ziegler-Birling, C.; Dam, M.; Gomar-Alba, M.; Carey, L.B.; Mendoza, M. Budding Yeast Complete DNA Synthesis after Chromosome Segregation Begins. Nat. Commun. 2020, 11, 2267. [Google Scholar] [CrossRef]
- Smith, J.A.; Martin, L. Do Cells Cycle? Proc. Natl. Acad. Sci. USA 1973, 70, 1263–1267. [Google Scholar] [CrossRef] [Green Version]
- Dowling, M.R.; Kan, A.; Heinzel, S.; Zhou, J.H.S.; Marchingo, J.M.; Wellard, C.J.; Markham, J.F.; Hodgkin, P.D. Stretched Cell Cycle Model for Proliferating Lymphocytes. Proc. Natl. Acad. Sci. USA 2014, 111, 6377–6382. [Google Scholar] [CrossRef] [Green Version]
- Masui, Y.; Markert, C.L. Cytoplasmic Control of Nuclear Behavior during Meiotic Maturation of Frog Oocytes. J. Exp. Zool. 1971, 177, 129–145. [Google Scholar] [CrossRef]
- Hartwell, L.H.; Culotti, J.; Pringle, J.R.; Reid, B.J. Genetic Control of the Cell Division Cycle in Yeast. Science 1974, 183, 46–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradbury, E.M.; Inglis, R.J.; Matthews, H.R. Control of Cell Division by Very Lysine Rich Histone (F1) Phosphorylation. Nature 1974, 247, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, E.M.; Inglis, R.J.; Matthews, H.R.; Langan, T.A. Molecular Basis of Control of Mitotic Cell Division in Eukaryotes. Nature 1974, 249, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Pines, J.; Hunt, T. Molecular Cloning and Characterization of the MRNA for Cyclin from Sea Urchin Eggs. EMBO J. 1987, 6, 2987–2995. [Google Scholar] [CrossRef] [PubMed]
- Hindley, J.; Phear, G.A. Sequence of the Cell Division Gene CDC2 from Schizosaccharomyces Pombe; Patterns of Splicing and Homology to Protein Kinases. Gene 1984, 31, 129–134. [Google Scholar] [CrossRef]
- Lörincz, A.T.; Reed, S.I. Primary Structure Homology between the Product of Yeast Cell Division Control Gene CDC28 and Vertebrate Oncogenes. Nature 1984, 307, 183–185. [Google Scholar] [CrossRef]
- Labbe, J.C.; Lee, M.G.; Nurse, P.; Picard, A.; Doree, M. Activation at M-Phase of a Protein Kinase Encoded by a Starfish Homologue of the Cell Cycle Control Gene Cdc2+. Nature 1988, 335, 251–254. [Google Scholar] [CrossRef]
- Gautier, J.; Norbury, C.; Lohka, M.; Nurse, P.; Maller, J. Purified Maturation-Promoting Factor Contains the Product of a Xenopus Homolog of the Fission Yeast Cell Cycle Control Gene Cdc2+. Cell 1988, 54, 433–439. [Google Scholar] [CrossRef]
- Meijer, L.; Arion, D.; Golsteyn, R.; Pines, J.; Brizuela, L.; Hunt, T.; Beach, D. Cyclin Is a Component of the Sea Urchin Egg M-Phase Specific Histone H1 Kinase. EMBO J. 1989, 8, 2275–2282. [Google Scholar] [CrossRef]
- Labbe, J.C.; Capony, J.P.; Caput, D.; Cavadore, J.C.; Derancourt, J.; Kaghad, M.; Lelias, J.M.; Picard, A.; Doree, M. MPF from Starfish Oocytes at First Meiotic Metaphase Is a Heterodimer Containing One Molecule of Cdc2 and One Molecule of Cyclin B. EMBO J. 1989, 8, 3053–3058. [Google Scholar] [CrossRef]
- Paris, J.; Le, G.R.; Couturier, A.; Le, G.K.; Omilli, F.; Camonis, J.; MacNeill, S.; Philippe, M. Cloning by Differential Screening of a Xenopus CDNA Coding for a Protein Highly Homologous to Cdc2. Proc. Natl. Acad. Sci. USA 1991, 88, 1039–1043. [Google Scholar] [CrossRef] [Green Version]
- Meyerson, M.; Enders, G.H.; Wu, C.L.; Su, L.K.; Gorka, C.; Nelson, C.; Harlow, E.; Tsai, L.H. A Family of Human Cdc2-Related Protein Kinases. EMBO J. 1992, 11, 2909–2917. [Google Scholar] [CrossRef]
- Lew, D.J.; Dulic, V.; Reed, S.I. Isolation of Three Novel Human Cyclins by Rescue of G1 Cyclin (Cln) Function in Yeast. Cell 1991, 66, 1197–1206. [Google Scholar] [CrossRef]
- Koff, A.; Cross, F.; Fisher, A.; Schumacher, J.; Leguellec, K.; Philippe, M.; Roberts, J.M. Human Cyclin E, a New Cyclin That Interacts with Two Members of the CDC2 Gene Family. Cell 1991, 66, 1217–1228. [Google Scholar] [CrossRef]
- Sherr, C.J. Mammalian G1 Cyclins. Cell 1993, 73, 1059–1065. [Google Scholar] [CrossRef]
- Fang, F.; Newport, J.W. Evidence That the G1-S and G2-M Transitions Are Controlled by Different Cdc2 Proteins in Higher Eukaryotes. Cell 1991, 66, 731–742. [Google Scholar] [CrossRef]
- Tsai, L.H.; Lees, E.; Faha, B.; Harlow, E.; Riabowol, K. The Cdk2 Kinase Is Required for the G1-to-S Transition in Mammalian Cells. Oncogene 1993, 8, 1593–1602. [Google Scholar]
- Van den Heuvel, S.; Harlow, E. Distinct Roles for Cyclin-Dependent Kinases in Cell Cycle Control. Science 1993, 262, 2050–2054. [Google Scholar] [CrossRef]
- Braun, K.; Holzl, G.; Soucek, T.; Geisen, C.; Moroy, T.; Hengstschlager, M. Investigation of the Cell Cycle Regulation of Cdk3-Associated Kinase Activity and the Role of Cdk3 in Proliferation and Transformation. Oncogene 1998, 17, 2259–2269. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Zhu, C.; Harper, J.W. A Premature-Termination Mutation in the Mus Musculus Cyclin-Dependent Kinase 3 Gene. Proc. Natl. Acad. Sci. USA 2001, 98, 1682–1686. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclin-Dependent Kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.; Kaldis, P. Cdks, Cyclins and CKIs: Roles beyond Cell Cycle Regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [Green Version]
- Hydbring, P.; Malumbres, M.; Sicinski, P. Non-Canonical Functions of Cell Cycle Cyclins and Cyclin-Dependent Kinases. Nat. Rev. Mol. Cell Biol. 2016, 17, 280–292. [Google Scholar] [CrossRef]
- Krasinska, L.; Fisher, D. Non-Cell Cycle Functions of the CDK Network in Ciliogenesis: Recycling the Cell Cycle Oscillator. Bioessays 2018, 40, e1800016. [Google Scholar] [CrossRef]
- Ewen, M.E.; Sluss, H.K.; Sherr, C.J.; Matsushime, H.; Kato, J.; Livingston, D.M. Functional Interactions of the Retinoblastoma Protein with Mammalian D-Type Cyclins. Cell 1993, 73, 487–497. [Google Scholar] [CrossRef]
- Kato, J.; Matsushime, H.; Hiebert, S.W.; Ewen, M.E.; Sherr, C.J. Direct Binding of Cyclin-D to the Retinoblastoma Gene Product (PRb) and PRb Phosphorylation by the Cyclin D- Dependent Kinase CDK4. Genes Dev. 1993, 7, 331–342. [Google Scholar]
- Meyerson, M.; Harlow, E. Identification of G1 Kinase Activity for Cdk6, a Novel Cyclin D Partner. Mol. Cell Biol. 1994, 14, 2077–2086. [Google Scholar]
- Ren, S.; Rollins, B.J. Cyclin C/Cdk3 Promotes Rb-Dependent G0 Exit. Cell 2004, 117, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Martinsson, H.-S.; Starborg, M.; Erlandsson, F.; Zetterberg, A. Single Cell Analysis of G1 Check Points-the Relationship between the Restriction Point and Phosphorylation of PRb. Exp. Cell Res. 2005, 305, 383–391. [Google Scholar] [CrossRef]
- Ezhevsky, S.A.; Ho, A.; Becker-Hapak, M.; Davis, P.K.; Dowdy, S.F. Differential Regulation of Retinoblastoma Tumor Suppressor Protein by G(1) Cyclin-Dependent Kinase Complexes in Vivo. Mol. Cell Biol. 2001, 21, 4773–4784. [Google Scholar] [CrossRef] [Green Version]
- Narasimha, A.M.; Kaulich, M.; Shapiro, G.S.; Choi, Y.J.; Sicinski, P.; Dowdy, S.F. Cyclin D Activates the Rb Tumor Suppressor by Mono-Phosphorylation. Elife 2014, 3, e02872. [Google Scholar] [CrossRef]
- Connell-Crowley, L.; Harper, J.W.; Goodrich, D.W. Cyclin D1/Cdk4 Regulates Retinoblastoma Protein-Mediated Cell Cycle Arrest by Site-Specific Phosphorylation. Mol. Biol. Cell 1997, 8, 287–301. [Google Scholar] [CrossRef] [Green Version]
- Lossaint, G.; Horvat, A.; Gire, V.; Bačević, K.; Mrouj, K.; Charrier-Savournin, F.; Georget, V.; Fisher, D.; Dulić, V. Reciprocal Regulation of P21 and Chk1 Controls the Cyclin D1-RB Pathway to Mediate Senescence Onset after G2 Arrest. J. Cell Sci. 2022, 135, jcs259114. [Google Scholar] [CrossRef] [PubMed]
- Argüello-Miranda, O.; Marchand, A.J.; Kennedy, T.; Russo, M.A.X.; Noh, J. Cell Cycle-Independent Integration of Stress Signals by Xbp1 Promotes Non-G1/G0 Quiescence Entry. J. Cell Biol. 2022, 221, e202103171. [Google Scholar] [CrossRef] [PubMed]
- Datar, S.A.; Jacobs, H.W.; de la Cruz, A.F.; Lehner, C.F.; Edgar, B.A. The Drosophila Cyclin D-Cdk4 Complex Promotes Cellular Growth. EMBO J. 2000, 19, 4543–4554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, C.A.; Jacobs, H.W.; Datar, S.A.; Du, W.; Edgar, B.A.; Lehner, C.F. Drosophila Cdk4 Is Required for Normal Growth and Is Dispensable for Cell Cycle Progression. EMBO J. 2000, 19, 4533–4542. [Google Scholar] [CrossRef] [Green Version]
- Frei, C.; Galloni, M.; Hafen, E.; Edgar, B.A. The Drosophila Mitochondrial Ribosomal Protein MRpL12 Is Required for Cyclin D/Cdk4-Driven Growth. EMBO J. 2005, 24, 623–634. [Google Scholar] [CrossRef] [Green Version]
- Malumbres, M.; Sotillo, R.; Santamaria, D.; Galan, J.; Cerezo, A.; Ortega, S.; Dubus, P.; Barbacid, M. Mammalian Cells Cycle without the D-Type Cyclin-Dependent Kinases Cdk4 and Cdk6. Cell 2004, 118, 493–504. [Google Scholar] [CrossRef] [Green Version]
- Kozar, K.; Ciemerych, M.A.; Rebel, V.I.; Shigematsu, H.; Zagozdzon, A.; Sicinska, E.; Geng, Y.; Yu, Q.; Bhattacharya, S.; Bronson, R.T.; et al. Mouse Development and Cell Proliferation in the Absence of D-Cyclins. Cell 2004, 118, 477–491. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Michowski, W.; Inuzuka, H.; Shimizu, K.; Nihira, N.T.; Chick, J.M.; Li, N.; Geng, Y.; Meng, A.Y.; Ordureau, A.; et al. G1 Cyclins Link Proliferation, Pluripotency and Differentiation of Embryonic Stem Cells. Nat. Cell Biol. 2017, 19, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Ohtsubo, M.; Roberts, J.M. Cyclin-Dependent Regulation of G1 in Mammalian Fibroblasts. Science 1993, 259, 1908–1912. [Google Scholar] [CrossRef]
- Quelle, D.E.; Ashmun, R.A.; Shurtleff, S.A.; Kato, J.Y.; Bar-Sagi, D.; Roussel, M.F.; Sherr, C.J. Overexpression of Mouse D-Type Cyclins Accelerates G1 Phase in Rodent Fibroblasts. Genes Dev. 1993, 7, 1559–1571. [Google Scholar] [CrossRef] [Green Version]
- Min, M.; Rong, Y.; Tian, C.; Spencer, S.L. Temporal Integration of Mitogen History in Mother Cells Controls Proliferation of Daughter Cells. Science 2020, 368, 1261–1265. [Google Scholar] [CrossRef]
- Chen, K.C.; Calzone, L.; Csikasz-Nagy, A.; Cross, F.R.; Novak, B.; Tyson, J.J. Integrative Analysis of Cell Cycle Control in Budding Yeast. Mol. Biol. Cell 2004, 15, 3841–3862. [Google Scholar] [CrossRef] [Green Version]
- Csikász-Nagy, A.; Battogtokh, D.; Chen, K.C.; Novák, B.; Tyson, J.J. Analysis of a Generic Model of Eukaryotic Cell-Cycle Regulation. Biophys. J. 2006, 90, 4361–4379. [Google Scholar] [CrossRef] [Green Version]
- Novák, B.; Tyson, J.J. Mechanisms of Signalling-Memory Governing Progression through the Eukaryotic Cell Cycle. Curr. Opin. Cell Biol. 2021, 69, 7–16. [Google Scholar] [CrossRef]
- Hayles, J.; Fisher, D.; Woollard, A.; Nurse, P. Temporal Order of s Phase and Mitosis in Fission Yeast Is Determined by the State of the P34(Cdc2) Mitotic b Cyclin Complex. Cell 1994, 78, 813–822. [Google Scholar] [CrossRef]
- Fisher, D.L.; Nurse, P. A Single Fission Yeast Mitotic Cyclin B P34cdc2 Kinase Promotes Both S-Phase and Mitosis in the Absence of G1 Cyclins. EMBO J. 1996, 15, 850–860. [Google Scholar] [CrossRef]
- Coudreuse, D.; Nurse, P. Driving the Cell Cycle with a Minimal CDK Control Network. Nature 2010, 468, 1074–1079. [Google Scholar] [CrossRef]
- Haase, S.B.; Reed, S.I. Evidence That a Free-Running Oscillator Drives G1 Events in the Budding Yeast Cell Cycle. Nature 1999, 401, 394–397. [Google Scholar] [CrossRef]
- Orlando, D.A.; Lin, C.Y.; Bernard, A.; Wang, J.Y.; Socolar, J.E.; Iversen, E.S.; Hartemink, A.J.; Haase, S.B. Global Control of Cell-Cycle Transcription by Coupled CDK and Network Oscillators. Nature 2008, 453, 944–947. [Google Scholar] [CrossRef] [Green Version]
- Banyai, G.; Baïdi, F.; Coudreuse, D.; Szilagyi, Z. Cdk1 Activity Acts as a Quantitative Platform for Coordinating Cell Cycle Progression with Periodic Transcription. Nat. Commun. 2016, 7, 11161. [Google Scholar] [CrossRef] [Green Version]
- Goldbeter, A.; Koshland, D.E., Jr. An Amplified Sensitivity Arising from Covalent Modification in Biological Systems. Proc. Natl. Acad. Sci. USA 1981, 78, 6840–6844. [Google Scholar] [CrossRef] [Green Version]
- Fisher, D.; Krasinska, L.; Coudreuse, D.; Novak, B. Phosphorylation Network Dynamics in Control of Cell Cycle Transitions. J. Cell Sci. 2012, 125, 4703–4711. [Google Scholar] [CrossRef] [Green Version]
- Krasinska, L.; Domingo-Sananes, M.R.; Kapuy, O.; Parisis, N.; Harker, B.; Moorhead, G.; Rossignol, M.; Novak, B.; Fisher, D. Protein Phosphatase 2A Controls the Order and Dynamics of Cell-Cycle Transitions. Mol. Cell 2011, 44, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Swaffer, M.P.; Jones, A.W.; Flynn, H.R.; Snijders, A.P.; Nurse, P. CDK Substrate Phosphorylation and Ordering the Cell Cycle. Cell 2016, 167, 1750–1761.e16. [Google Scholar] [CrossRef] [Green Version]
- Ferrigno, P.; Langan, T.A.; Cohen, P. Protein Phosphatase 2A1 Is the Major Enzyme in Vertebrate Cell Extracts That Dephosphorylates Several Physiological Substrates for Cyclin-Dependent Protein Kinases. Mol. Biol. Cell 1993, 4, 669–677. [Google Scholar] [CrossRef]
- Kamenz, J.; Gelens, L.; Ferrell, J.E. Bistable, Biphasic Regulation of PP2A-B55 Accounts for the Dynamics of Mitotic Substrate Phosphorylation. Curr. Biol. 2021, 31, 794–808.e6. [Google Scholar] [CrossRef]
- Sørensen, C.S.; Syljuåsen, R.G. Safeguarding Genome Integrity: The Checkpoint Kinases ATR, CHK1 and WEE1 Restrain CDK Activity during Normal DNA Replication. Nucleic Acids Res. 2012, 40, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Krasinska, L.; Besnard, E.; Cot, E.; Dohet, C.; Mechali, M.; Lemaitre, J.M.; Fisher, D. Cdk1 and Cdk2 Activity Levels Determine the Efficiency of Replication Origin Firing in Xenopus. EMBO J. 2008, 27, 758–769.e6. [Google Scholar] [CrossRef] [Green Version]
- Echalier, A.; Cot, E.; Camasses, A.; Hodimont, E.; Hoh, F.; Jay, P.; Sheinerman, F.B.; Krasinska, L.; Fisher, D. An Integrated Chemical Biology Approach Provides Insight into Cdk2 Functional Redundancy and Inhibitor Sensitivity. Chem. Biol. 2012, 19, 1028–1040. [Google Scholar] [CrossRef] [Green Version]
- Ortega, S.; Prieto, I.; Odajima, J.; Martin, A.; Dubus, P.; Sotillo, R.; Barbero, J.L.; Malumbres, M.; Barbacid, M. Cyclin-Dependent Kinase 2 Is Essential for Meiosis but Not for Mitotic Cell Division in Mice. Nat. Genet. 2003, 35, 25–31. [Google Scholar] [CrossRef]
- Berthet, C.; Aleem, E.; Coppola, V.; Tessarollo, L.; Kaldis, P. Cdk2 Knockout Mice Are Viable. Curr Biol 2003, 13, 1775–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aleem, E.; Kiyokawa, H.; Kaldis, P. Cdc2-Cyclin E Complexes Regulate the G1/S Phase Transition. Nat. Cell Biol. 2005, 7, 831–836. [Google Scholar] [CrossRef] [PubMed]
- Hochegger, H.; Dejsuphong, D.; Sonoda, E.; Saberi, A.; Rajendra, E.; Kirk, J.; Hunt, T.; Takeda, S. An Essential Role for Cdk1 in S Phase Control Is Revealed via Chemical Genetics in Vertebrate Cells. J. Cell Biol. 2007, 178, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, D.; Barriere, C.; Cerqueira, A.; Hunt, S.; Tardy, C.; Newton, K.; Caceres, J.F.; Dubus, P.; Malumbres, M.; Barbacid, M. Cdk1 Is Sufficient to Drive the Mammalian Cell Cycle. Nature 2007, 448, 811–815. [Google Scholar] [CrossRef]
- Suski, J.M.; Ratnayeke, N.; Braun, M.; Zhang, T.; Strmiska, V.; Michowski, W.; Can, G.; Simoneau, A.; Snioch, K.; Cup, M.; et al. CDC7-Independent G1/S Transition Revealed by Targeted Protein Degradation. Nature 2022, 605, 357–365. [Google Scholar] [CrossRef]
- Adhikari, D.; Zheng, W.; Shen, Y.; Gorre, N.; Ning, Y.; Halet, G.; Kaldis, P.; Liu, K. Cdk1, but Not Cdk2, Is the Sole Cdk That Is Essential and Sufficient to Drive Resumption of Meiosis in Mouse Oocytes. Hum. Mol. Genet. 2012, 21, 2476–2484. [Google Scholar] [CrossRef] [Green Version]
- Satyanarayana, A.; Berthet, C.; Lopez-Molina, J.; Coppola, V.; Tessarollo, L.; Kaldis, P. Genetic Substitution of Cdk1 by Cdk2 Leads to Embryonic Lethality and Loss of Meiotic Function of Cdk2. Development 2008, 135, 3389–3400. [Google Scholar] [CrossRef] [Green Version]
- Lau, H.W.; Ma, H.T.; Yeung, T.K.; Tam, M.Y.; Zheng, D.; Chu, S.K.; Poon, R.Y.C. Quantitative Differences between Cyclin-Dependent Kinases Underlie the Unique Functions of CDK1 in Human Cells. Cell Rep. 2021, 37, 109808. [Google Scholar] [CrossRef]
- Pickering, M.; Magner, M.; Keifenheim, D.; Rhind, N. The Fission Yeast S-Phase Cyclin Cig2 Can Drive Mitosis. Genetics 2021, 217, 1–12. [Google Scholar] [CrossRef]
- Basu, S.; Greenwood, J.; Jones, A.W.; Nurse, P. Core Control Principles of the Eukaryotic Cell Cycle. Nature 2022, 1–6. [Google Scholar] [CrossRef]
- Vigneron, S.; Sundermann, L.; Labbé, J.-C.; Pintard, L.; Radulescu, O.; Castro, A.; Lorca, T. Cyclin A-Cdk1-Dependent Phosphorylation of Bora Is the Triggering Factor Promoting Mitotic Entry. Dev. Cell 2018, 45, 637–650.e7. [Google Scholar] [CrossRef]
- Abrieu, A.; Brassac, T.; Galas, S.; Fisher, D.; Labbe, J.C.; Doree, M. The Polo-like Kinase Plx1 Is a Component of the MPF Amplification Loop at the G2/M-Phase Transition of the Cell Cycle in Xenopus Eggs. J. Cell Sci. 1998, 111, 1751–1757. [Google Scholar] [CrossRef]
- Strausfeld, U.P.; Howell, M.; Descombes, P.; Chevalier, S.; Rempel, R.E.; Adamczewski, J.; Maller, J.L.; Hunt, T.; Blow, J.J. Both Cyclin A and Cyclin E Have S-Phase Promoting (SPF) Activity in Xenopus Egg Extracts. J. Cell Sci. 1996, 109, 1555–1563. [Google Scholar] [CrossRef]
- Kalaszczynska, I.; Geng, Y.; Iino, T.; Mizuno, S.; Choi, Y.; Kondratiuk, I.; Silver, D.P.; Wolgemuth, D.J.; Akashi, K.; Sicinski, P. Cyclin A Is Redundant in Fibroblasts but Essential in Hematopoietic and Embryonic Stem Cells. Cell 2009, 138, 352–365. [Google Scholar] [CrossRef] [Green Version]
- Geng, Y.; Yu, Q.; Sicinska, E.; Das, M.; Schneider, J.E.; Bhattacharya, S.; Rideout, W.M.; Bronson, R.T.; Gardner, H.; Sicinski, P. Cyclin E Ablation in the Mouse. Cell 2003, 114, 431–443. [Google Scholar] [CrossRef] [Green Version]
- Moore, J.D.; Kirk, J.A.; Hunt, T. Unmasking the S-Phase-Promoting Potential of Cyclin B1. Science 2003, 300, 987–990. [Google Scholar] [CrossRef]
- Hégarat, N.; Crncec, A.; Suarez Peredo Rodriguez, M.F.; Echegaray Iturra, F.; Gu, Y.; Busby, O.; Lang, P.F.; Barr, A.R.; Bakal, C.; Kanemaki, M.T.; et al. Cyclin A Triggers Mitosis Either via the Greatwall Kinase Pathway or Cyclin B. EMBO J. 2020, 39, e104419. [Google Scholar] [CrossRef]
- Lehner, C.F.; O’Farrell, P.H. The Roles of Drosophila Cyclins A and B in Mitotic Control. Cell 1990, 61, 535–547. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, H.W.; Knoblich, J.A.; Lehner, C.F. Drosophila Cyclin B3 Is Required for Female Fertility and Is Dispensable for Mitosis like Cyclin B. Genes Dev. 1998, 12, 3741–3751. [Google Scholar] [CrossRef] [Green Version]
- Knoblich, J.A.; Lehner, C.F. Synergistic Action of Drosophila Cyclins A and B during the G2-M Transition. EMBO J. 1993, 12, 65–74. [Google Scholar] [CrossRef]
- Burkard, M.E.; Randall, C.L.; Larochelle, S.; Zhang, C.; Shokat, K.M.; Fisher, R.P.; Jallepalli, P.V. Chemical Genetics Reveals the Requirement for Polo-like Kinase 1 Activity in Positioning RhoA and Triggering Cytokinesis in Human Cells. Proc. Natl. Acad. Sci. USA 2007, 104, 4383–4388. [Google Scholar] [CrossRef] [Green Version]
- Fenton, B.; Glover, D.M. A Conserved Mitotic Kinase Active at Late Anaphase-Telophase in Syncytial Drosophila Embryos. Nature 1993, 363, 637–640. [Google Scholar] [CrossRef]
- Rossi, A.; Kontarakis, Z.; Gerri, C.; Nolte, H.; Hölper, S.; Krüger, M.; Stainier, D.Y.R. Genetic Compensation Induced by Deleterious Mutations but Not Gene Knockdowns. Nature 2015, 524, 230–233. [Google Scholar] [CrossRef]
- Loog, M.; Morgan, D.O. Cyclin Specificity in the Phosphorylation of Cyclin-Dependent Kinase Substrates. Nature 2005, 434, 104–108. [Google Scholar] [CrossRef]
- Pagliuca, F.W.; Collins, M.O.; Lichawska, A.; Zegerman, P.; Choudhary, J.S.; Pines, J. Quantitative Proteomics Reveals the Basis for the Biochemical Specificity of the Cell-Cycle Machinery. Mol. Cell 2011, 43, 406–417. [Google Scholar] [CrossRef] [Green Version]
- Örd, M.; Möll, K.; Agerova, A.; Kivi, R.; Faustova, I.; Venta, R.; Valk, E.; Loog, M. Multisite Phosphorylation Code of CDK. Nat. Struct. Mol. Biol. 2019, 26, 649–658. [Google Scholar] [CrossRef]
- Lopez-Aviles, S.; Kapuy, O.; Novak, B.; Uhlmann, F. Irreversibility of Mitotic Exit Is the Consequence of Systems-Level Feedback. Nature 2009, 459, 592–595. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fisher, D.; Krasinska, L. Explaining Redundancy in CDK-Mediated Control of the Cell Cycle: Unifying the Continuum and Quantitative Models. Cells 2022, 11, 2019. https://doi.org/10.3390/cells11132019
Fisher D, Krasinska L. Explaining Redundancy in CDK-Mediated Control of the Cell Cycle: Unifying the Continuum and Quantitative Models. Cells. 2022; 11(13):2019. https://doi.org/10.3390/cells11132019
Chicago/Turabian StyleFisher, Daniel, and Liliana Krasinska. 2022. "Explaining Redundancy in CDK-Mediated Control of the Cell Cycle: Unifying the Continuum and Quantitative Models" Cells 11, no. 13: 2019. https://doi.org/10.3390/cells11132019
APA StyleFisher, D., & Krasinska, L. (2022). Explaining Redundancy in CDK-Mediated Control of the Cell Cycle: Unifying the Continuum and Quantitative Models. Cells, 11(13), 2019. https://doi.org/10.3390/cells11132019