
Microglia Phenotypes in Aging and Neurodegenerative Diseases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Microglia in Physiology
1.1. Microglia Origin, Development, and Maturation
1.2. Microglia Functions in Physiology
1.3. Microglia Morphologies
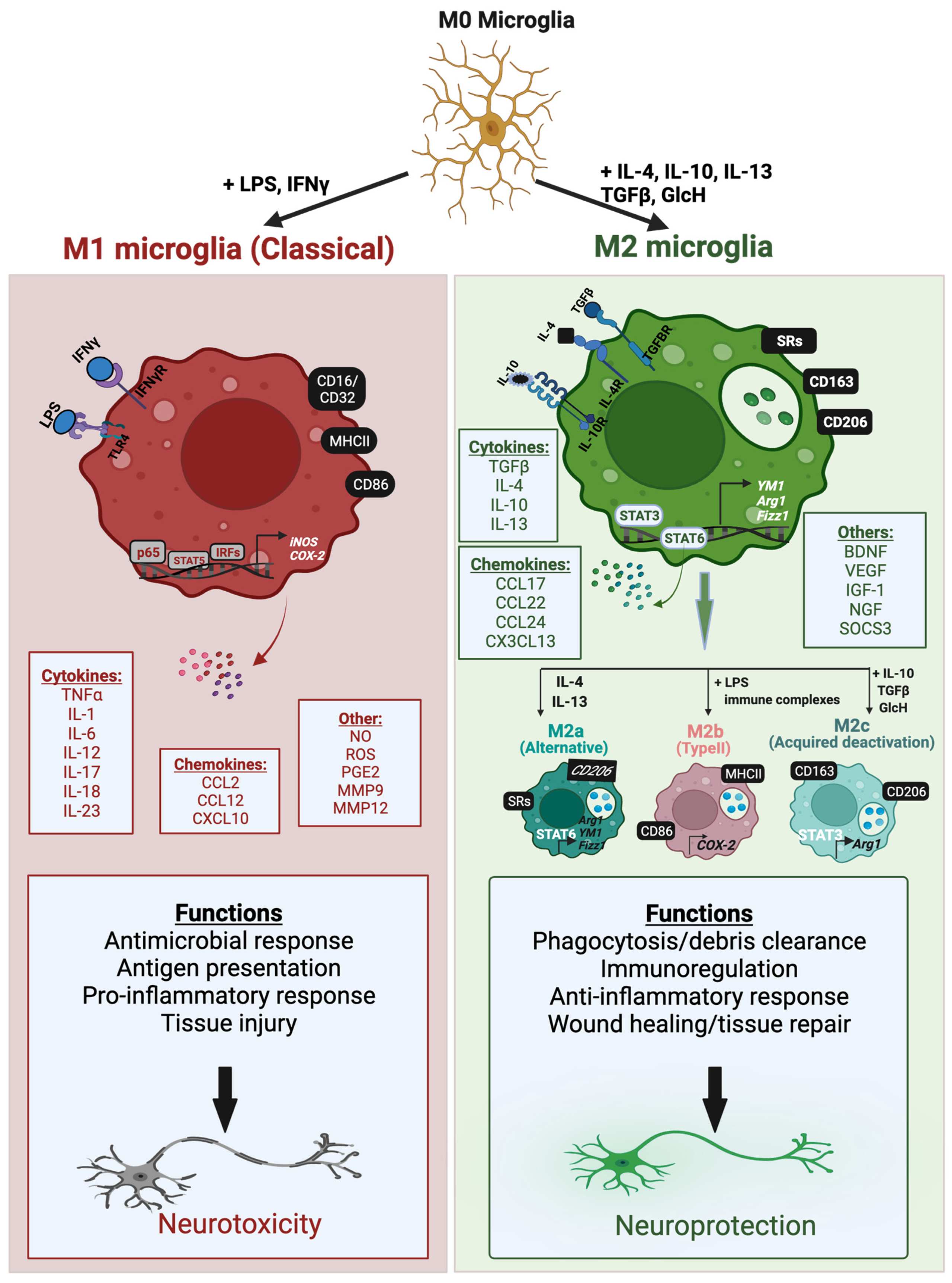
2. Microglia Activation and Polarization
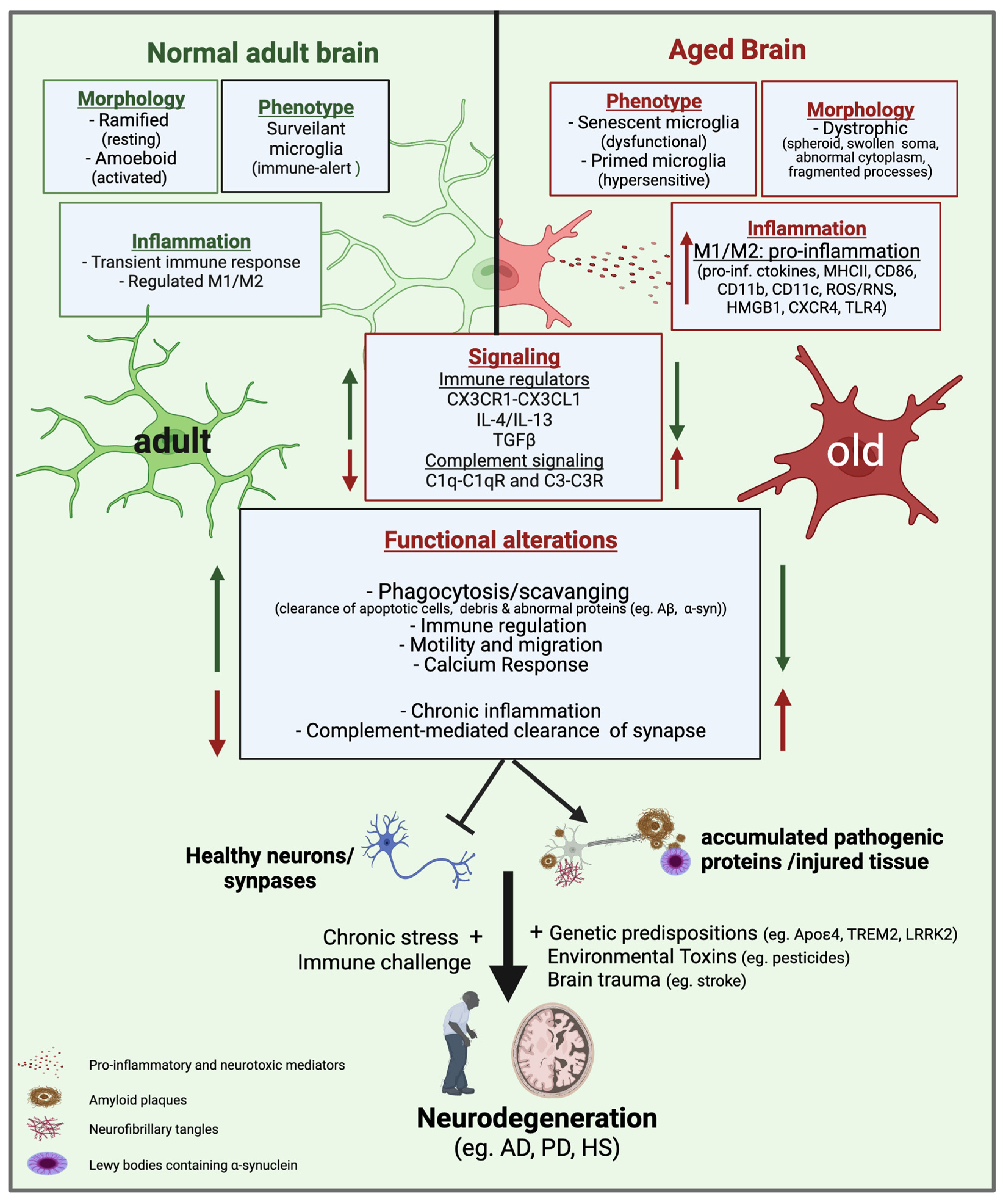
3. Microglia Phenotypes in Aging
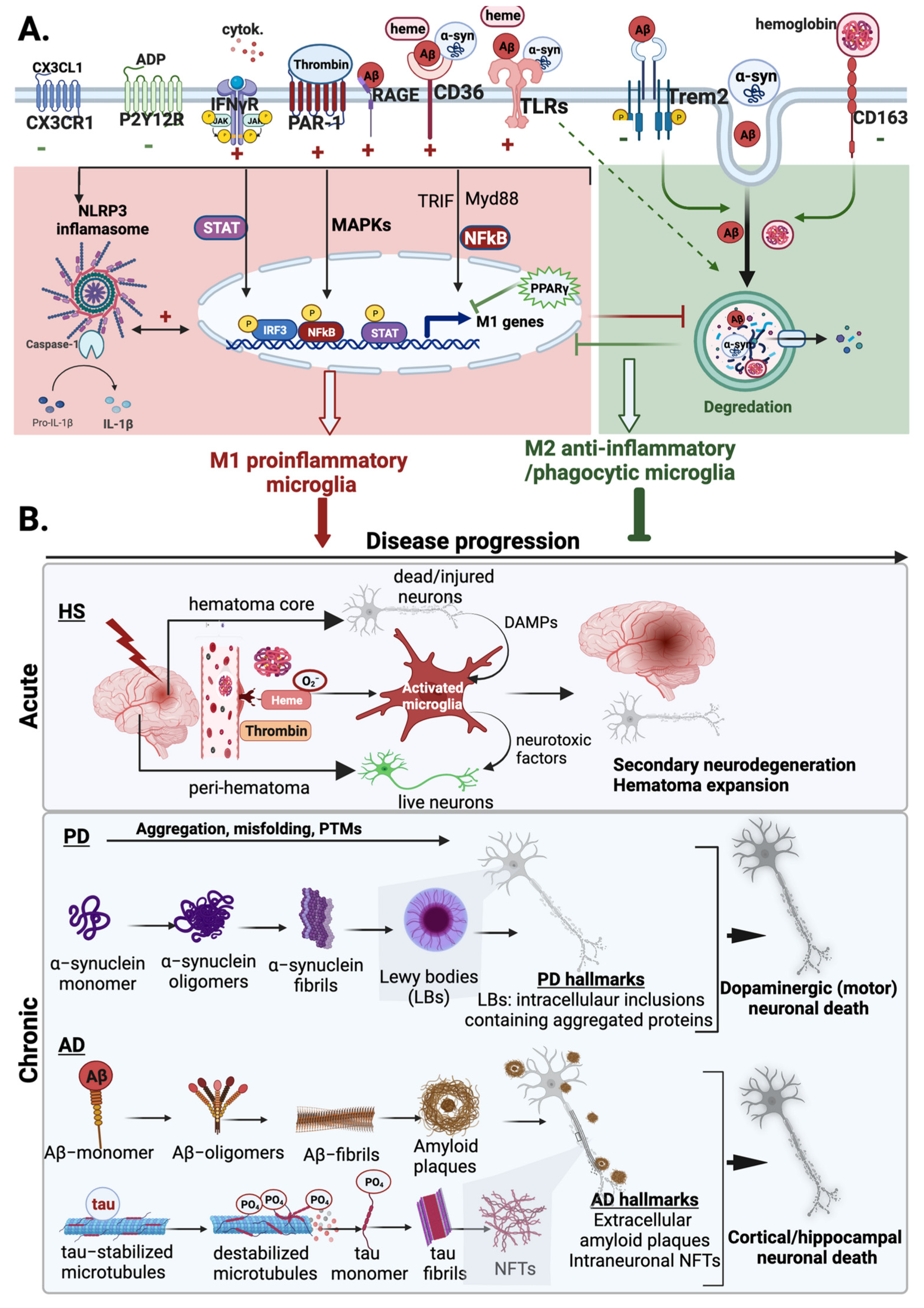
4. Microglia in Acute and Chronic Neurodegenerative Diseases
4.1. Microglia in Hemorrhagic Stroke
4.1.1. Hemorrhagic Stroke (HS)
4.1.2. Microglia Activation in HS
4.1.3. Microglia Activation by Hematoma Components
4.1.4. Microglia Activation by Thrombin
4.1.5. M1 and M2 Microglia in HS
4.1.6. Temporal Microglia Polarization in HS
4.2. Microglia in Alzheimer’s Disease
4.2.1. Alzheimer’s Disease (AD)
4.2.2. Microglia Activation in AD
4.2.3. Microglia Response to Amyloid-β
4.2.4. Microglia Response to Tau
4.2.5. Microglia Morphologies and Phenotypes in AD
4.2.6. M1 and M2 Microglia in AD
4.2.7. Timeline of Microglia Phenotypes in AD
4.2.8. Diversity of Microglia in AD Inferred from Transcriptome Studies: DAM and HAM
4.3. Microglia in Parkinson’s Disease
4.3.1. Parkinson’s Disease (PD)
4.3.2. Microglia Activation in PD
4.3.3. Microglia Response to α-Synuclein
4.3.4. Microglia Phenotypes in PD
M1 and M2 Microglia in PD
Microglia Diversity in PD Inferred from Transcriptome Studies
5. Modulators of Microglia Phenotypes in NDs
5.1. TLR Signaling Pathway
5.2. JAK/STAT Pathway
5.3. CX3CR1-CX3CL1 Signaling
5.4. Triggering Receptors Expressed on Myeloid Cells 2 (TREM2)
5.5. P2Y Receptors
5.6. Peroxisome Proliferator-Activated Receptor γ (PPARγ)
6. Challenges in Microglia Research
6.1. Specific Microglia Markers
6.2. Sex-Specific Heterogeneity of Microglia
6.3. Limitation and Heterogeneity of Human Microglia
7. Conclusions and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Ajami, B.; Bennett, J.L.; Krieger, C.; Tetzlaff, W.; Rossi, F.M. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 2007, 10, 1538–1543. [Google Scholar] [CrossRef]
- Alliot, F.; Godin, I.; Pessac, B. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Dev. Brain Res. 1999, 117, 145–152. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Goldmann, T.; Wieghofer, P.; Jordão, M.J.C.; Prutek, F.; Hagemeyer, N.; Frenzel, K.; Amann, L.; Staszewski, O.; Kierdorf, K.; Krueger, M.; et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 2016, 17, 797–805. [Google Scholar] [CrossRef]
- Ginhoux, F.; Garel, S. The mysterious origins of microglia. Nat. Neurosci. 2018, 21, 897–899. [Google Scholar] [CrossRef]
- Fogg, D.K.; Sibon, C.; Miled, C.; Jung, S.; Aucouturier, P.; Littman, D.R.; Cumano, A.; Geissmann, F. A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science 2006, 311, 83–87. [Google Scholar] [CrossRef]
- Monier, A.; Adle-Biassette, H.; Delezoide, A.L.; Evrard, P.; Gressens, P.; Verney, C. Entry and distribution of microglial cells in human embryonic and fetal cerebral cortex. J. Neuropathol. Exp. Neurol. 2007, 66, 372–382. [Google Scholar] [CrossRef]
- Perdiguero, G.E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Mittelbronn, M.; Dietz, K.; Schluesener, H.J.; Meyermann, R. Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol. 2001, 101, 249–255. [Google Scholar] [CrossRef]
- Askew, K.; Li, K.; Olmos-Alonso, A.; Garcia-Moreno, F.; Liang, Y.; Richardson, P.; Tipton, T.; Chapman, M.A.; Riecken, K.; Beccari, S.; et al. Coupled Proliferation and Apoptosis Maintain the Rapid Turnover of Microglia in the Adult Brain. Cell Rep. 2017, 18, 391–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Santos, S.E.; Medeiros, M.; Porfirio, J.; Tavares, W.; Pessoa, L.; Grinberg, L.; Leite, R.E.P.; Ferretti-Rebustini, R.E.L.; Suemoto, C.K.; Filho, W.J.; et al. Similar Microglial Cell Densities across Brain Structures and Mammalian Species: Implications for Brain Tissue Function. J. Neurosci. 2020, 40, 4622–4643. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef] [Green Version]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [Green Version]
- Buttgereit, A.; Lelios, I.; Yu, X.; Vrohlings, M.; Krakoski, N.R.; Gautier, E.L.; Nishinakamura, R.; Becher, B.; Greter, M. Sall1 is a transcriptional regulator defining microglia identity and function. Nat. Immunol. 2016, 17, 1397–1406. [Google Scholar] [CrossRef]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef]
- Hatori, K.; Nagai, A.; Heisel, R.; Ryu, J.K.; Kim, S.U. Fractalkine and fractalkine receptors in human neurons and glial cells. J. Neurosci. Res. 2002, 69, 418–426. [Google Scholar] [CrossRef]
- Pawelec, P.; Ziemka-Nalecz, M.; Sypecka, J.; Zalewska, T. The Impact of the CX3CL1/CX3CR1 Axis in Neurological Disorders. Cells 2020, 9, 2277. [Google Scholar] [CrossRef]
- Easley-Neal, C.; Foreman, O.; Sharma, N.; Zarrin, A.A.; Weimer, R.M. CSF1R Ligands IL-34 and CSF1 Are Differentially Required for Microglia Development and Maintenance in White and Gray Matter Brain Regions. Front. Immunol. 2019, 10, 2199. [Google Scholar] [CrossRef] [Green Version]
- Elmore, M.R.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Szretter, K.J.; Vermi, W.; Gilfillan, S.; Rossini, C.; Cella, M.; Barrow, A.D.; Diamond, M.S.; Colonna, M. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat. Immunol. 2012, 13, 753–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vainchtein, I.D.; Chin, G.; Cho, F.S.; Kelley, K.W.; Miller, J.G.; Chien, E.C.; Liddelow, S.A.; Nguyen, P.T.; Nakao-Inoue, H.; Dorman, L.C.; et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science 2018, 359, 1269–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Waard, D.M.; Bugiani, M. Astrocyte-Oligodendrocyte-Microglia Crosstalk in Astrocytopathies. Front. Cell. Neurosci. 2020, 14, 608073. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, M.; Kipnis, J.; Rivest, S.; Prat, A. How do immune cells support and shape the brain in health, disease, and aging? J. Neurosci. 2013, 33, 17587–17596. [Google Scholar] [CrossRef] [Green Version]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., 3rd; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [Green Version]
- Ueno, M.; Fujita, Y.; Tanaka, T.; Nakamura, Y.; Kikuta, J.; Ishii, M.; Yamashita, T. Layer V cortical neurons require microglial support for survival during postnatal development. Nat. Neurosci. 2013, 16, 543–551. [Google Scholar] [CrossRef]
- Butovsky, O.; Weiner, H.L. Microglial signatures and their role in health and disease. Nat. Rev. Neurosci. 2018, 19, 622–635. [Google Scholar] [CrossRef]
- Boche, D.; Perry, V.H.; Nicoll, J.A. Review: Activation patterns of microglia and their identification in the human brain. Neuropathol. Appl. Neurobiol. 2013, 39, 3–18. [Google Scholar] [CrossRef]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef]
- Parakalan, R.; Jiang, B.; Nimmi, B.; Janani, M.; Jayapal, M.; Lu, J.; Tay, S.S.; Ling, E.A.; Dheen, S.T. Transcriptome analysis of amoeboid and ramified microglia isolated from the corpus callosum of rat brain. BMC Neurosci. 2012, 13, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cserep, C.; Posfai, B.; Lenart, N.; Fekete, R.; Laszlo, Z.I.; Lele, Z.; Orsolits, B.; Molnar, G.; Heindl, S.; Schwarcz, A.D.; et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science 2020, 367, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Kaur, C.; Ling, E.A.; Wong, W.C. Transformation of amoeboid microglial cells into microglia in the corpus callosum of the postnatal rat brain. An electron microscopical study. Arch. Histol. Jpn. 1985, 48, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Madore, C.; Joffre, C.; Delpech, J.C.; De Smedt-Peyrusse, V.; Aubert, A.; Coste, L.; Laye, S.; Nadjar, A. Early morphofunctional plasticity of microglia in response to acute lipopolysaccharide. Brain Behav. Immun. 2013, 34, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Sierra, A.; de Castro, F.; Del Rio-Hortega, J.; Rafael Iglesias-Rozas, J.; Garrosa, M.; Kettenmann, H. The “Big-Bang” for modern glial biology: Translation and comments on Pio del Rio-Hortega 1919 series of papers on microglia. Glia 2016, 64, 1801–1840. [Google Scholar] [CrossRef]
- Savage, J.C.; Picard, K.; Gonzalez-Ibanez, F.; Tremblay, M.E. A Brief History of Microglial Ultrastructure: Distinctive Features, Phenotypes, and Functions Discovered Over the Past 60 Years by Electron Microscopy. Front. Immunol. 2018, 9, 803. [Google Scholar] [CrossRef]
- Au, N.P.B.; Ma, C.H.E. Recent Advances in the Study of Bipolar/Rod-Shaped Microglia and their Roles in Neurodegeneration. Front. Aging Neurosci. 2017, 9, 128. [Google Scholar] [CrossRef] [Green Version]
- Tam, W.Y.; Ma, C.H. Bipolar/rod-shaped microglia are proliferating microglia with distinct M1/M2 phenotypes. Sci. Rep. 2014, 4, 7279. [Google Scholar] [CrossRef]
- Ziebell, J.M.; Taylor, S.E.; Cao, T.; Harrison, J.L.; Lifshitz, J. Rod microglia: Elongation, alignment, and coupling to form trains across the somatosensory cortex after experimental diffuse brain injury. J. Neuroinflamm. 2012, 9, 247. [Google Scholar] [CrossRef] [Green Version]
- Bachstetter, A.D.; Ighodaro, E.T.; Hassoun, Y.; Aldeiri, D.; Neltner, J.H.; Patel, E.; Abner, E.L.; Nelson, P.T. Rod-shaped microglia morphology is associated with aging in 2 human autopsy series. Neurobiol. Aging 2017, 52, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Bachstetter, A.D.; Van Eldik, L.J.; Schmitt, F.A.; Neltner, J.H.; Ighodaro, E.T.; Webster, S.J.; Patel, E.; Abner, E.L.; Kryscio, R.J.; Nelson, P.T. Disease-related microglia heterogeneity in the hippocampus of Alzheimer’s disease, dementia with Lewy bodies, and hippocampal sclerosis of aging. Acta Neuropathol. Commun. 2015, 3, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, S.E.; Morganti-Kossmann, C.; Lifshitz, J.; Ziebell, J.M. Rod microglia: A morphological definition. PLoS ONE 2014, 9, e97096. [Google Scholar] [CrossRef] [PubMed]
- Doens, D.; Fernandez, P.L. Microglia receptors and their implications in the response to amyloid beta for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2014, 11, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zindel, J.; Kubes, P. DAMPs, PAMPs, and LAMPs in Immunity and Sterile Inflammation. Annu. Rev. Pathol. 2020, 15, 493–518. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, R.M.; Brown, M.A. Innate immunity in the central nervous system. J. Clin. Investig. 2012, 122, 1164–1171. [Google Scholar] [CrossRef]
- Broggi, A.; Granucci, F. Microbe- and danger-induced inflammation. Mol. Immunol. 2015, 63, 127–133. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Platanitis, E.; Decker, T. Regulatory Networks Involving STATs, IRFs, and NFkappaB in Inflammation. Front. Immunol. 2018, 9, 2542. [Google Scholar] [CrossRef] [Green Version]
- Goulopoulou, S.; McCarthy, C.G.; Webb, R.C. Toll-like Receptors in the Vascular System: Sensing the Dangers Within. Pharmacol. Rev. 2016, 68, 142–167. [Google Scholar] [CrossRef]
- Ivashkiv, L.B. IFNgamma: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558. [Google Scholar] [CrossRef]
- Chhor, V.; Le Charpentier, T.; Lebon, S.; Ore, M.V.; Celador, I.L.; Josserand, J.; Degos, V.; Jacotot, E.; Hagberg, H.; Savman, K.; et al. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav. Immun 2013, 32, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Chiba, K. Diversity and plasticity of microglial cells in psychiatric and neurological disorders. Pharmacol. Ther. 2015, 154, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Fernandez-Suarez, D. Alternatively activated microglia and macrophages in the central nervous system. Prog. Neurobiol. 2015, 131, 65–86. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, P.; Sheng, W.S.; Hu, S.; Prasad, S.; Lokensgard, J.R. Differential Cytokine-Induced Responses of Polarized Microglia. Brain Sci. 2021, 11, 1482. [Google Scholar] [CrossRef] [PubMed]
- Lehnardt, S. Innate immunity and neuroinflammation in the CNS: The role of microglia in Toll-like receptor-mediated neuronal injury. Glia 2010, 58, 253–263. [Google Scholar] [CrossRef]
- Song, G.J.; Suk, K. Pharmacological Modulation of Functional Phenotypes of Microglia in Neurodegenerative Diseases. Front Aging Neurosci. 2017, 9, 139. [Google Scholar] [CrossRef] [Green Version]
- Subramaniam, S.R.; Federoff, H.J. Targeting Microglial Activation States as a Therapeutic Avenue in Parkinson’s Disease. Front. Aging Neurosci. 2017, 9, 176. [Google Scholar] [CrossRef]
- Diaz-Jimenez, D.; Kolb, J.P.; Cidlowski, J.A. Glucocorticoids as Regulators of Macrophage-Mediated Tissue Homeostasis. Front. Immunol. 2021, 12, 669891. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, H.; Sun, G.; Zhang, J.; Edwards, N.J.; Aronowski, J. Neuronal Interleukin-4 as a Modulator of Microglial Pathways and Ischemic Brain Damage. J. Neurosci. 2015, 35, 11281–11291. [Google Scholar] [CrossRef] [Green Version]
- Jurga, A.M.; Paleczna, M.; Kuter, K.Z. Overview of General and Discriminating Markers of Differential Microglia Phenotypes. Front. Cell Neurosci. 2020, 14, 198. [Google Scholar] [CrossRef]
- Gadani, S.P.; Cronk, J.C.; Norris, G.T.; Kipnis, J. IL-4 in the brain: A cytokine to remember. J. Immunol. 2012, 189, 4213–4219. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.F.; Mosser, D.M. Cutting edge: Biasing immune responses by directing antigen to macrophage Fc gamma receptors. J. Immunol. 2002, 168, 3697–3701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clynes, R.; Maizes, J.S.; Guinamard, R.; Ono, M.; Takai, T.; Ravetch, J.V. Modulation of immune complex-induced inflammation in vivo by the coordinate expression of activation and inhibitory Fc receptors. J. Exp. Med. 1999, 189, 179–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swisher, J.F.; Haddad, D.A.; McGrath, A.G.; Boekhoudt, G.H.; Feldman, G.M. IgG4 can induce an M2-like phenotype in human monocyte-derived macrophages through FcgammaRI. MAbs 2014, 6, 1377–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, J.P.; Zhang, X.; Frauwirth, K.A.; Mosser, D.M. Biochemical and functional characterization of three activated macrophage populations. J. Leukoc. Biol. 2006, 80, 1298–1307. [Google Scholar] [CrossRef] [Green Version]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Mecha, M.; Feliu, A.; Carrillo-Salinas, F.J.; Rueda-Zubiaurre, A.; Ortega-Gutierrez, S.; de Sola, R.G.; Guaza, C. Endocannabinoids drive the acquisition of an alternative phenotype in microglia. Brain Behav. Immun. 2015, 49, 233–245. [Google Scholar] [CrossRef]
- Chiu, I.M.; Morimoto, E.T.; Goodarzi, H.; Liao, J.T.; O’Keeffe, S.; Phatnani, H.P.; Muratet, M.; Carroll, M.C.; Levy, S.; Tavazoie, S.; et al. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 2013, 4, 385–401. [Google Scholar] [CrossRef] [Green Version]
- Grabert, K.; Michoel, T.; Karavolos, M.H.; Clohisey, S.; Baillie, J.K.; Stevens, M.P.; Freeman, T.C.; Summers, K.M.; McColl, B.W. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 2016, 19, 504–516. [Google Scholar] [CrossRef] [Green Version]
- Wes, P.D.; Holtman, I.R.; Boddeke, E.W.; Moller, T.; Eggen, B.J. Next generation transcriptomics and genomics elucidate biological complexity of microglia in health and disease. Glia 2016, 64, 197–213. [Google Scholar] [CrossRef]
- Kumar, A.; Alvarez-Croda, D.M.; Stoica, B.A.; Faden, A.I.; Loane, D.J. Microglial/Macrophage Polarization Dynamics following Traumatic Brain Injury. J. Neurotrauma 2016, 33, 1732–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, T.; Huang, Z.; Sun, X.; Zhu, X.; Zhou, L.; Li, M.; Cheng, B.; Liu, X.; He, C. Microglia Polarization with M1/M2 Phenotype Changes in rd1 Mouse Model of Retinal Degeneration. Front. Neuroanat. 2017, 11, 77. [Google Scholar] [CrossRef] [Green Version]
- Ferrante, C.J.; Leibovich, S.J. Regulation of Macrophage Polarization and Wound Healing. Adv. Wound Care 2012, 1, 10–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrante, C.J.; Pinhal-Enfield, G.; Elson, G.; Cronstein, B.N.; Hasko, G.; Outram, S.; Leibovich, S.J. The adenosine-dependent angiogenic switch of macrophages to an M2-like phenotype is independent of interleukin-4 receptor alpha (IL-4Ralpha) signaling. Inflammation 2013, 36, 921–931. [Google Scholar] [CrossRef] [PubMed]
- L’Episcopo, F.; Tirolo, C.; Serapide, M.F.; Caniglia, S.; Testa, N.; Leggio, L.; Vivarelli, S.; Iraci, N.; Pluchino, S.; Marchetti, B. Microglia Polarization, Gene-Environment Interactions and Wnt/beta-Catenin Signaling: Emerging Roles of Glia-Neuron and Glia-Stem/Neuroprogenitor Crosstalk for Dopaminergic Neurorestoration in Aged Parkinsonian Brain. Front. Aging Neurosci. 2018, 10, 12. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S. Microglial activation after ischaemic stroke. Stroke Vasc. Neurol. 2019, 4, 71–74. [Google Scholar] [CrossRef] [Green Version]
- Katsuno, M.; Sahashi, K.; Iguchi, Y.; Hashizume, A. Preclinical progression of neurodegenerative diseases. Nagoya J. Med. Sci. 2018, 80, 289–298. [Google Scholar] [CrossRef]
- Gerrits, E.; Heng, Y.; Boddeke, E.; Eggen, B.J.L. Transcriptional profiling of microglia; current state of the art and future perspectives. Glia 2020, 68, 740–755. [Google Scholar] [CrossRef] [Green Version]
- Eme-Scolan, E.; Dando, S.J. Tools and Approaches for Studying Microglia In vivo. Front. Immunol. 2020, 11, 583647. [Google Scholar] [CrossRef]
- Streit, W.J.; Sammons, N.W.; Kuhns, A.J.; Sparks, D.L. Dystrophic microglia in the aging human brain. Glia 2004, 45, 208–212. [Google Scholar] [CrossRef]
- Spittau, B. Aging Microglia-Phenotypes, Functions and Implications for Age-Related Neurodegenerative Diseases. Front. Aging Neurosci. 2017, 9, 194. [Google Scholar] [CrossRef] [PubMed]
- Koellhoffer, E.C.; McCullough, L.D.; Ritzel, R.M. Old Maids: Aging and Its Impact on Microglia Function. Int. J. Mol. Sci. 2017, 18, 769. [Google Scholar] [CrossRef] [PubMed]
- Schuitemaker, A.; van der Doef, T.F.; Boellaard, R.; van der Flier, W.M.; Yaqub, M.; Windhorst, A.D.; Barkhof, F.; Jonker, C.; Kloet, R.W.; Lammertsma, A.A.; et al. Microglial activation in healthy aging. Neurobiol. Aging 2012, 33, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Norden, D.M.; Godbout, J.P. Review: Microglia of the aged brain: Primed to be activated and resistant to regulation. Neuropathol. Appl. Neurobiol. 2013, 39, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Holtman, I.R.; Raj, D.D.; Miller, J.A.; Schaafsma, W.; Yin, Z.; Brouwer, N.; Wes, P.D.; Moller, T.; Orre, M.; Kamphuis, W.; et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: A co-expression meta-analysis. Acta Neuropathol. Commun. 2015, 3, 31. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Zhou, Y.; Wang, Y.; Li, D.; Liu, J.; Zhang, F. Age-Associated Dopaminergic Neuron Loss and Midbrain Glia Cell Phenotypic Polarization. Neuroscience 2019, 415, 89–96. [Google Scholar] [CrossRef]
- Lee, D.C.; Ruiz, C.R.; Lebson, L.; Selenica, M.L.; Rizer, J.; Hunt, J.B., Jr.; Rojiani, R.; Reid, P.; Kammath, S.; Nash, K.; et al. Aging enhances classical activation but mitigates alternative activation in the central nervous system. Neurobiol. Aging 2013, 34, 1610–1620. [Google Scholar] [CrossRef] [Green Version]
- Frank, M.G.; Barrientos, R.M.; Biedenkapp, J.C.; Rudy, J.W.; Watkins, L.R.; Maier, S.F. mRNA up-regulation of MHC II and pivotal pro-inflammatory genes in normal brain aging. Neurobiol. Aging 2006, 27, 717–722. [Google Scholar] [CrossRef]
- Godbout, J.P.; Chen, J.; Abraham, J.; Richwine, A.F.; Berg, B.M.; Kelley, K.W.; Johnson, R.W. Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J. 2005, 19, 1329–1331. [Google Scholar] [CrossRef]
- Letiembre, M.; Hao, W.; Liu, Y.; Walter, S.; Mihaljevic, I.; Rivest, S.; Hartmann, T.; Fassbender, K. Innate immune receptor expression in normal brain aging. Neuroscience 2007, 146, 248–254. [Google Scholar] [CrossRef]
- Hart, A.D.; Wyttenbach, A.; Perry, V.H.; Teeling, J.L. Age related changes in microglial phenotype vary between CNS regions: Grey versus white matter differences. Brain Behav. Immun. 2012, 26, 754–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanGuilder, H.D.; Bixler, G.V.; Brucklacher, R.M.; Farley, J.A.; Yan, H.; Warrington, J.P.; Sonntag, W.E.; Freeman, W.M. Concurrent hippocampal induction of MHC II pathway components and glial activation with advanced aging is not correlated with cognitive impairment. J. Neuroinflamm. 2011, 8, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoller, T.; Schneider, A.; Kleimeyer, C.; Masuda, T.; Potru, P.S.; Pfeifer, D.; Blank, T.; Prinz, M.; Spittau, B. Silencing of TGFbeta signalling in microglia results in impaired homeostasis. Nat. Commun. 2018, 9, 4011. [Google Scholar] [CrossRef] [PubMed]
- Tichauer, J.E.; Flores, B.; Soler, B.; Eugenin-von Bernhardi, L.; Ramirez, G.; von Bernhardi, R. Age-dependent changes on TGFbeta1 Smad3 pathway modify the pattern of microglial cell activation. Brain Behav. Immun. 2014, 37, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Lopes, K.P.; Snijders, G.J.L.; Humphrey, J.; Allan, A.; Sneeboer, M.A.M.; Navarro, E.; Schilder, B.M.; Vialle, R.A.; Parks, M.; Missall, R.; et al. Genetic analysis of the human microglial transcriptome across brain regions, aging and disease pathologies. Nat. Genet. 2022, 54, 4–17. [Google Scholar] [CrossRef]
- Bachstetter, A.D.; Morganti, J.M.; Jernberg, J.; Schlunk, A.; Mitchell, S.H.; Brewster, K.W.; Hudson, C.E.; Cole, M.J.; Harrison, J.K.; Bickford, P.C.; et al. Fractalkine and CX 3 CR1 regulate hippocampal neurogenesis in adult and aged rats. Neurobiol. Aging 2011, 32, 2030–2044. [Google Scholar] [CrossRef] [Green Version]
- Soreq, L.; Consortium, U.K.B.E.; North American Brain Expression, C.; Rose, J.; Soreq, E.; Hardy, J.; Trabzuni, D.; Cookson, M.R.; Smith, C.; Ryten, M.; et al. Major Shifts in Glial Regional Identity Are a Transcriptional Hallmark of Human Brain Aging. Cell Rep. 2017, 18, 557–570. [Google Scholar] [CrossRef]
- Orre, M.; Kamphuis, W.; Osborn, L.M.; Melief, J.; Kooijman, L.; Huitinga, I.; Klooster, J.; Bossers, K.; Hol, E.M. Acute isolation and transcriptome characterization of cortical astrocytes and microglia from young and aged mice. Neurobiol. Aging 2014, 35, 1–14. [Google Scholar] [CrossRef]
- Fonken, L.K.; Frank, M.G.; Kitt, M.M.; D’Angelo, H.M.; Norden, D.M.; Weber, M.D.; Barrientos, R.M.; Godbout, J.P.; Watkins, L.R.; Maier, S.F. The Alarmin HMGB1 Mediates Age-Induced Neuroinflammatory Priming. J. Neurosci. 2016, 36, 7946–7956. [Google Scholar] [CrossRef]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef] [Green Version]
- Schartz, N.D.; Tenner, A.J. The good, the bad, and the opportunities of the complement system in neurodegenerative disease. J. Neuroinflamm. 2020, 17, 354. [Google Scholar] [CrossRef] [PubMed]
- Stephan, A.H.; Madison, D.V.; Mateos, J.M.; Fraser, D.A.; Lovelett, E.A.; Coutellier, L.; Kim, L.; Tsai, H.H.; Huang, E.J.; Rowitch, D.H.; et al. A dramatic increase of C1q protein in the CNS during normal aging. J. Neurosci. 2013, 33, 13460–13474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichwald, J.; Danner, S.; Wiederhold, K.H.; Staufenbiel, M. Expression of complement system components during aging and amyloid deposition in APP transgenic mice. J. Neuroinflamm. 2009, 6, 35. [Google Scholar] [CrossRef] [Green Version]
- Rutar, M.; Valter, K.; Natoli, R.; Provis, J.M. Synthesis and propagation of complement C3 by microglia/monocytes in the aging retina. PLoS ONE 2014, 9, e93343. [Google Scholar] [CrossRef] [Green Version]
- Rupprecht, C.; Rupprecht, R.; Rammes, G. C1q, a small molecule with high impact on brain development: Putative role for aging processes and the occurrence of Alzheimer’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 2021, 271, 809–812. [Google Scholar] [CrossRef] [PubMed]
- Olmedillas Del Moral, M.; Asavapanumas, N.; Uzcategui, N.L.; Garaschuk, O. Healthy Brain Aging Modifies Microglial Calcium Signaling In Vivo. Int. J. Mol. Sci. 2019, 20, 589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandran, R.; Kumar, M.; Kesavan, L.; Jacob, R.S.; Gunasekaran, S.; Lakshmi, S.; Sadasivan, C.; Omkumar, R.V. Cellular calcium signaling in the aging brain. J. Chem. Neuroanat. 2019, 95, 95–114. [Google Scholar] [CrossRef]
- Eyo, U.B.; Miner, S.A.; Ahlers, K.E.; Wu, L.J.; Dailey, M.E. P2X7 receptor activation regulates microglial cell death during oxygen-glucose deprivation. Neuropharmacology 2013, 73, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Koizumi, S.; Shigemoto-Mogami, Y.; Nasu-Tada, K.; Shinozaki, Y.; Ohsawa, K.; Tsuda, M.; Joshi, B.V.; Jacobson, K.A.; Kohsaka, S.; Inoue, K. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature 2007, 446, 1091–1095. [Google Scholar] [CrossRef] [Green Version]
- Brawek, B.; Garaschuk, O. Microglial calcium signaling in the adult, aged and diseased brain. Cell Calcium 2013, 53, 159–169. [Google Scholar] [CrossRef]
- Farber, K.; Kettenmann, H. Functional role of calcium signals for microglial function. Glia 2006, 54, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Eichhoff, G.; Brawek, B.; Garaschuk, O. Microglial calcium signal acts as a rapid sensor of single neuron damage in vivo. Biochim. Biophys. Acta 2011, 1813, 1014–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brawek, B.; Liang, Y.; Savitska, D.; Li, K.; Fomin-Thunemann, N.; Kovalchuk, Y.; Zirdum, E.; Jakobsson, J.; Garaschuk, O. A new approach for ratiometric in vivo calcium imaging of microglia. Sci. Rep. 2017, 7, 6030. [Google Scholar] [CrossRef] [PubMed]
- Brawek, B.; Schwendele, B.; Riester, K.; Kohsaka, S.; Lerdkrai, C.; Liang, Y.; Garaschuk, O. Impairment of in vivo calcium signaling in amyloid plaque-associated microglia. Acta Neuropathol. 2014, 127, 495–505. [Google Scholar] [CrossRef]
- Pozner, A.; Xu, B.; Palumbos, S.; Gee, J.M.; Tvrdik, P.; Capecchi, M.R. Intracellular calcium dynamics in cortical microglia responding to focal laser injury in the PC::G5-tdT reporter mouse. Front. Mol. Neurosci. 2015, 8, 12. [Google Scholar] [CrossRef]
- Lan, X.; Han, X.; Li, Q.; Yang, Q.W.; Wang, J. Modulators of microglial activation and polarization after intracerebral haemorrhage. Nat. Rev. Neurol. 2017, 13, 420–433. [Google Scholar] [CrossRef] [Green Version]
- Montano, A.; Hanley, D.F.; Hemphill, J.C., 3rd. Hemorrhagic stroke. Handb. Clin. Neurol. 2021, 176, 229–248. [Google Scholar] [CrossRef]
- Unnithan, A.K.A.; Mehta, P. Hemorrhagic Stroke. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Hemphill, J.C., 3rd; Bonovich, D.C.; Besmertis, L.; Manley, G.T.; Johnston, S.C. The ICH score: A simple, reliable grading scale for intracerebral hemorrhage. Stroke 2001, 32, 891–897. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zeng, L.; Hu, Z. Progressing haemorrhagic stroke: Categories, causes, mechanisms and managements. J. Neurol. 2014, 261, 2061–2078. [Google Scholar] [CrossRef]
- Aronowski, J.; Zhao, X. Molecular pathophysiology of cerebral hemorrhage: Secondary brain injury. Stroke 2011, 42, 1781–1786. [Google Scholar] [CrossRef]
- Thabet, A.M.; Kottapally, M.; Hemphill, J.C., 3rd. Management of intracerebral hemorrhage. Handb. Clin. Neurol. 2017, 140, 177–194. [Google Scholar] [CrossRef] [PubMed]
- Hemphill, J.C., 3rd; Greenberg, S.M.; Anderson, C.S.; Becker, K.; Bendok, B.R.; Cushman, M.; Fung, G.L.; Goldstein, J.N.; Macdonald, R.L.; Mitchell, P.H.; et al. Guidelines for the Management of Spontaneous Intracerebral Hemorrhage: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke 2015, 46, 2032–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tschoe, C.; Bushnell, C.D.; Duncan, P.W.; Alexander-Miller, M.A.; Wolfe, S.Q. Neuroinflammation after Intracerebral Hemorrhage and Potential Therapeutic Targets. J. Stroke 2020, 22, 29–46. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.A.; Sansing, L.H. Microglial responses after ischemic stroke and intracerebral hemorrhage. Clin. Dev. Immunol 2013, 2013, 746068. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, Y.; Wang, J.; Anne Stetler, R.; Yang, Q.W. Inflammation in intracerebral hemorrhage: From mechanisms to clinical translation. Prog. Neurobiol. 2014, 115, 25–44. [Google Scholar] [CrossRef] [PubMed]
- Babu, R.; Bagley, J.H.; Di, C.; Friedman, A.H.; Adamson, C. Thrombin and hemin as central factors in the mechanisms of intracerebral hemorrhage-induced secondary brain injury and as potential targets for intervention. Neurosurg. Focus 2012, 32, E8. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Rogove, A.D.; Tsirka, A.E.; Tsirka, S.E. Protective role of tuftsin fragment 1-3 in an animal model of intracerebral hemorrhage. Ann. Neurol. 2003, 54, 655–664. [Google Scholar] [CrossRef]
- Wang, J.; Tsirka, S.E. Tuftsin fragment 1-3 is beneficial when delivered after the induction of intracerebral hemorrhage. Stroke 2005, 36, 613–618. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Dore, S. Inflammation after intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 2007, 27, 894–908. [Google Scholar] [CrossRef] [Green Version]
- Xue, M.; Del Bigio, M.R. Intracerebral injection of autologous whole blood in rats: Time course of inflammation and cell death. Neurosci. Lett. 2000, 283, 230–232. [Google Scholar] [CrossRef]
- Xue, M.; Del Bigio, M.R. Comparison of brain cell death and inflammatory reaction in three models of intracerebral hemorrhage in adult rats. J. Stroke Cerebrovasc. Dis. 2003, 12, 152–159. [Google Scholar] [CrossRef]
- Gong, C.; Hoff, J.T.; Keep, R.F. Acute inflammatory reaction following experimental intracerebral hemorrhage in rat. Brain Res 2000, 871, 57–65. [Google Scholar] [CrossRef]
- Robinson, S.R.; Dang, T.N.; Dringen, R.; Bishop, G.M. Hemin toxicity: A preventable source of brain damage following hemorrhagic stroke. Redox Rep. 2009, 14, 228–235. [Google Scholar] [CrossRef]
- Righy, C.; Turon, R.; Freitas, G.; Japiassu, A.M.; Faria Neto, H.C.C.; Bozza, M.; Oliveira, M.F.; Bozza, F.A. Hemoglobin metabolism by-products are associated with an inflammatory response in patients with hemorrhagic stroke. Rev. Bras. Ter. Intensiv. 2018, 30, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lan, X.; Han, X.; Durham, F.; Wan, J.; Weiland, A.; Koehler, R.C.; Wang, J. Microglia-derived interleukin-10 accelerates post-intracerebral hemorrhage hematoma clearance by regulating CD36. Brain Behav. Immun. 2021, 94, 437–457. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Chen, J.; Lin, S.; Wang, P.; Wang, Y.; Xiong, X.; Yang, Q. CD36-mediated hematoma absorption following intracerebral hemorrhage: Negative regulation by TLR4 signaling. J. Immunol. 2014, 192, 5984–5992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Zhong, S.; Liu, Y.; Shen, H.; Yuan, B. Scavenger receptor SRA attenuates microglia activation and protects neuroinflammatory injury in intracerebral hemorrhage. J. Neuroimmunol. 2015, 278, 232–238. [Google Scholar] [CrossRef]
- Lin, S.; Yin, Q.; Zhong, Q.; Lv, F.L.; Zhou, Y.; Li, J.Q.; Wang, J.Z.; Su, B.Y.; Yang, Q.W. Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. J. Neuroinflamm. 2012, 9, 46. [Google Scholar] [CrossRef] [Green Version]
- Teng, W.; Wang, L.; Xue, W.; Guan, C. Activation of TLR4-mediated NFkappaB signaling in hemorrhagic brain in rats. Mediat. Inflamm. 2009, 2009, 473276. [Google Scholar] [CrossRef] [Green Version]
- Matz, P.; Turner, C.; Weinstein, P.R.; Massa, S.M.; Panter, S.S.; Sharp, F.R. Heme-oxygenase-1 induction in glia throughout rat brain following experimental subarachnoid hemorrhage. Brain Res. 1996, 713, 211–222. [Google Scholar] [CrossRef]
- Li, Q.Q.; Li, L.J.; Wang, X.Y.; Sun, Y.Y.; Wu, J. Research Progress in Understanding the Relationship Between Heme Oxygenase-1 and Intracerebral Hemorrhage. Front. Neurol. 2018, 9, 682. [Google Scholar] [CrossRef] [PubMed]
- Nakaso, K.; Kitayama, M.; Mizuta, E.; Fukuda, H.; Ishii, T.; Nakashima, K.; Yamada, K. Co-induction of heme oxygenase-1 and peroxiredoxin I in astrocytes and microglia around hemorrhagic region in the rat brain. Neurosci. Lett. 2000, 293, 49–52. [Google Scholar] [CrossRef]
- Zhang, Z.; Song, Y.; Zhang, Z.; Li, D.; Zhu, H.; Liang, R.; Gu, Y.; Pang, Y.; Qi, J.; Wu, H.; et al. Distinct role of heme oxygenase-1 in early- and late-stage intracerebral hemorrhage in 12-month-old mice. J. Cereb. Blood Flow Metab. 2017, 37, 25–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen-Roetling, J.; Song, W.; Schipper, H.M.; Regan, C.S.; Regan, R.F. Astrocyte overexpression of heme oxygenase-1 improves outcome after intracerebral hemorrhage. Stroke 2015, 46, 1093–1098. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Wu, T.; Xu, X.; Wang, J.; Wang, J. Iron toxicity in mice with collagenase-induced intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 2011, 31, 1243–1250. [Google Scholar] [CrossRef]
- Zhao, F.; Hua, Y.; He, Y.; Keep, R.F.; Xi, G. Minocycline-induced attenuation of iron overload and brain injury after experimental intracerebral hemorrhage. Stroke 2011, 42, 3587–3593. [Google Scholar] [CrossRef] [Green Version]
- Kasuya, H.; Shimizu, T.; Takakura, K. Thrombin activity in CSF after SAH is correlated with the degree of SAH the persistence of subarachnoid clot and the development of vasospasm. Acta Neurochir. 1998, 140, 579–584. [Google Scholar] [CrossRef]
- Cheng, Y.; Xi, G.; Jin, H.; Keep, R.F.; Feng, J.; Hua, Y. Thrombin-induced cerebral hemorrhage: Role of protease-activated receptor-1. Transl. Stroke Res. 2014, 5, 472–475. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhu, Z.; Gao, S.; Zhang, L.; Cheng, X.; Li, S.; Li, M. Inhibition of fibrin formation reduces neuroinflammation and improves long-term outcome after intracerebral hemorrhage. Int. Immunopharmacol. 2019, 72, 473–478. [Google Scholar] [CrossRef]
- Carreno-Muller, E.; Herrera, A.J.; de Pablos, R.M.; Tomas-Camardiel, M.; Venero, J.L.; Cano, J.; Machado, A. Thrombin induces in vivo degeneration of nigral dopaminergic neurones along with the activation of microglia. J. Neurochem. 2003, 84, 1201–1214. [Google Scholar] [CrossRef]
- Choi, S.H.; Joe, E.H.; Kim, S.U.; Jin, B.K. Thrombin-induced microglial activation produces degeneration of nigral dopaminergic neurons in vivo. J. Neurosci. 2003, 23, 5877–5886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Ma, R.; Sun, S.; Wei, G.; Fang, Y.; Liu, R.; Li, G. JAK2-STAT3 signaling pathway mediates thrombin-induced proinflammatory actions of microglia in vitro. J. Neuroimmunol. 2008, 204, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.; Pyo, H.; Jou, I.; Joe, E. Thrombin induces NO release from cultured rat microglia via protein kinase C, mitogen-activated protein kinase, and NF-kappa B. J. Biol. Chem. 2000, 275, 29955–29959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suo, Z.; Wu, M.; Ameenuddin, S.; Anderson, H.E.; Zoloty, J.E.; Citron, B.A.; Andrade-Gordon, P.; Festoff, B.W. Participation of protease-activated receptor-1 in thrombin-induced microglial activation. J. Neurochem. 2002, 80, 655–666. [Google Scholar] [CrossRef]
- Fujimoto, S.; Katsuki, H.; Ohnishi, M.; Takagi, M.; Kume, T.; Akaike, A. Thrombin induces striatal neurotoxicity depending on mitogen-activated protein kinase pathways in vivo. Neuroscience 2007, 144, 694–701. [Google Scholar] [CrossRef] [Green Version]
- Ohnishi, M.; Katsuki, H.; Fujimoto, S.; Takagi, M.; Kume, T.; Akaike, A. Involvement of thrombin and mitogen-activated protein kinase pathways in hemorrhagic brain injury. Exp. Neurol. 2007, 206, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Coughlin, S.R. Thrombin signalling and protease-activated receptors. Nature 2000, 407, 258–264. [Google Scholar] [CrossRef]
- Nakano, T.; Nakamura, Y.; Irie, K.; Okano, S.; Morimoto, M.; Yamashita, Y.; Kozako, T.; Hayashi, T.; Honda, S.I.; Matsuo, K.; et al. Antithrombin gamma attenuates macrophage/microglial activation and brain damage after transient focal cerebral ischemia in mice. Life Sci. 2020, 252, 117665. [Google Scholar] [CrossRef]
- Wan, S.; Cheng, Y.; Jin, H.; Guo, D.; Hua, Y.; Keep, R.F.; Xi, G. Microglia Activation and Polarization After Intracerebral Hemorrhage in Mice: The Role of Protease-Activated Receptor-1. Transl. Stroke Res. 2016, 7, 478–487. [Google Scholar] [CrossRef] [Green Version]
- Moller, T.; Hanisch, U.K.; Ransom, B.R. Thrombin-induced activation of cultured rodent microglia. J. Neurochem. 2000, 75, 1539–1547. [Google Scholar] [CrossRef] [Green Version]
- Ducruet, A.F.; Zacharia, B.E.; Hickman, Z.L.; Grobelny, B.T.; Yeh, M.L.; Sosunov, S.A.; Connolly, E.S., Jr. The complement cascade as a therapeutic target in intracerebral hemorrhage. Exp. Neurol. 2009, 219, 398–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Song, L.; Pedersen, D.V.; Li, A.; Lambris, J.D.; Andersen, G.R.; Mollnes, T.E.; Ma, Y.J.; Garred, P. Soluble collectin-12 mediates C3-independent docking of properdin that activates the alternative pathway of complement. Elife 2020, 9, e60908. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Nakamura, T.; Hua, Y.; Keep, R.F.; Younger, J.G.; Hoff, J.T.; Xi, G. Intracerebral hemorrhage in complement C3-deficient mice. Acta Neurochir. Suppl. 2006, 96, 227–231. [Google Scholar] [CrossRef]
- Huber-Lang, M.; Sarma, J.V.; Zetoune, F.S.; Rittirsch, D.; Neff, T.A.; McGuire, S.R.; Lambris, J.D.; Warner, R.L.; Flierl, M.A.; Hoesel, L.M.; et al. Generation of C5a in the absence of C3: A new complement activation pathway. Nat. Med. 2006, 12, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Fan, R.M.; Chen, J.L.; Wang, C.M.; Zeng, Y.C.; Han, C.; Jiao, S.; Xia, X.P.; Chen, W.; Yao, S.T. Neuroprotective effects of argatroban and C5a receptor antagonist (PMX53) following intracerebral haemorrhage. Clin. Exp. Immunol. 2014, 175, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.V.; Wong, K.C.G. Microglial activation and polarization after subarachnoid hemorrhage. Neuroimmunol. Neuroinflammation 2019, 6, e60908. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Zhou, W.; Yan, Z.; Qu, M.; Bu, X. Toll-like Receptor 4 (TLR4) is Associated with Cerebral Vasospasm and Delayed Cerebral Ischemia in Aneurysmal Subarachnoid Hemorrhage. Neurol. Med. Chir. 2015, 55, 878–884. [Google Scholar] [CrossRef] [Green Version]
- Sansing, L.H.; Harris, T.H.; Welsh, F.A.; Kasner, S.E.; Hunter, C.A.; Kariko, K. Toll-like receptor 4 contributes to poor outcome after intracerebral hemorrhage. Ann. Neurol. 2011, 70, 646–656. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Yanez, M.; Brea, D.; Arias, S.; Blanco, M.; Pumar, J.M.; Castillo, J.; Sobrino, T. Increased expression of Toll-like receptors 2 and 4 is associated with poor outcome in intracerebral hemorrhage. J. Neuroimmunol. 2012, 247, 75–80. [Google Scholar] [CrossRef]
- Murakami, K.; Koide, M.; Dumont, T.M.; Russell, S.R.; Tranmer, B.I.; Wellman, G.C. Subarachnoid Hemorrhage Induces Gliosis and Increased Expression of the Pro-inflammatory Cytokine High Mobility Group Box 1 Protein. Transl. Stroke Res. 2011, 2, 72–79. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.F.; Wan, J.; Li, Q.; Renfroe, S.C.; Heller, N.M.; Wang, J. Alternative activation-skewed microglia/macrophages promote hematoma resolution in experimental intracerebral hemorrhage. Neurobiol. Dis. 2017, 103, 54–69. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ding, S.; Huang, W.; Hu, J.; Huang, S.; Zhang, Y.; Zhuge, Q. Interleukin-4 Ameliorates the Functional Recovery of Intracerebral Hemorrhage Through the Alternative Activation of Microglia/Macrophage. Front. Neurosci. 2016, 10, 61. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Sun, G.; Zhang, J.; Strong, R.; Song, W.; Gonzales, N.; Grotta, J.C.; Aronowski, J. Hematoma resolution as a target for intracerebral hemorrhage treatment: Role for peroxisome proliferator-activated receptor gamma in microglia/macrophages. Ann. Neurol. 2007, 61, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.A.; Chang, C.F.; Goods, B.A.; Hammond, M.D.; Mac Grory, B.; Ai, Y.; Steinschneider, A.F.; Renfroe, S.C.; Askenase, M.H.; McCullough, L.D.; et al. TGF-beta1 modulates microglial phenotype and promotes recovery after intracerebral hemorrhage. J. Clin. Investig. 2017, 127, 280–292. [Google Scholar] [CrossRef] [Green Version]
- Leclerc, J.L.; Lampert, A.S.; Loyola Amador, C.; Schlakman, B.; Vasilopoulos, T.; Svendsen, P.; Moestrup, S.K.; Dore, S. The absence of the CD163 receptor has distinct temporal influences on intracerebral hemorrhage outcomes. J. Cereb. Blood Flow Metab. 2018, 38, 262–273. [Google Scholar] [CrossRef] [Green Version]
- Bi, R.; Fang, Z.; You, M.; He, Q.; Hu, B. Microglia Phenotype and Intracerebral Hemorrhage: A Balance of Yin and Yang. Front Cell Neurosci. 2021, 15, 765205. [Google Scholar] [CrossRef]
- Kanazawa, M.; Ninomiya, I.; Hatakeyama, M.; Takahashi, T.; Shimohata, T. Microglia and Monocytes/Macrophages Polarization Reveal Novel Therapeutic Mechanism against Stroke. Int. J. Mol. Sci. 2017, 18, 2135. [Google Scholar] [CrossRef]
- Li, R.; Liu, W.; Yin, J.; Chen, Y.; Guo, S.; Fan, H.; Li, X.; Zhang, X.; He, X.; Duan, C. TSG-6 attenuates inflammation-induced brain injury via modulation of microglial polarization in SAH rats through the SOCS3/STAT3 pathway. J. Neuroinflamm. 2018, 15, 231. [Google Scholar] [CrossRef]
- Lin, L.; Yihao, T.; Zhou, F.; Yin, N.; Qiang, T.; Haowen, Z.; Qianwei, C.; Jun, T.; Yuan, Z.; Gang, Z.; et al. Inflammatory Regulation by Driving Microglial M2 Polarization: Neuroprotective Effects of Cannabinoid Receptor-2 Activation in Intracerebral Hemorrhage. Front. Immunol. 2017, 8, 112. [Google Scholar] [CrossRef] [Green Version]
- Lan, X.; Han, X.; Li, Q.; Li, Q.; Gao, Y.; Cheng, T.; Wan, J.; Zhu, W.; Wang, J. Pinocembrin protects hemorrhagic brain primarily by inhibiting toll-like receptor 4 and reducing M1 phenotype microglia. Brain Behav. Immun. 2017, 61, 326–339. [Google Scholar] [CrossRef] [Green Version]
- Liesz, A.; Middelhoff, M.; Zhou, W.; Karcher, S.; Illanes, S.; Veltkamp, R. Comparison of humoral neuroinflammation and adhesion molecule expression in two models of experimental intracerebral hemorrhage. Exp. Transl. Stroke Med. 2011, 3, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manaenko, A.; Chen, H.; Zhang, J.H.; Tang, J. Comparison of different preclinical models of intracerebral hemorrhage. Acta Neurochir. Suppl. 2011, 111, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, A.; Iliffe, S. Alzheimer’s disease. BMJ 2009, 338, b158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorszewska, J.; Prendecki, M.; Oczkowska, A.; Dezor, M.; Kozubski, W. Molecular Basis of Familial and Sporadic Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 952–963. [Google Scholar] [CrossRef]
- Crews, L.; Masliah, E. Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum. Mol. Genet. 2010, 19, R12–R20. [Google Scholar] [CrossRef]
- Wilquet, V.; De Strooper, B. Amyloid-beta precursor protein processing in neurodegeneration. Curr. Opin. Neurobiol. 2004, 14, 582–588. [Google Scholar] [CrossRef]
- Olsson, F.; Schmidt, S.; Althoff, V.; Munter, L.M.; Jin, S.; Rosqvist, S.; Lendahl, U.; Multhaup, G.; Lundkvist, J. Characterization of intermediate steps in amyloid beta (Abeta) production under near-native conditions. J. Biol. Chem. 2014, 289, 1540–1550. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Bertram, L. Alzheimer’s disease genetics current status and future perspectives. Int. Rev. Neurobiol. 2009, 84, 167–184. [Google Scholar] [CrossRef]
- Lanoiselee, H.M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef] [Green Version]
- Safieh, M.; Korczyn, A.D.; Michaelson, D.M. ApoE4: An emerging therapeutic target for Alzheimer’s disease. BMC Med. 2019, 17, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makin, S. The amyloid hypothesis on trial. Nature 2018, 559, S4–S7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, B.; Stein, P.; Cavazzoni, P. Approval of Aducanumab for Alzheimer Disease-The FDA’s Perspective. JAMA Intern Med 2021, 181, 1276–1278. [Google Scholar] [CrossRef]
- Tampi, R.R.; Forester, B.P.; Agronin, M. Aducanumab: Evidence from clinical trial data and controversies. Drugs Context 2021, 10, 1–9. [Google Scholar] [CrossRef]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gratuze, M.; Leyns, C.E.G.; Holtzman, D.M. New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 66. [Google Scholar] [CrossRef] [Green Version]
- Hamelin, L.; Lagarde, J.; Dorothee, G.; Leroy, C.; Labit, M.; Comley, R.A.; de Souza, L.C.; Corne, H.; Dauphinot, L.; Bertoux, M.; et al. Early and protective microglial activation in Alzheimer’s disease: A prospective study using 18F-DPA-714 PET imaging. Brain 2016, 139, 1252–1264. [Google Scholar] [CrossRef] [Green Version]
- Condello, C.; Yuan, P.; Schain, A.; Grutzendler, J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Abeta42 hotspots around plaques. Nat. Commun. 2015, 6, 6176. [Google Scholar] [CrossRef] [Green Version]
- Feng, W.; Zhang, Y.; Wang, Z.; Xu, H.; Wu, T.; Marshall, C.; Gao, J.; Xiao, M. Microglia prevent beta-amyloid plaque formation in the early stage of an Alzheimer’s disease mouse model with suppression of glymphatic clearance. Alzheimers Res. Ther. 2020, 12, 125. [Google Scholar] [CrossRef]
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 2016, 213, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Merlo, S.; Spampinato, S.F.; Beneventano, M.; Sortino, M.A. The contribution of microglia to early synaptic compensatory responses that precede beta-amyloid-induced neuronal death. Sci. Rep. 2018, 8, 7297. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Brooks, D.J.; Okello, A.; Edison, P. An early and late peak in microglial activation in Alzheimer’s disease trajectory. Brain 2017, 140, 792–803. [Google Scholar] [CrossRef] [PubMed]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loving, B.A.; Bruce, K.D. Lipid and Lipoprotein Metabolism in Microglia. Front. Physiol. 2020, 11, 393. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Landreth, G.E. The role of microglia in amyloid clearance from the AD brain. J. Neural. Transm. 2010, 117, 949–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Ye, R.D. Microglial Abeta receptors in Alzheimer’s disease. Cell Mol. Neurobiol. 2015, 35, 71–83. [Google Scholar] [CrossRef]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Deane, R.; Singh, I.; Sagare, A.P.; Bell, R.D.; Ross, N.T.; LaRue, B.; Love, R.; Perry, S.; Paquette, N.; Deane, R.J.; et al. A multimodal RAGE-specific inhibitor reduces amyloid beta-mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Investig. 2012, 122, 1377–1392. [Google Scholar] [CrossRef] [Green Version]
- Hanslik, K.L.; Ulland, T.K. The Role of Microglia and the Nlrp3 Inflammasome in Alzheimer’s Disease. Front. Neurol. 2020, 11, 570711. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Ostapchenko, V.G.; Chen, M.; Guzman, M.S.; Xie, Y.F.; Lavine, N.; Fan, J.; Beraldo, F.H.; Martyn, A.C.; Belrose, J.C.; Mori, Y.; et al. The Transient Receptor Potential Melastatin 2 (TRPM2) Channel Contributes to beta-Amyloid Oligomer-Related Neurotoxicity and Memory Impairment. J. Neurosci. 2015, 35, 15157–15169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alawieyah Syed Mortadza, S.; Sim, J.A.; Neubrand, V.E.; Jiang, L.H. A critical role of TRPM2 channel in Abeta42 -induced microglial activation and generation of tumor necros.sis factor-alpha. Glia 2018, 66, 562–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aminzadeh, M.; Roghani, M.; Sarfallah, A.; Riazi, G.H. TRPM2 dependence of ROS-induced NLRP3 activation in Alzheimer’s disease. Int. Immunopharmacol. 2018, 54, 78–85. [Google Scholar] [CrossRef]
- Hu, Y.; Fryatt, G.L.; Ghorbani, M.; Obst, J.; Menassa, D.A.; Martin-Estebane, M.; Muntslag, T.A.O.; Olmos-Alonso, A.; Guerrero-Carrasco, M.; Thomas, D.; et al. Replicative senescence dictates the emergence of disease-associated microglia and contributes to Abeta pathology. Cell Rep. 2021, 35, 109228. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kiyota, T.; Walsh, S.M.; Liu, J.; Kipnis, J.; Ikezu, T. Cytokine-mediated inhibition of fibrillar amyloid-beta peptide degradation by human mononuclear phagocytes. J. Immunol. 2008, 181, 3877–3886. [Google Scholar] [CrossRef]
- Michelucci, A.; Heurtaux, T.; Grandbarbe, L.; Morga, E.; Heuschling, P. Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: Effects of oligomeric and fibrillar amyloid-beta. J. Neuroimmunol. 2009, 210, 3–12. [Google Scholar] [CrossRef]
- Vodovotz, Y.; Lucia, M.S.; Flanders, K.C.; Chesler, L.; Xie, Q.W.; Smith, T.W.; Weidner, J.; Mumford, R.; Webber, R.; Nathan, C.; et al. Inducible nitric oxide synthase in tangle-bearing neurons of patients with Alzheimer’s disease. J. Exp. Med. 1996, 184, 1425–1433. [Google Scholar] [CrossRef]
- Goodwin, J.L.; Uemura, E.; Cunnick, J.E. Microglial release of nitric oxide by the synergistic action of beta-amyloid and IFN-gamma. Brain Res. 1995, 692, 207–214. [Google Scholar] [CrossRef]
- Kummer, M.P.; Hermes, M.; Delekarte, A.; Hammerschmidt, T.; Kumar, S.; Terwel, D.; Walter, J.; Pape, H.C.; Konig, S.; Roeber, S.; et al. Nitration of tyrosine 10 critically enhances amyloid beta aggregation and plaque formation. Neuron 2011, 71, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Kummer, M.P.; Vogl, T.; Axt, D.; Griep, A.; Vieira-Saecker, A.; Jessen, F.; Gelpi, E.; Roth, J.; Heneka, M.T. Mrp14 deficiency ameliorates amyloid beta burden by increasing microglial phagocytosis and modulation of amyloid precursor protein processing. J. Neurosci. 2012, 32, 17824–17829. [Google Scholar] [CrossRef] [PubMed]
- Taupenot, L.; Ciesielski-Treska, J.; Ulrich, G.; Chasserot-Golaz, S.; Aunis, D.; Bader, M.F. Chromogranin A triggers a phenotypic transformation and the generation of nitric oxide in brain microglial cells. Neuroscience 1996, 72, 377–389. [Google Scholar] [CrossRef]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucin, K.M.; O’Brien, C.E.; Bieri, G.; Czirr, E.; Mosher, K.I.; Abbey, R.J.; Mastroeni, D.F.; Rogers, J.; Spencer, B.; Masliah, E.; et al. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron 2013, 79, 873–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef] [PubMed]
- Caldeira, C.; Cunha, C.; Vaz, A.R.; Falcao, A.S.; Barateiro, A.; Seixas, E.; Fernandes, A.; Brites, D. Key Aging-Associated Alterations in Primary Microglia Response to Beta-Amyloid Stimulation. Front. Aging Neurosci. 2017, 9, 277. [Google Scholar] [CrossRef] [PubMed]
- Daria, A.; Colombo, A.; Llovera, G.; Hampel, H.; Willem, M.; Liesz, A.; Haass, C.; Tahirovic, S. Young microglia restore amyloid plaque clearance of aged microglia. EMBO J. 2017, 36, 583–603. [Google Scholar] [CrossRef] [Green Version]
- Shibuya, Y.; Kumar, K.K.; Mader, M.M.; Yoo, Y.; Ayala, L.A.; Zhou, M.; Mohr, M.A.; Neumayer, G.; Kumar, I.; Yamamoto, R.; et al. Treatment of a genetic brain disease by CNS-wide microglia replacement. Sci. Transl. Med. 2022, 14, eabl9945. [Google Scholar] [CrossRef]
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front. Aging Neurosci. 2019, 11, 204. [Google Scholar] [CrossRef] [Green Version]
- Sheffield, L.G.; Marquis, J.G.; Berman, N.E. Regional distribution of cortical microglia parallels that of neurofibrillary tangles in Alzheimer’s disease. Neurosci. Lett. 2000, 285, 165–168. [Google Scholar] [CrossRef]
- Bolos, M.; Llorens-Martin, M.; Jurado-Arjona, J.; Hernandez, F.; Rabano, A.; Avila, J. Direct Evidence of Internalization of Tau by Microglia In Vitro and In Vivo. J. Alzheimers Dis. 2016, 50, 77–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kugler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopp, S.C.; Lin, Y.; Oakley, D.; Roe, A.D.; DeVos, S.L.; Hanlon, D.; Hyman, B.T. The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, R.; Zubko, O.; Bradley, R.; Harper, E.; Pank, L.; O’Brien, J.; Fox, C.; Tabet, N.; Livingston, G.; Bentham, P.; et al. Minocycline at 2 Different Dosages vs Placebo for Patients With Mild Alzheimer Disease: A Randomized Clinical Trial. JAMA Neurol. 2020, 77, 164–174. [Google Scholar] [CrossRef]
- Garwood, C.J.; Cooper, J.D.; Hanger, D.P.; Noble, W. Anti-inflammatory impact of minocycline in a mouse model of tauopathy. Front. Psychiatry 2010, 1, 136. [Google Scholar] [CrossRef] [Green Version]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018, 562, 578–582. [Google Scholar] [CrossRef]
- Ruan, Z.; Pathak, D.; Venkatesan Kalavai, S.; Yoshii-Kitahara, A.; Muraoka, S.; Bhatt, N.; Takamatsu-Yukawa, K.; Hu, J.; Wang, Y.; Hersh, S.; et al. Alzheimer’s disease brain-derived extracellular vesicles spread tau pathology in interneurons. Brain 2021, 144, 288–309. [Google Scholar] [CrossRef]
- Ramirez, A.I.; de Hoz, R.; Salobrar-Garcia, E.; Salazar, J.J.; Rojas, B.; Ajoy, D.; Lopez-Cuenca, I.; Rojas, P.; Trivino, A.; Ramirez, J.M. The Role of Microglia in Retinal Neurodegeneration: Alzheimer’s Disease, Parkinson, and Glaucoma. Front. Aging Neurosci. 2017, 9, 214. [Google Scholar] [CrossRef] [Green Version]
- Bhaskar, K.; Konerth, M.; Kokiko-Cochran, O.N.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T. Regulation of tau pathology by the microglial fractalkine receptor. Neuron 2010, 68, 19–31. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl Med. 2015, 3, 136. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Morimoto, H.; Ohgidani, M.; Ueki, T. Modulation of inflammatory responses by fractalkine signaling in microglia. PLoS ONE 2021, 16, e0252118. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Harrison, J.; Deaton, C.A.; Johnson, G.V.W. Tau Clearance Mechanisms. Adv. Exp. Med. Biol. 2019, 1184, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Bolos, M.; Llorens-Martin, M.; Perea, J.R.; Jurado-Arjona, J.; Rabano, A.; Hernandez, F.; Avila, J. Absence of CX3CR1 impairs the internalization of Tau by microglia. Mol. Neurodegener. 2017, 12, 59. [Google Scholar] [CrossRef] [Green Version]
- Chidambaram, H.; Das, R.; Chinnathambi, S. Interaction of Tau with the chemokine receptor, CX3CR1 and its effect on microglial activation, migration and proliferation. Cell Biosci. 2020, 10, 109. [Google Scholar] [CrossRef]
- Davies, D.S.; Ma, J.; Jegathees, T.; Goldsbury, C. Microglia show altered morphology and reduced arborization in human brain during aging and Alzheimer’s disease. Brain Pathol. 2017, 27, 795–808. [Google Scholar] [CrossRef]
- Plescher, M.; Seifert, G.; Hansen, J.N.; Bedner, P.; Steinhauser, C.; Halle, A. Plaque-dependent morphological and electrophysiological heterogeneity of microglia in an Alzheimer’s disease mouse model. Glia 2018, 66, 1464–1480. [Google Scholar] [CrossRef]
- Franco-Bocanegra, D.K.; Gourari, Y.; McAuley, C.; Chatelet, D.S.; Johnston, D.A.; Nicoll, J.A.R.; Boche, D. Microglial morphology in Alzheimer’s disease and after Abeta immunotherapy. Sci. Rep. 2021, 11, 15955. [Google Scholar] [CrossRef]
- Wierzba-Bobrowicz, T.; Gwiazda, E.; Kosno-Kruszewska, E.; Lewandowska, E.; Lechowicz, W.; Bertrand, E.; Szpak, G.M.; Schmidt-Sidor, B. Morphological analysis of active microglia--rod and ramified microglia in human brains affected by some neurological diseases (SSPE, Alzheimer’s disease and Wilson’s disease). Folia Neuropathol. 2002, 40, 125–131. [Google Scholar]
- Suzumura, A.; Marunouchi, T.; Yamamoto, H. Morphological transformation of microglia in vitro. Brain Res. 1991, 545, 301–306. [Google Scholar] [CrossRef]
- Tam, W.Y.; Au, N.P.; Ma, C.H. The association between laminin and microglial morphology in vitro. Sci. Rep. 2016, 6, 28580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisht, K.; Sharma, K.; Lacoste, B.; Tremblay, M.E. Dark microglia: Why are they dark? Commun. Integr. Biol. 2016, 9, e1230575. [Google Scholar] [CrossRef] [PubMed]
- Bisht, K.; Sharma, K.P.; Lecours, C.; Sanchez, M.G.; El Hajj, H.; Milior, G.; Olmos-Alonso, A.; Gomez-Nicola, D.; Luheshi, G.; Vallieres, L.; et al. Dark microglia: A new phenotype predominantly associated with pathological states. Glia 2016, 64, 826–839. [Google Scholar] [CrossRef] [Green Version]
- Elgayar, S.A.M.; Abdel-Hafez, A.A.M.; Gomaa, A.M.S.; Elsherif, R. Vulnerability of glia and vessels of rat substantia nigra in rotenone Parkinson model. Ultrastruct. Pathol. 2018, 42, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Uranova, N.A.; Vikhreva, O.V.; Rakhmanova, V.I.; Orlovskaya, D.D. Ultrastructural pathology of oligodendrocytes adjacent to microglia in prefrontal white matter in schizophrenia. NPJ Schizophr. 2018, 4, 26. [Google Scholar] [CrossRef] [Green Version]
- St-Pierre, M.K.; Simoncicova, E.; Bogi, E.; Tremblay, M.E. Shedding Light on the Dark Side of the Microglia. ASN Neuro 2020, 12, 1759091420925335. [Google Scholar] [CrossRef]
- Yao, K.; Zu, H.B. Microglial polarization: Novel therapeutic mechanism against Alzheimer’s disease. Inflammopharmacology 2020, 28, 95–110. [Google Scholar] [CrossRef]
- Kuwar, R.; Rolfe, A.; Di, L.; Blevins, H.; Xu, Y.; Sun, X.; Bloom, G.S.; Zhang, S.; Sun, D. A Novel Inhibitor Targeting NLRP3 Inflammasome Reduces Neuropathology and Improves Cognitive Function in Alzheimer’s Disease Transgenic Mice. J. Alzheimers Dis. 2021, 82, 1769–1783. [Google Scholar] [CrossRef]
- Jimenez, S.; Baglietto-Vargas, D.; Caballero, C.; Moreno-Gonzalez, I.; Torres, M.; Sanchez-Varo, R.; Ruano, D.; Vizuete, M.; Gutierrez, A.; Vitorica, J. Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer’s disease: Age-dependent switch in the microglial phenotype from alternative to classic. J. Neurosci. 2008, 28, 11650–11661. [Google Scholar] [CrossRef]
- Wang, Q.; Yao, H.; Liu, W.; Ya, B.; Cheng, H.; Xing, Z.; Wu, Y. Microglia Polarization in Alzheimer’s Disease: Mechanisms and a Potential Therapeutic Target. Front. Aging Neurosci. 2021, 13, 772717. [Google Scholar] [CrossRef]
- Balce, D.R.; Li, B.; Allan, E.R.; Rybicka, J.M.; Krohn, R.M.; Yates, R.M. Alternative activation of macrophages by IL-4 enhances the proteolytic capacity of their phagosomes through synergistic mechanisms. Blood 2011, 118, 4199–4208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majumdar, A.; Cruz, D.; Asamoah, N.; Buxbaum, A.; Sohar, I.; Lobel, P.; Maxfield, F.R. Activation of microglia acidifies lysosomes and leads to degradation of Alzheimer amyloid fibrils. Mol. Biol. Cell 2007, 18, 1490–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahara, K.; Suenobu, M.; Yoshida, A.; Koga, K.; Hyodo, A.; Ohtsuka, H.; Kuniyasu, A.; Tamamaki, N.; Sugimoto, Y.; Nakayama, H. Intracerebral microinjection of interleukin-4/interleukin-13 reduces beta-amyloid accumulation in the ipsilateral side and improves cognitive deficits in young amyloid precursor protein 23 mice. Neuroscience 2012, 207, 243–260. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, P.; Tianbai, L.; Herring, A.; Ceballos-Diaz, C.; Das, P.; Golde, T.E. Hippocampal expression of murine IL-4 results in exacerbation of amyloid deposition. Mol. Neurodegener. 2012, 7, 36. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarty, P.; Ceballos-Diaz, C.; Beccard, A.; Janus, C.; Dickson, D.; Golde, T.E.; Das, P. IFN-gamma promotes complement expression and attenuates amyloid plaque deposition in amyloid beta precursor protein transgenic mice. J. Immunol. 2010, 184, 5333–5343. [Google Scholar] [CrossRef]
- Chakrabarty, P.; Jansen-West, K.; Beccard, A.; Ceballos-Diaz, C.; Levites, Y.; Verbeeck, C.; Zubair, A.C.; Dickson, D.; Golde, T.E.; Das, P. Massive gliosis induced by interleukin-6 suppresses Abeta deposition in vivo: Evidence against inflammation as a driving force for amyloid deposition. FASEB J. 2010, 24, 548–559. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarty, P.; Herring, A.; Ceballos-Diaz, C.; Das, P.; Golde, T.E. Hippocampal expression of murine TNFalpha results in attenuation of amyloid deposition in vivo. Mol. Neurodegener. 2011, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Shen, Z.; Bao, X.; Wang, R. Clinical PET Imaging of Microglial Activation: Implications for Microglial Therapeutics in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 314. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Parhizkar, S.; Arzberger, T.; Brendel, M.; Kleinberger, G.; Deussing, M.; Focke, C.; Nuscher, B.; Xiong, M.; Ghasemigharagoz, A.; Katzmarski, N.; et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat. Neurosci. 2019, 22, 191–204. [Google Scholar] [CrossRef]
- Colton, C.A.; Mott, R.T.; Sharpe, H.; Xu, Q.; Van Nostrand, W.E.; Vitek, M.P. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J. Neuroinflamm. 2006, 3, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, J.B.; Ossenkoppele, R.; Palmqvist, S.; Strandberg, T.O.; Smith, R.; Westman, E.; Hansson, O. Amyloid and tau accumulate across distinct spatial networks and are differentially associated with brain connectivity. Elife 2019, 8, e50830. [Google Scholar] [CrossRef] [PubMed]
- Dionisio-Santos, D.A.; Olschowka, J.A.; O’Banion, M.K. Exploiting microglial and peripheral immune cell crosstalk to treat Alzheimer’s disease. J. Neuroinflamm. 2019, 16, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, K.; Friedman, B.A.; Larson, J.L.; Lauffer, B.E.; Goldstein, L.D.; Appling, L.L.; Borneo, J.; Poon, C.; Ho, T.; Cai, F.; et al. Untangling the brain’s neuroinflammatory and neurodegenerative transcriptional responses. Nat. Commun. 2016, 7, 11295. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e569. [Google Scholar] [CrossRef] [Green Version]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e1217. [Google Scholar] [CrossRef]
- Grubman, A.; Choo, X.Y.; Chew, G.; Ouyang, J.F.; Sun, G.; Croft, N.P.; Rossello, F.J.; Simmons, R.; Buckberry, S.; Landin, D.V.; et al. Transcriptional signature in microglia associated with Abeta plaque phagocytosis. Nat. Commun. 2021, 12, 3015. [Google Scholar] [CrossRef]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Olah, M.; Menon, V.; Habib, N.; Taga, M.F.; Ma, Y.; Yung, C.J.; Cimpean, M.; Khairallah, A.; Coronas-Samano, G.; Sankowski, R.; et al. Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat. Commun. 2020, 11, 6129. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, K.; Friedman, B.A.; Etxeberria, A.; Huntley, M.A.; van der Brug, M.P.; Foreman, O.; Paw, J.S.; Modrusan, Z.; Beach, T.G.; Serrano, G.E.; et al. Alzheimer’s Patient Microglia Exhibit Enhanced Aging and Unique Transcriptional Activation. Cell Rep. 2020, 31, 107843. [Google Scholar] [CrossRef] [PubMed]
- Hasselmann, J.; Coburn, M.A.; England, W.; Figueroa Velez, D.X.; Kiani Shabestari, S.; Tu, C.H.; McQuade, A.; Kolahdouzan, M.; Echeverria, K.; Claes, C.; et al. Development of a Chimeric Model to Study and Manipulate Human Microglia In Vivo. Neuron 2019, 103, 1016–1033.e1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Böttcher, C.; Amann, L.; Sagar; Scheiwe, C.; Nessler, S.; Kunz, P.; Van Loo, G.; et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 2019, 566, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Vitek, M.P.; Araujo, J.A.; Fossel, M.; Greenberg, B.D.; Howell, G.R.; Rizzo, S.J.S.; Seyfried, N.T.; Tenner, A.J.; Territo, P.R.; Windisch, M.; et al. Translational animal models for Alzheimer’s disease: An Alzheimer’s Association Business Consortium Think Tank. Alzheimers Dement. 2020, 6, e12114. [Google Scholar] [CrossRef]
- Stratoulias, V.; Venero, J.L.; Tremblay, M.E.; Joseph, B. Microglial subtypes: Diversity within the microglial community. EMBO J. 2019, 38, e101997. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Dijkstra, A.A.; Voorn, P.; Berendse, H.W.; Groenewegen, H.J.; Netherlands Brain, B.; Rozemuller, A.J.; van de Berg, W.D. Stage-dependent nigral neuronal loss in incidental Lewy body and Parkinson’s disease. Mov. Disord. 2014, 29, 1244–1251. [Google Scholar] [CrossRef]
- Iacono, D.; Geraci-Erck, M.; Rabin, M.L.; Adler, C.H.; Serrano, G.; Beach, T.G.; Kurlan, R. Parkinson disease and incidental Lewy body disease: Just a question of time? Neurology 2015, 85, 1670–1679. [Google Scholar] [CrossRef] [Green Version]
- Kouli, A.; Torsney, K.M.; Kuan, W.L. Parkinson’s Disease: Etiology, Neuropathology, and Pathogenesis. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018. [Google Scholar] [CrossRef]
- Maiti, P.; Manna, J.; Dunbar, G.L. Current understanding of the molecular mechanisms in Parkinson’s disease: Targets for potential treatments. Transl. Neurodegener. 2017, 6, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhi, K.R.; Saadabadi, A. Levodopa (L-Dopa). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Iwai, A.; Masliah, E.; Yoshimoto, M.; Ge, N.; Flanagan, L.; de Silva, H.A.; Kittel, A.; Saitoh, T. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995, 14, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Li, X.; Li, J.D. The Roles of Post-translational Modifications on alpha-Synuclein in the Pathogenesis of Parkinson’s Diseases. Front. Neurosci. 2019, 13, 381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borghi, R.; Marchese, R.; Negro, A.; Marinelli, L.; Forloni, G.; Zaccheo, D.; Abbruzzese, G.; Tabaton, M. Full length alpha-synuclein is present in cerebrospinal fluid from Parkinson’s disease and normal subjects. Neurosci. Lett. 2000, 287, 65–67. [Google Scholar] [CrossRef]
- El-Agnaf, O.M.; Salem, S.A.; Paleologou, K.E.; Cooper, L.J.; Fullwood, N.J.; Gibson, M.J.; Curran, M.D.; Court, J.A.; Mann, D.M.; Ikeda, S.; et al. Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J. 2003, 17, 1945–1947. [Google Scholar] [CrossRef]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Alpha-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [Green Version]
- Kiffin, R.; Kaushik, S.; Zeng, M.; Bandyopadhyay, U.; Zhang, C.; Massey, A.C.; Martinez-Vicente, M.; Cuervo, A.M. Altered dynamics of the lysosomal receptor for chaperone-mediated autophagy with age. J. Cell Sci. 2007, 120, 782–791. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.C.; Krainc, D. alpha-synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef]
- O’Hara, D.M.; Pawar, G.; Kalia, S.K.; Kalia, L.V. LRRK2 and alpha-Synuclein: Distinct or Synergistic Players in Parkinson’s Disease? Front. Neurosci. 2020, 14, 577. [Google Scholar] [CrossRef]
- He, J.; Zhu, G.; Wang, G.; Zhang, F. Oxidative Stress and Neuroinflammation Potentiate Each Other to Promote Progression of Dopamine Neurodegeneration. Oxid. Med. Cell. Longev. 2020, 2020, 6137521. [Google Scholar] [CrossRef]
- Kim, W.G.; Mohney, R.P.; Wilson, B.; Jeohn, G.H.; Liu, B.; Hong, J.S. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: Role of microglia. J. Neurosci. 2000, 20, 6309–6316. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Forno, L.S.; Tetrud, J.; Reeves, A.G.; Kaplan, J.A.; Karluk, D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann. Neurol. 1999, 46, 598–605. [Google Scholar] [CrossRef]
- Nonnekes, J.; Post, B.; Tetrud, J.W.; Langston, J.W.; Bloem, B.R. MPTP-induced parkinsonism: An historical case series. Lancet Neurol. 2018, 17, 300–301. [Google Scholar] [CrossRef] [Green Version]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef]
- Ouchi, Y.; Yagi, S.; Yokokura, M.; Sakamoto, M. Neuroinflammation in the living brain of Parkinson’s disease. Parkinsonism Relat. Disord. 2009, 15 (Suppl. S3), S200–S204. [Google Scholar] [CrossRef]
- Pradhan, S.; Andreasson, K. Commentary: Progressive inflammation as a contributing factor to early development of Parkinson’s disease. Exp. Neurol. 2013, 241, 148–155. [Google Scholar] [CrossRef]
- Terada, T.; Yokokura, M.; Yoshikawa, E.; Futatsubashi, M.; Kono, S.; Konishi, T.; Miyajima, H.; Hashizume, T.; Ouchi, Y. Extrastriatal spreading of microglial activation in Parkinson’s disease: A positron emission tomography study. Ann. Nucl. Med. 2016, 30, 579–587. [Google Scholar] [CrossRef]
- Stokholm, M.G.; Iranzo, A.; Ostergaard, K.; Serradell, M.; Otto, M.; Svendsen, K.B.; Garrido, A.; Vilas, D.; Borghammer, P.; Santamaria, J.; et al. Assessment of neuroinflammation in patients with idiopathic rapid-eye-movement sleep behaviour disorder: A case-control study. Lancet Neurol. 2017, 16, 789–796. [Google Scholar] [CrossRef] [Green Version]
- Doorn, K.J.; Moors, T.; Drukarch, B.; van de Berg, W.; Lucassen, P.J.; van Dam, A.M. Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson’s disease patients. Acta Neuropathol. Commun. 2014, 2, 90. [Google Scholar] [CrossRef] [Green Version]
- Imamura, K.; Hishikawa, N.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol. 2003, 106, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Xu, L.; Pan, X.; Sun, L.; Ding, J.; Xie, C.; Koliatsos, V.E.; Cai, H. LRRK2 modulates microglial activity through regulation of chemokine (C-X3-C) receptor 1 -mediated signalling pathways. Hum. Mol. Genet. 2016, 25, 3515–3523. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.S.; Heng, Y.; Yuan, Y.H.; Chen, N.H. Pathological alpha-synuclein exacerbates the progression of Parkinson’s disease through microglial activation. Toxicol. Lett. 2017, 265, 30–37. [Google Scholar] [CrossRef]
- Croisier, E.; Moran, L.B.; Dexter, D.T.; Pearce, R.K.; Graeber, M.B. Microglial inflammation in the parkinsonian substantia nigra: Relationship to alpha-synuclein deposition. J. Neuroinflamm. 2005, 2, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, C.Y.; Koprich, J.B.; Siddiqi, H.; Isacson, O. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. J. Neurosci. 2009, 29, 3365–3373. [Google Scholar] [CrossRef] [Green Version]
- Marinova-Mutafchieva, L.; Sadeghian, M.; Broom, L.; Davis, J.B.; Medhurst, A.D.; Dexter, D.T. Relationship between microglial activation and dopaminergic neuronal loss in the substantia nigra: A time course study in a 6-hydroxydopamine model of Parkinson’s disease. J. Neurochem. 2009, 110, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.M.; Zhang, F.; Zhou, H.; Kam, W.; Wilson, B.; Hong, J.S. Neuroinflammation and alpha-synuclein dysfunction potentiate each other, driving chronic progression of neurodegeneration in a mouse model of Parkinson’s disease. Environ. Health Perspect. 2011, 119, 807–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.S.; et al. Aggregated alpha-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef]
- Shavali, S.; Combs, C.K.; Ebadi, M. Reactive macrophages increase oxidative stress and alpha-synuclein nitration during death of dopaminergic neuronal cells in co-culture: Relevance to Parkinson’s disease. Neurochem. Res. 2006, 31, 85–94. [Google Scholar] [CrossRef]
- Ferreira, S.A.; Romero-Ramos, M. Microglia Response During Parkinson’s Disease: Alpha-Synuclein Intervention. Front. Cell Neurosci. 2018, 12, 247. [Google Scholar] [CrossRef] [Green Version]
- Hoenen, C.; Gustin, A.; Birck, C.; Kirchmeyer, M.; Beaume, N.; Felten, P.; Grandbarbe, L.; Heuschling, P.; Heurtaux, T. Alpha-Synuclein Proteins Promote Pro-Inflammatory Cascades in Microglia: Stronger Effects of the A53T Mutant. PLoS ONE 2016, 11, e0162717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trudler, D.; Nazor, K.L.; Eisele, Y.S.; Grabauskas, T.; Dolatabadi, N.; Parker, J.; Sultan, A.; Zhong, Z.; Goodwin, M.S.; Levites, Y.; et al. Soluble alpha-synuclein-antibody complexes activate the NLRP3 inflammasome in hiPSC-derived microglia. Proc. Natl. Acad. Sci. USA 2021, 118, e2025847118. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.; Zhang, Y.; Seegobin, S.P.; Pruvost, M.; Wang, Q.; Purtell, K.; Zhang, B.; Yue, Z. Microglia clear neuron-released alpha-synuclein via selective autophagy and prevent neurodegeneration. Nat. Commun. 2020, 11, 1386. [Google Scholar] [CrossRef] [Green Version]
- Kawabori, M.; Kacimi, R.; Kauppinen, T.; Calosing, C.; Kim, J.Y.; Hsieh, C.L.; Nakamura, M.C.; Yenari, M.A. Triggering receptor expressed on myeloid cells 2 (TREM2) deficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J. Neurosci. 2015, 35, 3384–3396. [Google Scholar] [CrossRef]
- Bido, S.; Muggeo, S.; Massimino, L.; Marzi, M.J.; Giannelli, S.G.; Melacini, E.; Nannoni, M.; Gambare, D.; Bellini, E.; Ordazzo, G.; et al. Microglia-specific overexpression of alpha-synuclein leads to severe dopaminergic neurodegeneration by phagocytic exhaustion and oxidative toxicity. Nat. Commun. 2021, 12, 6237. [Google Scholar] [CrossRef] [PubMed]
- Austin, S.A.; Floden, A.M.; Murphy, E.J.; Combs, C.K. Alpha-synuclein expression modulates microglial activation phenotype. J. Neurosci. 2006, 26, 10558–10563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melki, R. Role of Different Alpha-Synuclein Strains in Synucleinopathies, Similarities with other Neurodegenerative Diseases. J. Parkinsons Dis. 2015, 5, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Kwon, S.; Iba, M.; Spencer, B.; Rockenstein, E.; Mante, M.; Adame, A.; Shin, S.J.; Fields, J.A.; Rissman, R.A.; et al. Effects of innate immune receptor stimulation on extracellular alpha-synuclein uptake and degradation by brain resident cells. Exp. Mol. Med. 2021, 53, 281–290. [Google Scholar] [CrossRef]
- Watson, M.B.; Richter, F.; Lee, S.K.; Gabby, L.; Wu, J.; Masliah, E.; Effros, R.B.; Chesselet, M.F. Regionally-specific microglial activation in young mice over-expressing human wildtype alpha-synuclein. Exp. Neurol. 2012, 237, 318–334. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.J.; Woo, M.S.; Moon, P.G.; Baek, M.C.; Choi, I.Y.; Kim, W.K.; Junn, E.; Kim, H.S. Alpha-synuclein activates microglia by inducing the expressions of matrix metalloproteinases and the subsequent activation of protease-activated receptor-1. J. Immunol. 2010, 185, 615–623. [Google Scholar] [CrossRef] [Green Version]
- Dela Cruz, C.S.; Kang, M.J. Mitochondrial dysfunction and damage associated molecular patterns (DAMPs) in chronic inflammatory diseases. Mitochondrion 2018, 41, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Boka, G.; Anglade, P.; Wallach, D.; Javoy-Agid, F.; Agid, Y.; Hirsch, E.C. Immunocytochemical analysis of tumor necrosis factor and its receptors in Parkinson’s disease. Neurosci. Lett. 1994, 172, 151–154. [Google Scholar] [CrossRef]
- L’Episcopo, F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Impagnatiello, F.; Marchetti, B. Switching the microglial harmful phenotype promotes lifelong restoration of subtantia nigra dopaminergic neurons from inflammatory neurodegeneration in aged mice. Rejuvenation Res. 2011, 14, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Knott, C.; Stern, G.; Wilkin, G.P. Inflammatory regulators in Parkinson’s disease: iNOS, lipocortin-1, and cyclooxygenases-1 and -2. Mol. Cell. Neurosci. 2000, 16, 724–739. [Google Scholar] [CrossRef]
- Hunot, S.; Boissiere, F.; Faucheux, B.; Brugg, B.; Mouatt-Prigent, A.; Agid, Y.; Hirsch, E.C. Nitric oxide synthase and neuronal vulnerability in Parkinson’s disease. Neuroscience 1996, 72, 355–363. [Google Scholar] [CrossRef]
- Vijitruth, R.; Liu, M.; Choi, D.Y.; Nguyen, X.V.; Hunter, R.L.; Bing, G. Cyclooxygenase-2 mediates microglial activation and secondary dopaminergic cell death in the mouse MPTP model of Parkinson’s disease. J. Neuroinflamm. 2006, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- English, C.; Aloi, J.J. New FDA-Approved Disease-Modifying Therapies for Multiple Sclerosis. Clin. Ther. 2015, 37, 691–715. [Google Scholar] [CrossRef]
- Benner, E.J.; Mosley, R.L.; Destache, C.J.; Lewis, T.B.; Jackson-Lewis, V.; Gorantla, S.; Nemachek, C.; Green, S.R.; Przedborski, S.; Gendelman, H.E. Therapeutic immunization protects dopaminergic neurons in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 9435–9440. [Google Scholar] [CrossRef] [Green Version]
- Joniec-Maciejak, I.; Ciesielska, A.; Wawer, A.; Sznejder-Pacholek, A.; Schwenkgrub, J.; Cudna, A.; Hadaczek, P.; Bankiewicz, K.S.; Czlonkowska, A.; Czlonkowski, A. The influence of AAV2-mediated gene transfer of human IL-10 on neurodegeneration and immune response in a murine model of Parkinson’s disease. Pharmacol. Rep. 2014, 66, 660–669. [Google Scholar] [CrossRef]
- Arimoto, T.; Choi, D.Y.; Lu, X.; Liu, M.; Nguyen, X.V.; Zheng, N.; Stewart, C.A.; Kim, H.C.; Bing, G. Interleukin-10 protects against inflammation-mediated degeneration of dopaminergic neurons in substantia nigra. Neurobiol. Aging 2007, 28, 894–906. [Google Scholar] [CrossRef]
- Cockey, S.G.; McFarland, K.N.; Koller, E.J.; Brooks, M.M.T.; Gonzalez De La Cruz, E.; Cruz, P.E.; Ceballos-Diaz, C.; Rosario, A.M.; Levites, Y.R.; Borchelt, D.R.; et al. Il-10 signaling reduces survival in mouse models of synucleinopathy. NPJ Parkinsons Dis. 2021, 7, 30. [Google Scholar] [CrossRef]
- Karpenko, M.N.; Vasilishina, A.A.; Gromova, E.A.; Muruzheva, Z.M.; Miliukhina, I.V.; Bernadotte, A. Interleukin-1beta, interleukin-1 receptor antagonist, interleukin-6, interleukin-10, and tumor necrosis factor-alpha levels in CSF and serum in relation to the clinical diversity of Parkinson’s disease. Cell Immunol. 2018, 327, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Shu, L.; Liang, D.; Pan, H.; Xu, Q.; Yan, X.; Tang, B.; Sun, Q. Gastrointestinal Dysfunctions Are Associated with IL-10 Variants in Parkinson’s Disease. Parkinsons Dis. 2018, 2018, 5908359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Song, X.; Huang, H.; Huang, H.; Ye, Z. Association of Parkinson’s disease-related pain with plasma interleukin-1, interleukin-6, interleukin-10, and tumour necrosis factor-alpha. Neurosci. Lett. 2018, 683, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Huhner, L.; Rilka, J.; Gilsbach, R.; Zhou, X.; Machado, V.; Spittau, B. Interleukin-4 Protects Dopaminergic Neurons In vitro but Is Dispensable for MPTP-Induced Neurodegeneration In vivo. Front. Mol. Neurosci. 2017, 10, 62. [Google Scholar] [CrossRef] [Green Version]
- Tesseur, I.; Nguyen, A.; Chang, B.; Li, L.; Woodling, N.S.; Wyss-Coray, T.; Luo, J. Deficiency in Neuronal TGF-beta Signaling Leads to Nigrostriatal Degeneration and Activation of TGF-beta Signaling Protects against MPTP Neurotoxicity in Mice. J. Neurosci. 2017, 37, 4584–4592. [Google Scholar] [CrossRef]
- Zhou, X.; Zoller, T.; Krieglstein, K.; Spittau, B. TGFbeta1 inhibits IFNgamma-mediated microglia activation and protects mDA neurons from IFNgamma-driven neurotoxicity. J. Neurochem. 2015, 134, 125–134. [Google Scholar] [CrossRef]
- Polazzi, E.; Altamira, L.E.; Eleuteri, S.; Barbaro, R.; Casadio, C.; Contestabile, A.; Monti, B. Neuroprotection of microglial conditioned medium on 6-hydroxydopamine-induced neuronal death: Role of transforming growth factor beta-2. J. Neurochem. 2009, 110, 545–556. [Google Scholar] [CrossRef]
- Vawter, M.P.; Dillon-Carter, O.; Tourtellotte, W.W.; Carvey, P.; Freed, W.J. TGFbeta1 and TGFbeta2 concentrations are elevated in Parkinson’s disease in ventricular cerebrospinal fluid. Exp. Neurol. 1996, 142, 313–322. [Google Scholar] [CrossRef]
- Theodore, S.; Cao, S.; McLean, P.J.; Standaert, D.G. Targeted overexpression of human alpha-synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. J. Neuropathol. Exp. Neurol. 2008, 67, 1149–1158. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.M.; Jiang, J.; Wilson, B.; Zhang, W.; Hong, J.S.; Liu, B. Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: Relevance to Parkinson’s disease. J. Neurochem. 2002, 81, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Mastroeni, D.; Nolz, J.; Sekar, S.; Delvaux, E.; Serrano, G.; Cuyugan, L.; Liang, W.S.; Beach, T.G.; Rogers, J.; Coleman, P.D. Laser-captured microglia in the Alzheimer’s and Parkinson’s brain reveal unique regional expression profiles and suggest a potential role for hepatitis B in the Alzheimer’s brain. Neurobiol. Aging 2018, 63, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Uriarte Huarte, O.; Kyriakis, D.; Heurtaux, T.; Pires-Afonso, Y.; Grzyb, K.; Halder, R.; Buttini, M.; Skupin, A.; Mittelbronn, M.; Michelucci, A. Single-Cell Transcriptomics and In Situ Morphological Analyses Reveal Microglia Heterogeneity Across the Nigrostriatal Pathway. Front. Immunol. 2021, 12, 639613. [Google Scholar] [CrossRef] [PubMed]
- Smajic, S.; Prada-Medina, C.A.; Landoulsi, Z.; Ghelfi, J.; Delcambre, S.; Dietrich, C.; Jarazo, J.; Henck, J.; Balachandran, S.; Pachchek, S.; et al. Single-cell sequencing of human midbrain reveals glial activation and a Parkinson-specific neuronal state. Brain 2021, 145, 964–978. [Google Scholar] [CrossRef]
- El-Zayat, S.R.; Sibaii, H.; Mannaa, F.A. Toll-like receptors activation, signaling, and targeting: An overview. Bull. Natl. Res. Cent. 2019, 43, 187. [Google Scholar] [CrossRef] [Green Version]
- Fiebich, B.L.; Batista, C.R.A.; Saliba, S.W.; Yousif, N.M.; de Oliveira, A.C.P. Role of Microglia TLRs in Neurodegeneration. Front. Cell Neurosci. 2018, 12, 329. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Li, D.; Yang, K.; Hu, Y.; Zeng, Q. Toll-like receptor-4 and mitogen-activated protein kinase signal system are involved in activation of dendritic cells in patients with acute coronary syndrome. Immunology 2008, 125, 122–130. [Google Scholar] [CrossRef]
- Yao, X.; Liu, S.; Ding, W.; Yue, P.; Jiang, Q.; Zhao, M.; Hu, F.; Zhang, H. TLR4 signal ablation attenuated neurological deficits by regulating microglial M1/M2 phenotype after traumatic brain injury in mice. J. Neuroimmunol. 2017, 310, 38–45. [Google Scholar] [CrossRef]
- Liu, J.T.; Wu, S.X.; Zhang, H.; Kuang, F. Inhibition of MyD88 Signaling Skews Microglia/Macrophage Polarization and Attenuates Neuronal Apoptosis in the Hippocampus After Status Epilepticus in Mice. Neurotherapeutics 2018, 15, 1093–1111. [Google Scholar] [CrossRef] [Green Version]
- Ganbold, T.; Bao, Q.; Zandan, J.; Hasi, A.; Baigude, H. Modulation of Microglia Polarization through Silencing of NF-kappaB p65 by Functionalized Curdlan Nanoparticle-Mediated RNAi. ACS Appl. Mater. Interfaces 2020, 12, 11363–11374. [Google Scholar] [CrossRef]
- Yang, Z.; Liu, B.; Zhong, L.; Shen, H.; Lin, C.; Lin, L.; Zhang, N.; Yuan, B. Toll-like receptor-4-mediated autophagy contributes to microglial activation and inflammatory injury in mouse models of intracerebral haemorrhage. Neuropathol. Appl. Neurobiol. 2015, 41, e95–e106. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Wang, P.F.; Fang, H.; Chen, J.; Xiong, X.Y.; Yang, Q.W. Toll-like receptor 4 antagonist attenuates intracerebral hemorrhage-induced brain injury. Stroke 2013, 44, 2545–2552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, W.; Sun, C.; Ma, Y.; Wang, S.; Wang, X.; Zhang, Y. Inhibition of TLR4 Induces M2 Microglial Polarization and Provides Neuroprotection via the NLRP3 Inflammasome in Alzheimer’s Disease. Front. Neurosci. 2020, 14, 444. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chen, Y.; Xu, C.; Zhang, H.; Lin, C. TLR4 Targeting as a Promising Therapeutic Strategy for Alzheimer Disease Treatment. Front. Neurosci. 2020, 14, 602508. [Google Scholar] [CrossRef]
- Song, M.; Jin, J.; Lim, J.E.; Kou, J.; Pattanayak, A.; Rehman, J.A.; Kim, H.D.; Tahara, K.; Lalonde, R.; Fukuchi, K. TLR4 mutation reduces microglial activation, increases Abeta deposits and exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. J. Neuroinflamm. 2011, 8, 92. [Google Scholar] [CrossRef] [Green Version]
- Go, M.; Kou, J.; Lim, J.E.; Yang, J.; Fukuchi, K.I. Microglial response to LPS increases in wild-type mice during aging but diminishes in an Alzheimer’s mouse model: Implication of TLR4 signaling in disease progression. Biochem. Biophys. Res. Commun. 2016, 479, 331–337. [Google Scholar] [CrossRef] [Green Version]
- Stirling, D.P.; Cummins, K.; Mishra, M.; Teo, W.; Yong, V.W.; Stys, P. Toll-like receptor 2-mediated alternative activation of microglia is protective after spinal cord injury. Brain 2014, 137, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Jain, M.; Singh, M.K.; Shyam, H.; Mishra, A.; Kumar, S.; Kumar, A.; Kushwaha, J. Role of JAK/STAT in the Neuroinflammation and its Association with Neurological Disorders. Ann. Neurosci. 2022, 28, 191–200. [Google Scholar] [CrossRef]
- Butturini, E.; Boriero, D.; Carcereri de Prati, A.; Mariotto, S. STAT1 drives M1 microglia activation and neuroinflammation under hypoxia. Arch. Biochem. Biophys. 2019, 669, 22–30. [Google Scholar] [CrossRef]
- Qin, C.; Fan, W.H.; Liu, Q.; Shang, K.; Murugan, M.; Wu, L.J.; Wang, W.; Tian, D.S. Fingolimod Protects Against Ischemic White Matter Damage by Modulating Microglia Toward M2 Polarization via STAT3 Pathway. Stroke 2017, 48, 3336–3346. [Google Scholar] [CrossRef]
- Ding, Y.; Qian, J.; Li, H.; Shen, H.; Li, X.; Kong, Y.; Xu, Z.; Chen, G. Effects of SC99 on cerebral ischemia-perfusion injury in rats: Selective modulation of microglia polarization to M2 phenotype via inhibiting JAK2-STAT3 pathway. Neurosci. Res. 2019, 142, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.L.; Li, X.X.; Chen, Y.T.; Yu, L.J.; Zhang, H.; Lao, J.M.; Zhang, X.; Xu, Y. Neuronal Soluble Fas Ligand Drives M1-Microglia Polarization after Cerebral Ischemia. CNS Neurosci. Ther. 2016, 22, 771–781. [Google Scholar] [CrossRef]
- Wei, S.; Luo, C.; Yu, S.; Gao, J.; Liu, C.; Wei, Z.; Zhang, Z.; Wei, L.; Yi, B. Erythropoietin ameliorates early brain injury after subarachnoid haemorrhage by modulating microglia polarization via the EPOR/JAK2-STAT3 pathway. Exp. Cell Res. 2017, 361, 342–352. [Google Scholar] [CrossRef] [PubMed]
- An, J.Y.; Pang, H.G.; Huang, T.Q.; Song, J.N.; Li, D.D.; Zhao, Y.L.; Ma, X.D. AG490 ameliorates early brain injury via inhibition of JAK2/STAT3-mediated regulation of HMGB1 in subarachnoid hemorrhage. Exp. Ther. Med. 2018, 15, 1330–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, H.; Buckley, J.A.; Li, X.; Liu, Y.; Fox, T.H., 3rd; Meares, G.P.; Yu, H.; Yan, Z.; Harms, A.S.; Li, Y.; et al. Inhibition of the JAK/STAT Pathway Protects Against alpha-Synuclein-Induced Neuroinflammation and Dopaminergic Neurodegeneration. J. Neurosci. 2016, 36, 5144–5159. [Google Scholar] [CrossRef] [PubMed]
- Zujovic, V.; Schussler, N.; Jourdain, D.; Duverger, D.; Taupin, V. In vivo neutralization of endogenous brain fractalkine increases hippocampal TNFalpha and 8-isoprostane production induced by intracerebroventricular injection of LPS. J Neuroimmunol. 2001, 115, 135–143. [Google Scholar] [CrossRef]
- Cardona, A.E.; Pioro, E.P.; Sasse, M.E.; Kostenko, V.; Cardona, S.M.; Dijkstra, I.M.; Huang, D.; Kidd, G.; Dombrowski, S.; Dutta, R.; et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 2006, 9, 917–924. [Google Scholar] [CrossRef]
- Mizuno, T.; Kawanokuchi, J.; Numata, K.; Suzumura, A. Production and neuroprotective functions of fractalkine in the central nervous system. Brain Res. 2003, 979, 65–70. [Google Scholar] [CrossRef]
- Rogers, J.T.; Morganti, J.M.; Bachstetter, A.D.; Hudson, C.E.; Peters, M.M.; Grimmig, B.A.; Weeber, E.J.; Bickford, P.C.; Gemma, C. CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J. Neurosci. 2011, 31, 16241–16250. [Google Scholar] [CrossRef] [Green Version]
- Morganti, J.M.; Nash, K.R.; Grimmig, B.A.; Ranjit, S.; Small, B.; Bickford, P.C.; Gemma, C. The soluble isoform of CX3CL1 is necessary for neuroprotection in a mouse model of Parkinson’s disease. J. Neurosci. 2012, 32, 14592–14601. [Google Scholar] [CrossRef] [Green Version]
- Castro-Sanchez, S.; Garcia-Yague, A.J.; Lopez-Royo, T.; Casarejos, M.; Lanciego, J.L.; Lastres-Becker, I. Cx3cr1-deficiency exacerbates alpha-synuclein-A53T induced neuroinflammation and neurodegeneration in a mouse model of Parkinson’s disease. Glia 2018, 66, 1752–1762. [Google Scholar] [CrossRef] [PubMed]
- Nash, K.R.; Moran, P.; Finneran, D.J.; Hudson, C.; Robinson, J.; Morgan, D.; Bickford, P.C. Fractalkine over expression suppresses alpha-synuclein-mediated neurodegeneration. Mol. Ther. 2015, 23, 17–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pabon, M.M.; Bachstetter, A.D.; Hudson, C.E.; Gemma, C.; Bickford, P.C. CX3CL1 reduces neurotoxicity and microglial activation in a rat model of Parkinson’s disease. J. Neuroinflamm. 2011, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- Gyoneva, S.; Hosur, R.; Gosselin, D.; Zhang, B.; Ouyang, Z.; Cotleur, A.C.; Peterson, M.; Allaire, N.; Challa, R.; Cullen, P.; et al. Cx3cr1-deficient microglia exhibit a premature aging transcriptome. Life Sci. Alliance 2019, 2, e201900453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, L.; Zhang, Z.L.; Li, X.; Liao, C.; Mou, P.; Wang, T.; Wang, Z.; Wang, Z.; Wei, M.; Xu, H.; et al. TREM2/DAP12 Complex Regulates Inflammatory Responses in Microglia via the JNK Signaling Pathway. Front. Aging Neurosci. 2017, 9, 204. [Google Scholar] [CrossRef]
- Yeh, F.L.; Wang, Y.; Tom, I.; Gonzalez, L.C.; Sheng, M. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron 2016, 91, 328–340. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Pina-Crespo, J.C.; Zhang, M.; et al. TREM2 Is a Receptor for beta-Amyloid that Mediates Microglial Function. Neuron 2018, 97, 1023–1031.e1027. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Feng, S.; Nie, K.; Li, Y.; Gao, Y.; Gan, R.; Wang, L.; Li, B.; Sun, X.; Wang, L.; et al. TREM2 modulates microglia phenotypes in the neuroinflammation of Parkinson’s disease. Biochem. Biophys. Res. Commun. 2018, 499, 797–802. [Google Scholar] [CrossRef]
- Liu, W.; Taso, O.; Wang, R.; Bayram, S.; Graham, A.C.; Garcia-Reitboeck, P.; Mallach, A.; Andrews, W.D.; Piers, T.M.; Botia, J.A.; et al. Trem2 promotes anti-inflammatory responses in microglia and is suppressed under pro-inflammatory conditions. Hum. Mol. Genet. 2020, 29, 3224–3248. [Google Scholar] [CrossRef]
- Hu, Y.; Li, C.; Wang, X.; Chen, W.; Qian, Y.; Dai, X. TREM2, Driving the Microglial Polarization, Has a TLR4 Sensitivity Profile After Subarachnoid Hemorrhage. Front. Cell Dev. Biol. 2021, 9, 693342. [Google Scholar] [CrossRef]
- Sheng, L.; Chen, M.; Cai, K.; Song, Y.; Yu, D.; Zhang, H.; Xu, G. Microglial Trem2 induces synaptic impairment at early stage and prevents amyloidosis at late stage in APP/PS1 mice. FASEB J. 2019, 33, 10425–10442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovaszi, M.; Branco Haas, C.; Antonioli, L.; Pacher, P.; Hasko, G. The role of P2Y receptors in regulating immunity and metabolism. Biochem. Pharmacol 2021, 187, 114419. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.; Pons, V.; Rivest, S. Microglia Purinoceptor P2Y6: An Emerging Therapeutic Target in CNS Diseases. Cells 2020, 9, 1595. [Google Scholar] [CrossRef] [PubMed]
- Wen, R.X.; Shen, H.; Huang, S.X.; Wang, L.P.; Li, Z.W.; Peng, P.; Mamtilahun, M.; Tang, Y.H.; Shen, F.X.; Tian, H.L.; et al. P2Y6 receptor inhibition aggravates ischemic brain injury by reducing microglial phagocytosis. CNS Neurosci. Ther. 2020, 26, 416–429. [Google Scholar] [CrossRef]
- Puigdellivol, M.; Milde, S.; Vilalta, A.; Cockram, T.O.J.; Allendorf, D.H.; Lee, J.Y.; Dundee, J.M.; Pampuscenko, K.; Borutaite, V.; Nuthall, H.N.; et al. The microglial P2Y6 receptor mediates neuronal loss and memory deficits in neurodegeneration. Cell Rep. 2021, 37, 110148. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.; Rivest, S. Alzheimer’s disease: Microglia targets and their modulation to promote amyloid phagocytosis and mitigate neuroinflammation. Expert Opin. Ther. Targets 2020, 24, 331–344. [Google Scholar] [CrossRef]
- Yang, X.; Lou, Y.; Liu, G.; Wang, X.; Qian, Y.; Ding, J.; Chen, S.; Xiao, Q. Microglia P2Y6 receptor is related to Parkinson’s disease through neuroinflammatory process. J. Neuroinflamm. 2017, 14, 38. [Google Scholar] [CrossRef] [Green Version]
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.B.; Julius, D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 2006, 9, 1512–1519. [Google Scholar] [CrossRef]
- Maeda, J.; Minamihisamatsu, T.; Shimojo, M.; Zhou, X.; Ono, M.; Matsuba, Y.; Ji, B.; Ishii, H.; Ogawa, M.; Akatsu, H.; et al. Distinct microglial response against Alzheimer’s amyloid and tau pathologies characterized by P2Y12 receptor. Brain Commun. 2021, 3, fcab011. [Google Scholar] [CrossRef]
- Zrzavy, T.; Hametner, S.; Wimmer, I.; Butovsky, O.; Weiner, H.L.; Lassmann, H. Loss of ‘homeostatic’ microglia and patterns of their activation in active multiple sclerosis. Brain 2017, 140, 1900–1913. [Google Scholar] [CrossRef]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar-Colucci, S.; Karlo, J.C.; Landreth, G.E. Mechanisms underlying the rapid peroxisome proliferator-activated receptor-gamma-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer’s disease. J. Neurosci. 2012, 32, 10117–10128. [Google Scholar] [CrossRef] [Green Version]
- Wen, L.; You, W.; Wang, H.; Meng, Y.; Feng, J.; Yang, X. Polarization of Microglia to the M2 Phenotype in a Peroxisome Proliferator-Activated Receptor Gamma-Dependent Manner Attenuates Axonal Injury Induced by Traumatic Brain Injury in Mice. J. Neurotrauma 2018, 35, 2330–2340. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Jin, J.L.; Ge, H.M.; Yin, K.L.; Chen, X.; Han, L.J.; Chen, Y.; Qian, L.; Li, X.X.; Xu, Y. Malibatol A regulates microglia M1/M2 polarization in experimental stroke in a PPARgamma-dependent manner. J. Neuroinflamm. 2015, 12, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.C.; Ma, L.S.; Chu, Z.H.; Xu, H.; Wu, W.Q.; Liu, F. Regulation of microglial activation in stroke. Acta Pharmacol Sin. 2017, 38, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Saunders, A.M.; Burns, D.K.; Gottschalk, W.K. Reassessment of Pioglitazone for Alzheimer’s Disease. Front. Neurosci. 2021, 15, 666958. [Google Scholar] [CrossRef]
- Pisanu, A.; Lecca, D.; Mulas, G.; Wardas, J.; Simbula, G.; Spiga, S.; Carta, A.R. Dynamic changes in pro- and anti-inflammatory cytokines in microglia after PPAR-gamma agonist neuroprotective treatment in the MPTPp mouse model of progressive Parkinson’s disease. Neurobiol. Dis. 2014, 71, 280–291. [Google Scholar] [CrossRef]
- Carta, A.R.; Frau, L.; Pisanu, A.; Wardas, J.; Spiga, S.; Carboni, E. Rosiglitazone decreases peroxisome proliferator receptor-gamma levels in microglia and inhibits TNF-alpha production: New evidences on neuroprotection in a progressive Parkinson’s disease model. Neuroscience 2011, 194, 250–261. [Google Scholar] [CrossRef]
- Yamanaka, M.; Ishikawa, T.; Griep, A.; Axt, D.; Kummer, M.P.; Heneka, M.T. PPARgamma/RXRalpha-induced and CD36-mediated microglial amyloid-beta phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J. Neurosci. 2012, 32, 17321–17331. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Li, T.; Duan, S.N.; Dong, L.; Sun, X.G.; Xue, F. PPAR-gamma Promotes Hematoma Clearance through Haptoglobin-Hemoglobin-CD163 in a Rat Model of Intracerebral Hemorrhage. Behav. Neurol. 2018, 2018, 7646104. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Grotta, J.; Gonzales, N.; Aronowski, J. Hematoma resolution as a therapeutic target: The role of microglia/macrophages. Stroke 2009, 40, S92–S94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miron, V.E.; Priller, J. Investigating Microglia in Health and Disease: Challenges and Opportunities. Trends Immunol. 2020, 41, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Amici, S.A.; Dong, J.; Guerau-de-Arellano, M. Molecular Mechanisms Modulating the Phenotype of Macrophages and Microglia. Front. Immunol. 2017, 8, 1520. [Google Scholar] [CrossRef] [Green Version]
- Martin, E.; El-Behi, M.; Fontaine, B.; Delarasse, C. Analysis of Microglia and Monocyte-derived Macrophages from the Central Nervous System by Flow Cytometry. J. Vis. Exp. 2017, 124, 55781. [Google Scholar] [CrossRef] [PubMed]
- Honarpisheh, P.; Lee, J.; Banerjee, A.; Blasco-Conesa, M.P.; Honarpisheh, P.; d’Aigle, J.; Mamun, A.A.; Ritzel, R.M.; Chauhan, A.; Ganesh, B.P.; et al. Potential caveats of putative microglia-specific markers for assessment of age-related cerebrovascular neuroinflammation. J. Neuroinflamm. 2020, 17, 366. [Google Scholar] [CrossRef]
- van Wageningen, T.A.; Vlaar, E.; Kooij, G.; Jongenelen, C.A.M.; Geurts, J.J.G.; van Dam, A.M. Regulation of microglial TMEM119 and P2RY12 immunoreactivity in multiple sclerosis white and grey matter lesions is dependent on their inflammatory environment. Acta Neuropathol. Commun. 2019, 7, 206. [Google Scholar] [CrossRef] [Green Version]
- Yanguas-Casás, N. Physiological sex differences in microglia and their relevance in neurological disorders. Neuroimmunol. Neuroinflammation 2020, 7, 13–22. [Google Scholar] [CrossRef]
- Guneykaya, D.; Ivanov, A.; Hernandez, D.P.; Haage, V.; Wojtas, B.; Meyer, N.; Maricos, M.; Jordan, P.; Buonfiglioli, A.; Gielniewski, B.; et al. Transcriptional and Translational Differences of Microglia from Male and Female Brains. Cell Rep. 2018, 24, 2773–2783.e2776. [Google Scholar] [CrossRef] [Green Version]
- Villa, A.; Gelosa, P.; Castiglioni, L.; Cimino, M.; Rizzi, N.; Pepe, G.; Lolli, F.; Marcello, E.; Sironi, L.; Vegeto, E.; et al. Sex-Specific Features of Microglia from Adult Mice. Cell Rep. 2018, 23, 3501–3511. [Google Scholar] [CrossRef]
- Yanguas-Casas, N.; Crespo-Castrillo, A.; de Ceballos, M.L.; Chowen, J.A.; Azcoitia, I.; Arevalo, M.A.; Garcia-Segura, L.M. Sex differences in the phagocytic and migratory activity of microglia and their impairment by palmitic acid. Glia 2018, 66, 522–537. [Google Scholar] [CrossRef]
- Geirsdottir, L.; David, E.; Keren-Shaul, H.; Weiner, A.; Bohlen, S.C.; Neuber, J.; Balic, A.; Giladi, A.; Sheban, F.; Dutertre, C.A.; et al. Cross-Species Single-Cell Analysis Reveals Divergence of the Primate Microglia Program. Cell 2019, 179, 1609–1622.e1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosselin, D.; Skola, D.; Coufal, N.G.; Holtman, I.R.; Schlachetzki, J.C.M.; Sajti, E.; Jaeger, B.N.; O’Connor, C.; Fitzpatrick, C.; Pasillas, M.P.; et al. An environment-dependent transcriptional network specifies human microglia identity. Science 2017, 356, eaal3222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizee, M.R.; Miedema, S.; Van Der Poel, M.; Adelia; Schuurman, K.G.; Van Strien, M.E.; Melief, J.; Smolders, J.; Hendrickx, D.A.; Heutinck, K.M.; et al. Isolation of primary microglia from the human post-mortem brain: Effects of ante- and post-mortem variables. Acta Neuropathol. Commun. 2017, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Chen, Y.; Dai, R.; Jiang, Y.; Tian, J.; Liu, S.; Xu, M.; Li, M.; Zhou, J.; Liu, C.; et al. Agonal Factors Distort Gene-Expression Patterns in Human Postmortem Brains. Front. Neurosci. 2021, 15, 614142. [Google Scholar] [CrossRef] [PubMed]
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.M.; Nguyen, C.H.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.M.; Marsh, S.E.; Fimbres, C.; et al. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 2017, 94, 278–293.e279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ormel, P.R.; Vieira de Sa, R.; van Bodegraven, E.J.; Karst, H.; Harschnitz, O.; Sneeboer, M.A.M.; Johansen, L.E.; van Dijk, R.E.; Scheefhals, N.; Berdenis van Berlekom, A.; et al. Microglia innately develop within cerebral organoids. Nat. Commun. 2018, 9, 4167. [Google Scholar] [CrossRef]
- Bang, S.; Jeong, S.; Choi, N.; Kim, H.N. Brain-on-a-chip: A history of development and future perspective. Biomicrofluidics 2019, 13, 051301. [Google Scholar] [CrossRef]
- Svoboda, D.S.; Barrasa, M.I.; Shu, J.; Rietjens, R.; Zhang, S.; Mitalipova, M.; Berube, P.; Fu, D.; Shultz, L.D.; Bell, G.W.; et al. Human iPSC-derived microglia assume a primary microglia-like state after transplantation into the neonatal mouse brain. Proc. Natl. Acad. Sci. USA 2019, 116, 25293–25303. [Google Scholar] [CrossRef]
- Mancuso, R.; Van Den Daele, J.; Fattorelli, N.; Wolfs, L.; Balusu, S.; Burton, O.; Liston, A.; Sierksma, A.; Fourne, Y.; Poovathingal, S.; et al. Stem-cell-derived human microglia transplanted in mouse brain to study human disease. Nat. Neurosci. 2019, 22, 2111–2116. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wendimu, M.Y.; Hooks, S.B. Microglia Phenotypes in Aging and Neurodegenerative Diseases. Cells 2022, 11, 2091. https://doi.org/10.3390/cells11132091
Wendimu MY, Hooks SB. Microglia Phenotypes in Aging and Neurodegenerative Diseases. Cells. 2022; 11(13):2091. https://doi.org/10.3390/cells11132091
Chicago/Turabian StyleWendimu, Menbere Y., and Shelley B. Hooks. 2022. "Microglia Phenotypes in Aging and Neurodegenerative Diseases" Cells 11, no. 13: 2091. https://doi.org/10.3390/cells11132091
APA StyleWendimu, M. Y., & Hooks, S. B. (2022). Microglia Phenotypes in Aging and Neurodegenerative Diseases. Cells, 11(13), 2091. https://doi.org/10.3390/cells11132091