
Mitoribosomal Deregulation Drives Senescence via TPP1-Mediated Telomere Deprotection

 , , and
, , and 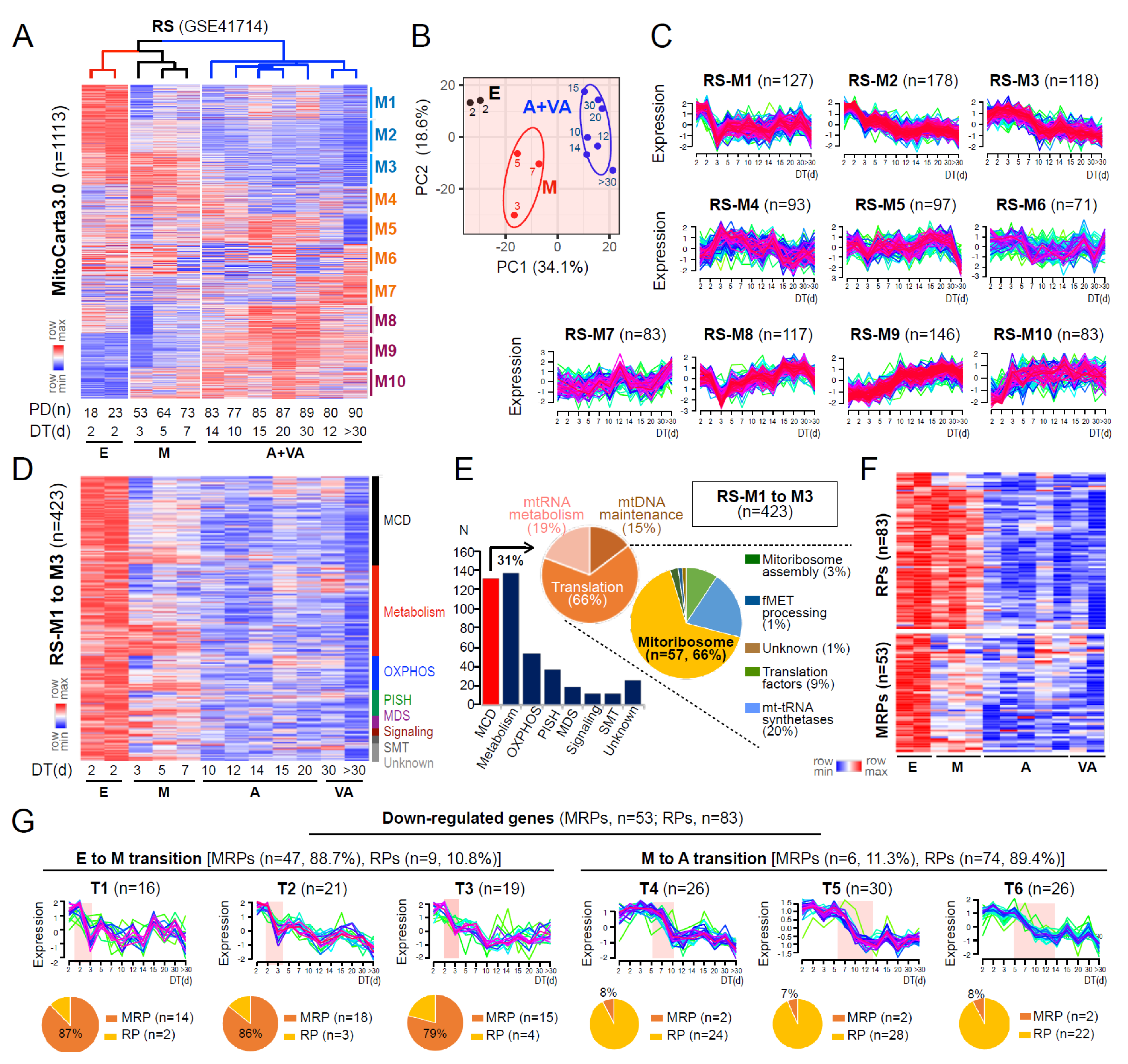{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Transcriptomic Analysis of Cellular Senescence Models
2.2. MitoCarta3.0 and Genes Encoding MRPs and RPs
2.3. Transcriptomic Analysis of Aging Skin from Mice and Humans
2.4. Single-Cell RNA-Seq Analysis of Murine Aging Dermal Fibroblasts
2.5. Cell Culture, Cell Growth, and Development of Cellular Senescence
2.6. Skin Tissues from Aging Animals
2.7. Determination of Mitochondrial ROS Level and Monitoring of Cell Size and Cell Granularity
2.8. SA-β-Gal Activity Staining
2.9. Transfection of siRNAs and Expression Plasmid into Cells
2.10. Western Blot Analysis
2.11. Quantitative Real-Time PCR (qPCR) for mRNA Expression
2.12. Estimation of Telomere Length
2.13. Monitoring Mitochondrial Translation Activity
2.14. Immunofluorescence–Fluorescence In Situ Hybridization (IF–FISH) Analysis for Telomeric DNA Damage
2.15. Statistical Analyses
3. Results
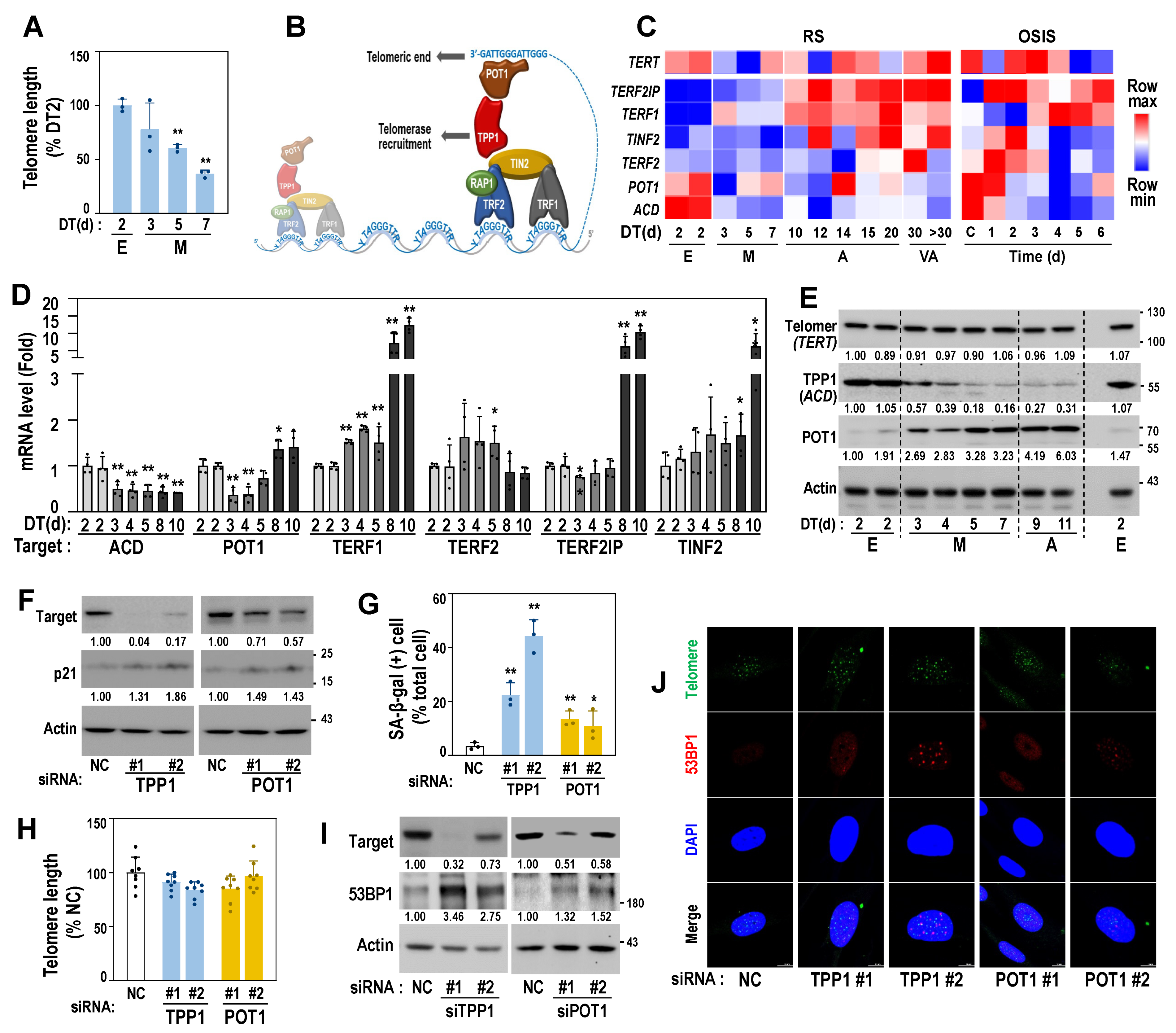
3.1. Deregulation of Mitoribosomal Genes Is an Initial Event of Replicative Senescence
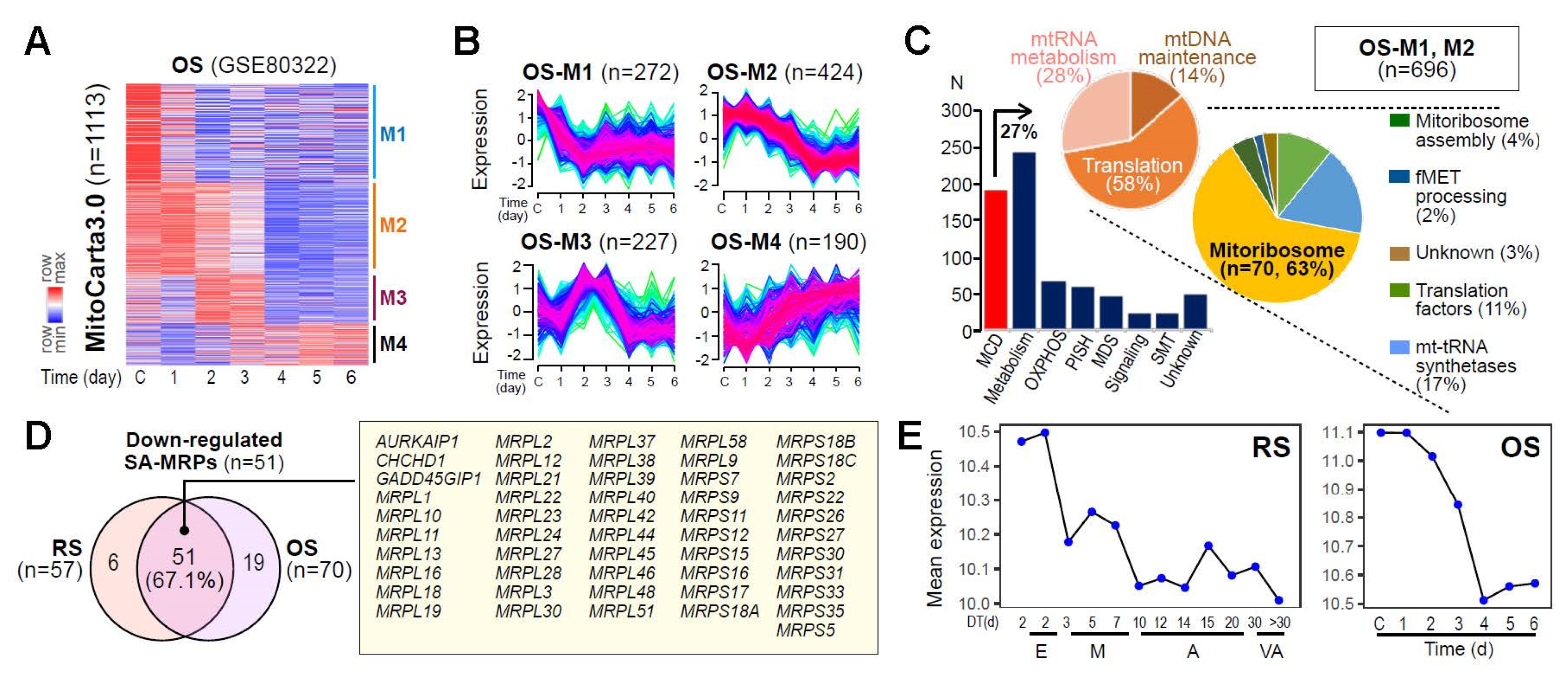
3.2. Identification of a Senescence-Associated Mitoribosomal Gene Signature
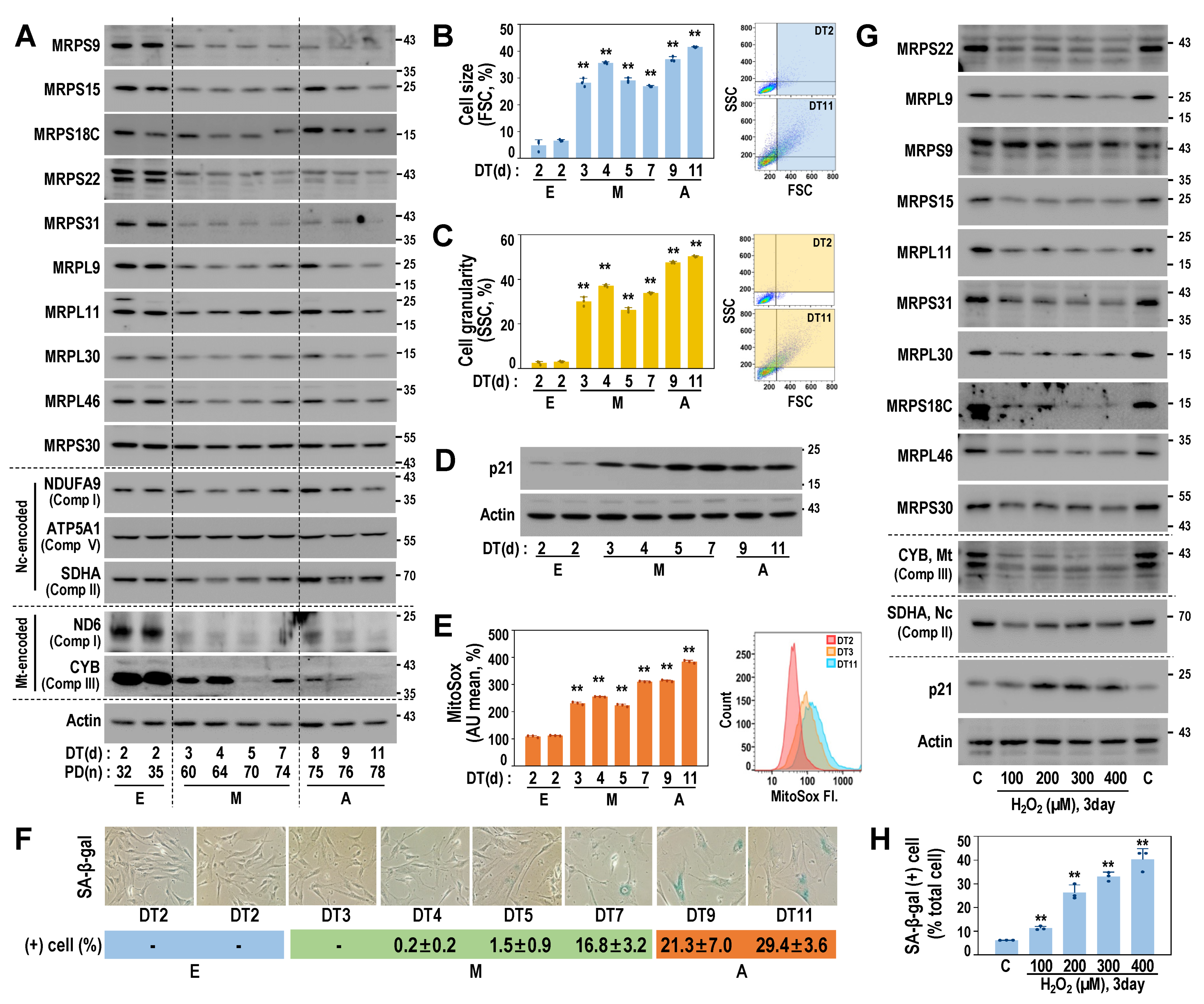
3.3. Downregulated Protein Expressions of MRPs Are Linked to Early Senescent Phenotypes and Mitochondrial ROS Generation
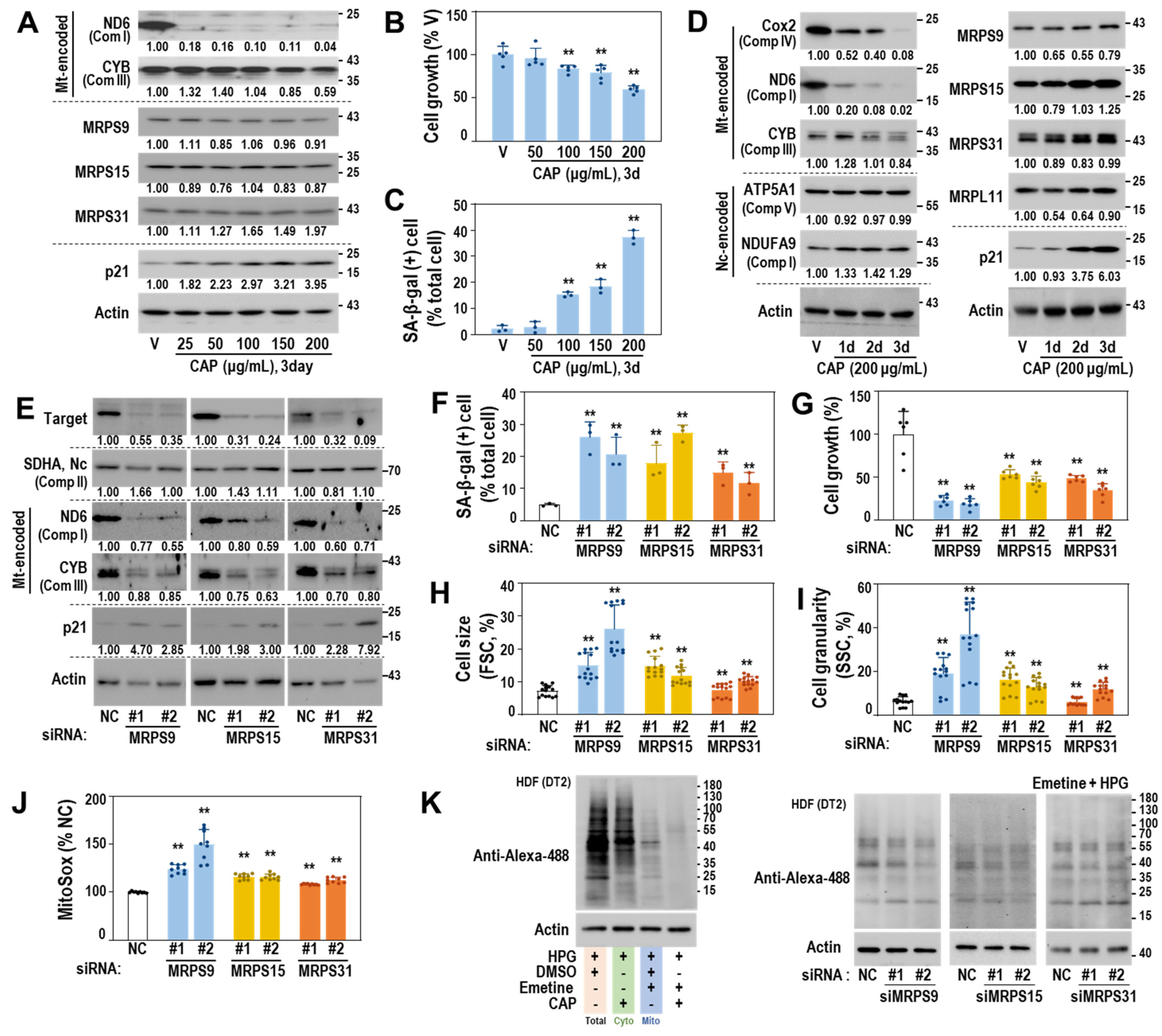
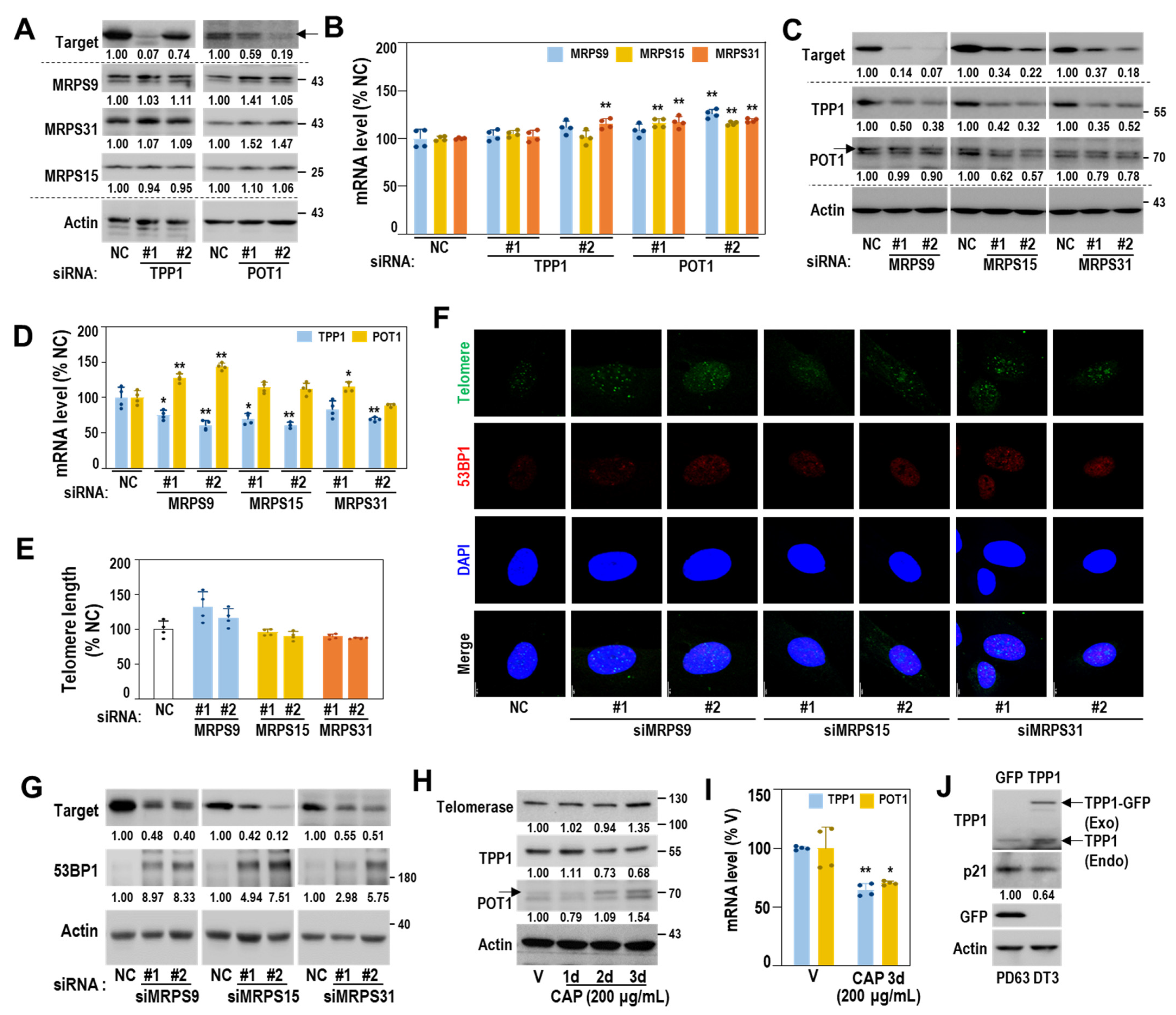
3.4. Mitoribosomal Perturbation Triggers Cellular Senescence
3.5. Decreased Protein Expression of TPP1 Is an Early Event Driving Cellular Senescence
3.6. Mitoribosomal Deregulation Functions Upstream of Transcriptional TPP1 Suppression
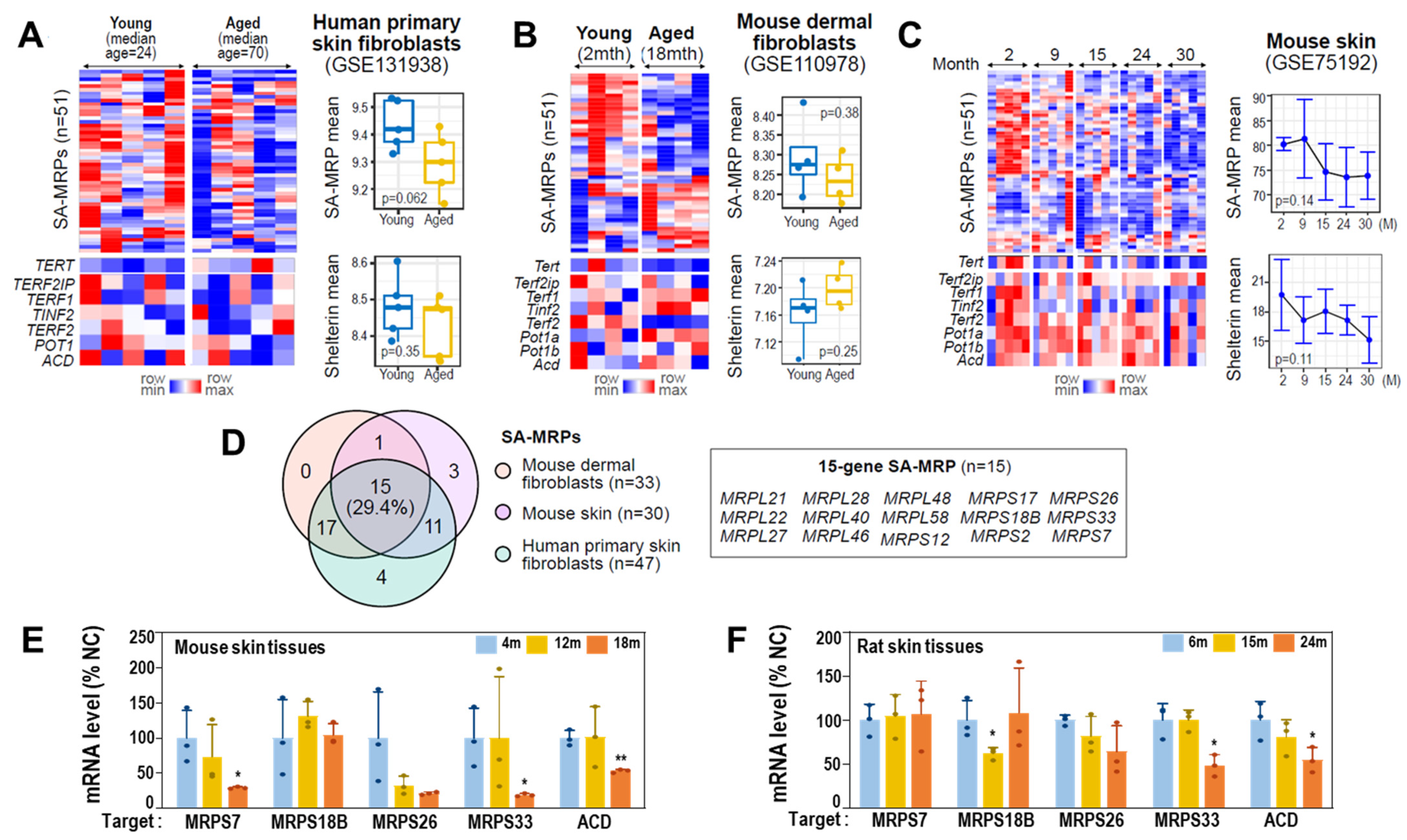
3.7. Gene Expressions of MRPs and Shelterin Complex Are Downregulated in Aged Human Cells and Rat and Mouse Skin Tissues
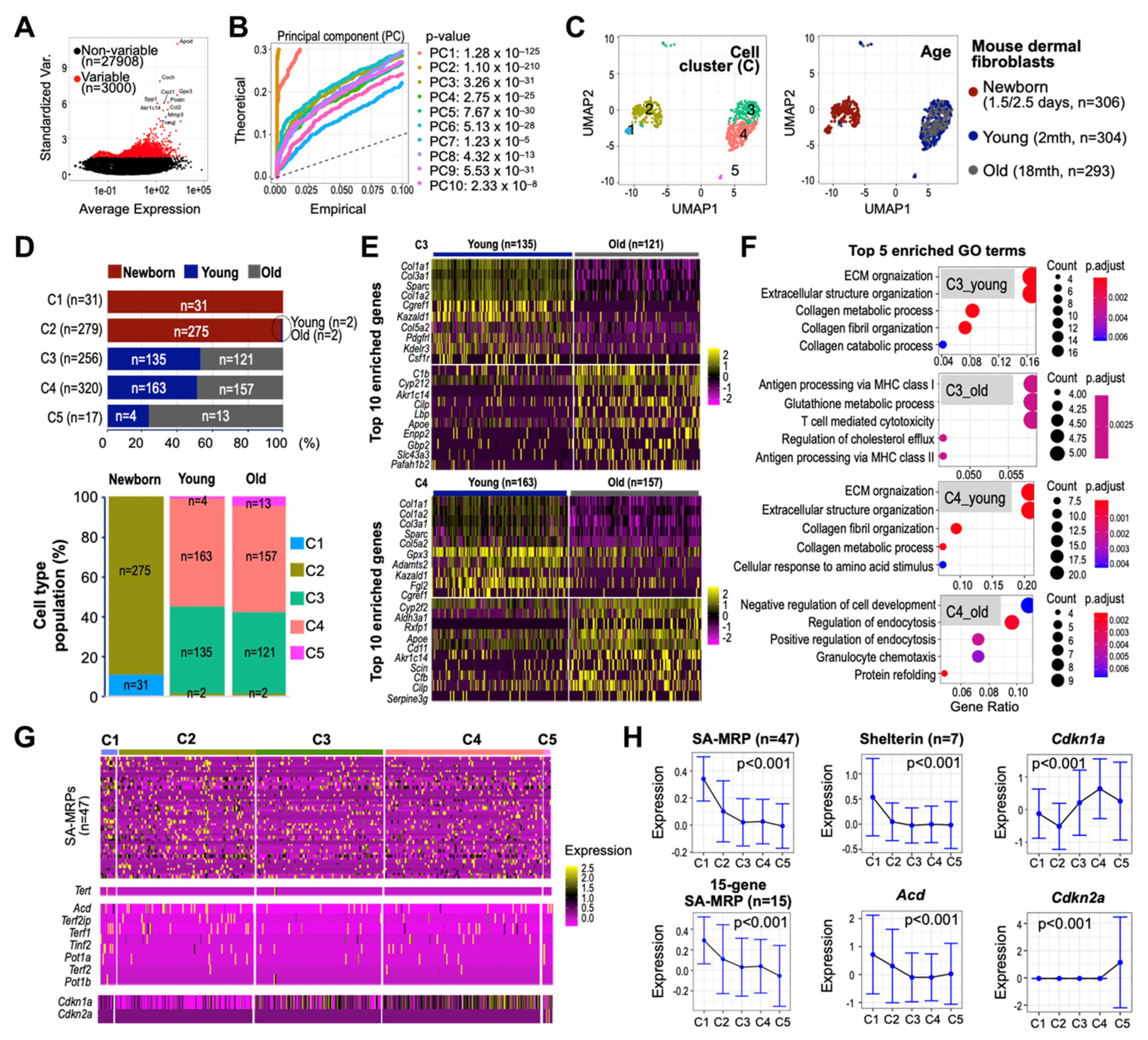
3.8. Single-Cell Transcriptomes of Human Skin Reveal Early Suppressed Expressions of SA-MRPs and Shelterin Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harman, D. The Free Radical Theory of Aging: Effect of Age on Serum Copper Levels. J. Gerontol. 1965, 20, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Raha, S.; Robinson, B.H. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem. Sci. 2000, 25, 502–508. [Google Scholar] [CrossRef]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021, 49, D1541–D1547. [Google Scholar] [CrossRef] [PubMed]
- Attardi, G.; Schatz, G. Biogenesis of mitochondria. Ann. Rev. Cell Biol. 1988, 4, 289–333. [Google Scholar] [CrossRef]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef] [Green Version]
- Linnane, A.W.; Marzuki, S.; Ozawa, T.; Tanaka, M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet 1989, 1, 642–645. [Google Scholar] [CrossRef]
- Shigenaga, M.K.; Hagen, T.M.; Ames, B.N. Oxidative damage and mitochondrial decay in aging. Proc. Natl. Acad. Sci. USA 1994, 91, 10771–10778. [Google Scholar] [CrossRef] [Green Version]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly, Y.M.; Gidlof, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef]
- Smeitink, J.; van den Heuvel, L.; DiMauro, S. The genetics and pathology of oxidative phosphorylation. Nat. Rev. Genet. 2001, 2, 342–352. [Google Scholar] [CrossRef] [PubMed]
- De Silva, D.; Tu, Y.T.; Amunts, A.; Fontanesi, F.; Barrientos, A. Mitochondrial ribosome assembly in health and disease. Cell Cycle 2015, 14, 2226–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayflick, L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Maier, A.B.; Westendorp, R.G. Relation between replicative senescence of human fibroblasts and life history characteristics. Ageing Res. Rev. 2009, 8, 237–243. [Google Scholar] [CrossRef]
- Olovnikov, A.M. A theory of marginotomy: The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J. Theor. Biol. 1973, 41, 181–190. [Google Scholar] [CrossRef]
- Olovnikov, A.M. Telomeres, telomerase, and aging: Origin of the theory. Exp. Gerontol. 1996, 31, 443–448. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, X.; Ding, X.; Wang, F.; Geng, X. Telomere and its role in the aging pathways: Telomere shortening, cell senescence and mitochondria dysfunction. Biogerontology 2019, 20, 1–16. [Google Scholar] [CrossRef]
- Henle, E.S.; Han, Z.; Tang, N.; Rai, P.; Luo, Y.; Linn, S. Sequence-specific DNA cleavage by Fe2+-mediated fenton reactions has possible biological implications. J. Biol. Chem. 1999, 274, 962–971. [Google Scholar] [CrossRef] [Green Version]
- Oikawa, S.; Kawanishi, S. Site-specific DNA damage at GGG sequence by oxidative stress may accelerate telomere shortening. FEBS Lett. 1999, 453, 365–368. [Google Scholar] [CrossRef] [Green Version]
- De Lange, T. How telomeres solve the end-protection problem. Science 2009, 326, 948–952. [Google Scholar] [CrossRef] [Green Version]
- Lazzerini-Denchi, E.; Sfeir, A. Stop pulling my strings—What telomeres taught us about the DNA damage response. Nat. Rev. Mol. Cell Biol. 2016, 17, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Horikawa, I.; Mondal, A.M.; Jenkins, L.M.; Appella, E.; Vojtesek, B.; Bourdon, J.C.; Lane, D.P.; Harris, C.C. Positive feedback between p53 and TRF2 during telomere-damage signalling and cellular senescence. Nat. Cell Biol. 2010, 12, 1205–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Jung, M.; Hong, J.; Kim, M.K.; Chung, I.K. Loss of RNA-binding protein HuR facilitates cellular senescence through posttranscriptional regulation of TIN2 mRNA. Nucleic Acids Res. 2018, 46, 4271–4285. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.M.; Byun, H.O.; Jee, B.A.; Cho, H.; Seo, Y.H.; Kim, Y.S.; Park, M.H.; Chung, H.Y.; Woo, H.G.; Yoon, G. Implications of time-series gene expression profiles of replicative senescence. Aging Cell 2013, 12, 622–634. [Google Scholar] [CrossRef]
- Holzscheck, N.; Sohle, J.; Kristof, B.; Gronniger, E.; Gallinat, S.; Wenck, H.; Winnefeld, M.; Falckenhayn, C.; Kaderali, L. Multi-omics network analysis reveals distinct stages in the human aging progression in epidermal tissue. Aging 2020, 12, 12393–12409. [Google Scholar] [CrossRef]
- Jung, H.J.; Byun, H.O.; Jee, B.A.; Min, S.; Jeoun, U.W.; Lee, Y.K.; Seo, Y.; Woo, H.G.; Yoon, G. The Ubiquitin-like with PHD and Ring Finger Domains 1 (UHRF1)/DNA Methyltransferase 1 (DNMT1) Axis Is a Primary Regulator of Cell Senescence. J. Biol. Chem. 2017, 292, 3729–3739. [Google Scholar] [CrossRef] [Green Version]
- Greber, B.J.; Ban, N. Structure and Function of the Mitochondrial Ribosome. Ann. Rev. BioChem. 2016, 85, 103–132. [Google Scholar] [CrossRef]
- Aramillo Irizar, P.; Schauble, S.; Esser, D.; Groth, M.; Frahm, C.; Priebe, S.; Baumgart, M.; Hartmann, N.; Marthandan, S.; Menzel, U.; et al. Transcriptomic alterations during ageing reflect the shift from cancer to degenerative diseases in the elderly. Nat. Commun. 2018, 9, 327. [Google Scholar] [CrossRef] [Green Version]
- Oblong, J.E.; Bowman, A.; Rovito, H.A.; Jarrold, B.B.; Sherrill, J.D.; Black, M.R.; Nelson, G.; Kimball, A.B.; Birch-Machin, M.A. Metabolic dysfunction in human skin: Restoration of mitochondrial integrity and metabolic output by nicotinamide (niacinamide) in primary dermal fibroblasts from older aged donors. Aging Cell 2020, 19, e13248. [Google Scholar] [CrossRef]
- Salzer, M.C.; Lafzi, A.; Berenguer-Llergo, A.; Youssif, C.; Castellanos, A.; Solanas, G.; Peixoto, F.O.; Stephan-Otto Attolini, C.; Prats, N.; Aguilera, M.; et al. Identity Noise and Adipogenic Traits Characterize Dermal Fibroblast Aging. Cell 2018, 175, 1575–1590.e22. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Song, M.C.; Kwak, I.H.; Park, T.J.; Lim, I.K. Constitutive induction of p-Erk1/2 accompanied by reduced activities of protein phosphatases 1 and 2A and MKP3 due to reactive oxygen species during cellular senescence. J. Biol. Chem. 2003, 278, 37497–37510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Callaghan, N.J.; Fenech, M. A quantitative PCR method for measuring absolute telomere length. Biol. Proced. Online 2011, 13, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousefi, R.; Fornasiero, E.F.; Cyganek, L.; Montoya, J.; Jakobs, S.; Rizzoli, S.O.; Rehling, P.; Pacheu-Grau, D. Monitoring mitochondrial translation in living cells. EMBO Rep. 2021, 22, e51635. [Google Scholar] [CrossRef] [PubMed]
- McKee, E.E.; Ferguson, M.; Bentley, A.T.T.; Marks, T.A. Inhibition of mammalian mitochondrial protein synthesis by oxazolidinones. Antimicrob. Agents Chemother. 2006, 50, 2042–2049. [Google Scholar] [CrossRef] [Green Version]
- Lototska, L.; Yue, J.X.; Li, J.; Giraud-Panis, M.J.; Songyang, Z.; Royle, N.J.; Liti, G.; Ye, J.; Gilson, E.; Mendez-Bermudez, A. Human RAP1 specifically protects telomeres of senescent cells from DNA damage. EMBO Rep. 2020, 21, e49076. [Google Scholar] [CrossRef]
- Mirza-Aghazadeh-Attari, M.; Mohammadzadeh, A.; Yousefi, B.; Mihanfar, A.; Karimian, A.; Majidinia, M. 53BP1: A key player of DNA damage response with critical functions in cancer. DNA Repair 2019, 73, 110–119. [Google Scholar] [CrossRef]
- Sole-Boldo, L.; Raddatz, G.; Schutz, S.; Mallm, J.P.; Rippe, K.; Lonsdorf, A.S.; Rodriguez-Paredes, M.; Lyko, F. Single-cell transcriptomes of the human skin reveal age-related loss of fibroblast priming. Commun. Biol. 2020, 3, 188. [Google Scholar] [CrossRef] [Green Version]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [Green Version]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Wang, C.; Jurk, D.; Maddick, M.; Nelson, G.; Martin-Ruiz, C.; von Zglinicki, T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 2009, 8, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.M.; Marquess, D.; Dananberg, J.; van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Mouchiroud, L.; Ryu, D.; Moullan, N.; Katsyuba, E.; Knott, G.; Williams, R.W.; Auwerx, J. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 2013, 497, 451–457. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Maiti, P.; Barrientos, A. Mitochondrial ribosomes in cancer. Semin. Cancer Biol. 2017, 47, 67–81. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.S.; Safari, A.; Xin, H.; Liu, D.; Songyang, Z. A critical role for TPP1 and TIN2 interaction in high-order telomeric complex assembly. Proc. Natl. Acad. Sci. USA 2006, 103, 11874–11879. [Google Scholar] [CrossRef] [Green Version]
- Palm, W.; de Lange, T. How shelterin protects mammalian telomeres. Ann. Rev. Genet. 2008, 42, 301–334. [Google Scholar] [CrossRef] [Green Version]
- Rajavel, M.; Mullins, M.R.; Taylor, D.J. Multiple facets of TPP1 in telomere maintenance. Biochim. Biophys. Acta 2014, 1844, 1550–1559. [Google Scholar] [CrossRef] [Green Version]
- Zaug, A.J.; Podell, E.R.; Nandakumar, J.; Cech, T.R. Functional interaction between telomere protein TPP1 and telomerase. Genes Dev. 2010, 24, 613–622. [Google Scholar] [CrossRef] [Green Version]
- Else, T.; Theisen, B.K.; Wu, Y.; Hutz, J.E.; Keegan, C.E.; Hammer, G.D.; Ferguson, D.O. Tpp1/Acd maintains genomic stability through a complex role in telomere protection. Chromosome Res. 2007, 15, 1001–1013. [Google Scholar] [CrossRef]
- Hockemeyer, D.; Palm, W.; Else, T.; Daniels, J.P.; Takai, K.K.; Ye, J.Z.; Keegan, C.E.; de Lange, T.; Hammer, G.D. Telomere protection by mammalian Pot1 requires interaction with Tpp1. Nat. Struct. Mol. Biol. 2007, 14, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Tejera, A.M.; Stagno d’Alcontres, M.; Thanasoula, M.; Marion, R.M.; Martinez, P.; Liao, C.; Flores, J.M.; Tarsounas, M.; Blasco, M.A. TPP1 is required for TERT recruitment, telomere elongation during nuclear reprogramming, and normal skin development in mice. Dev. Cell 2010, 18, 775–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kibe, T.; Osawa, G.A.; Keegan, C.E.; de Lange, T. Telomere protection by TPP1 is mediated by POT1a and POT1b. Mol. Cell Biol. 2010, 30, 1059–1066. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Deng, Y.; Lin, Y.; Cosme-Blanco, W.; Chan, S.; He, H.; Yuan, G.; Brown, E.J.; Chang, S. Dysfunctional telomeres activate an ATM-ATR-dependent DNA damage response to suppress tumorigenesis. EMBO J. 2007, 26, 4709–4719. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, T.; Sundar, I.K.; Tormos, A.M.; Lerner, C.A.; Gerloff, J.; Yao, H.; Rahman, I. Shelterin Telomere Protection Protein 1 Reduction Causes Telomere Attrition and Cellular Senescence via Sirtuin 1 Deacetylase in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Cell Mol. Biol. 2017, 56, 38–49. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Min, S.; Kwon, S.M.; Hong, J.; Lee, Y.-K.; Park, T.J.; Lim, S.B.; Yoon, G. Mitoribosomal Deregulation Drives Senescence via TPP1-Mediated Telomere Deprotection. Cells 2022, 11, 2079. https://doi.org/10.3390/cells11132079
Min S, Kwon SM, Hong J, Lee Y-K, Park TJ, Lim SB, Yoon G. Mitoribosomal Deregulation Drives Senescence via TPP1-Mediated Telomere Deprotection. Cells. 2022; 11(13):2079. https://doi.org/10.3390/cells11132079
Chicago/Turabian StyleMin, Seongki, So Mee Kwon, Jiwon Hong, Young-Kyoung Lee, Tae Jun Park, Su Bin Lim, and Gyesoon Yoon. 2022. "Mitoribosomal Deregulation Drives Senescence via TPP1-Mediated Telomere Deprotection" Cells 11, no. 13: 2079. https://doi.org/10.3390/cells11132079
APA StyleMin, S., Kwon, S. M., Hong, J., Lee, Y. -K., Park, T. J., Lim, S. B., & Yoon, G. (2022). Mitoribosomal Deregulation Drives Senescence via TPP1-Mediated Telomere Deprotection. Cells, 11(13), 2079. https://doi.org/10.3390/cells11132079