
mTOR-Dependent Autophagy Regulates Slit Diaphragm Density in Podocyte-like Drosophila Nephrocytes

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fly Strains and Husbandry
2.2. Immunofluorescence Studies, TUNEL Detection and LIVE/DEAD Staining Using Drosophila Tissue
2.3. Channel Diffusion Assay
2.4. Fluorescent Tracer Uptake
2.5. Nile Red Staining of Neutral Lipids
2.6. Electron Microscopy
2.7. Statistics
3. Results
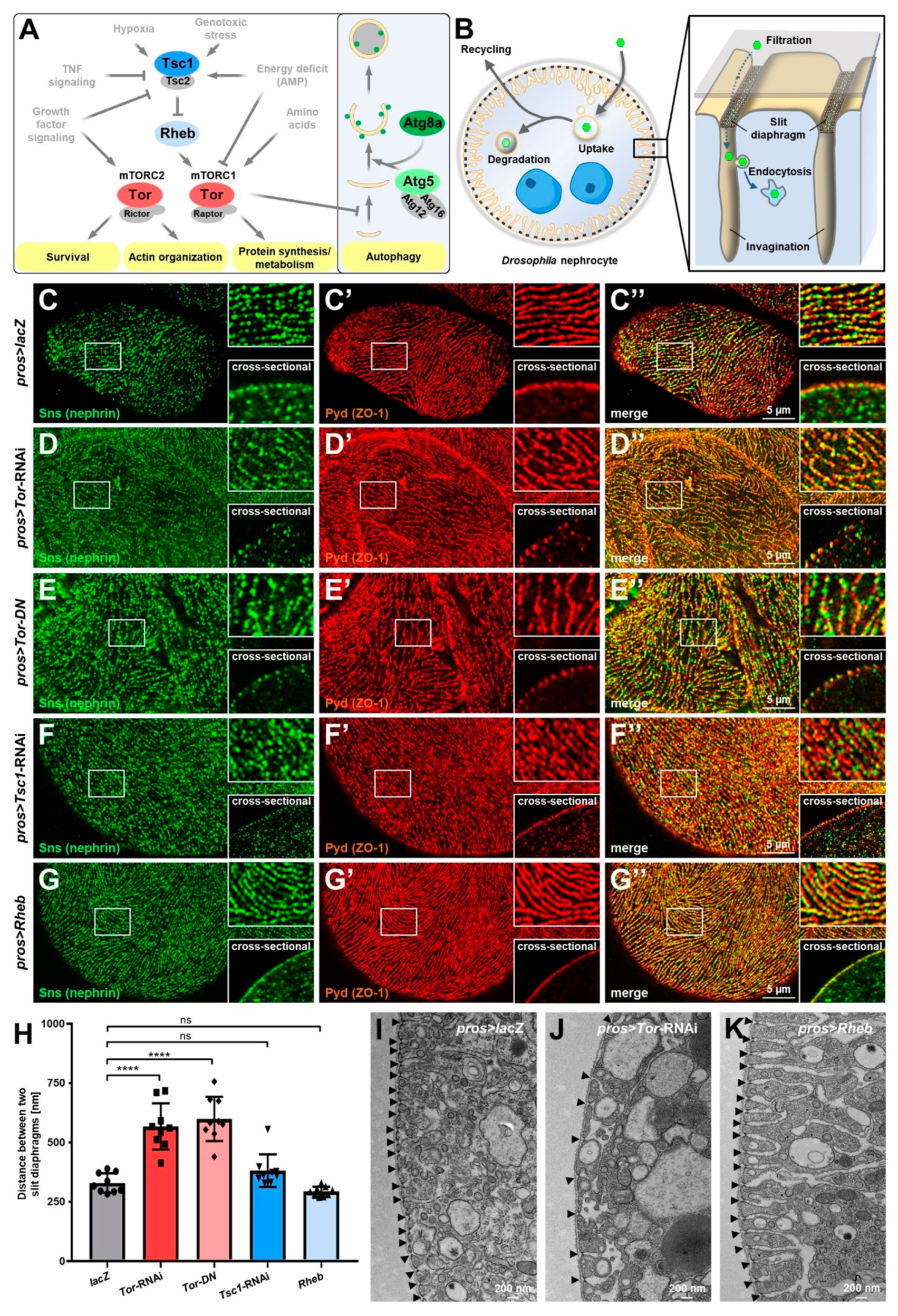
3.1. mTOR Signaling Controls Spacing of Slit Diaphragms in Drosophila Nephrocytes
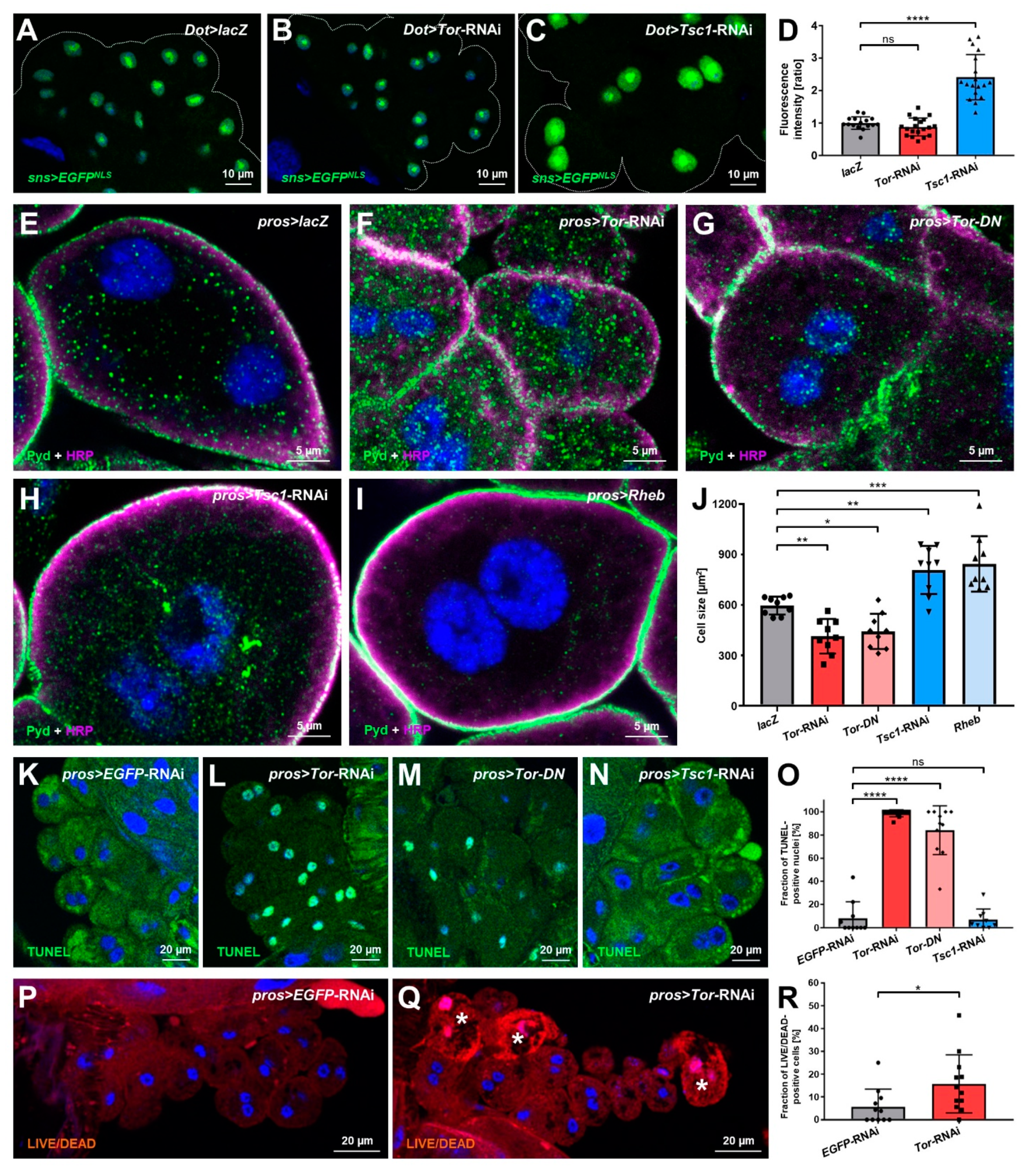
3.2. Manipulation of mTOR Signaling Affects Nephrocyte Cell Size, Survival and sns (Nephrin) Expression
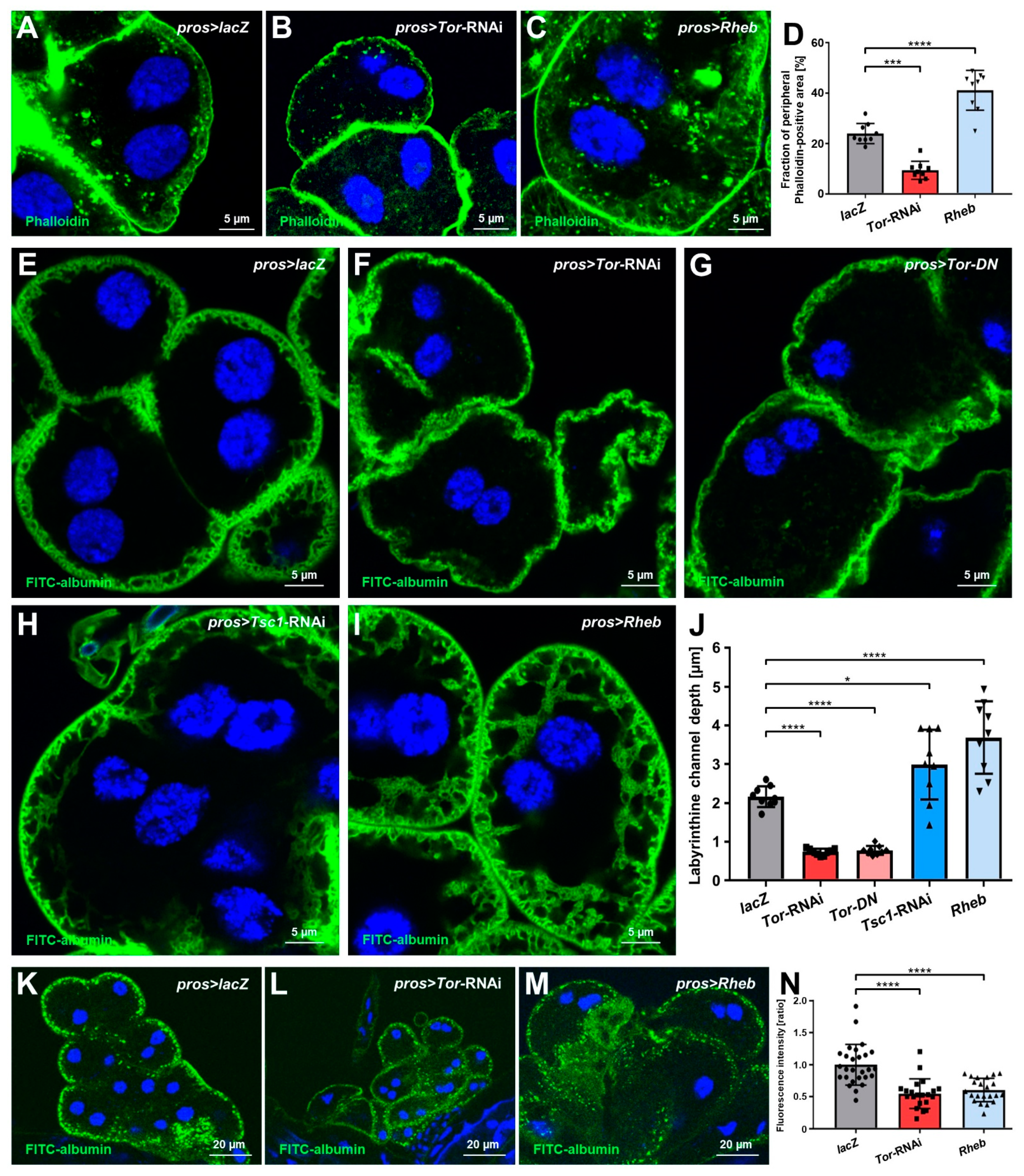
3.3. mTOR Signaling Regulates the Labyrinthine Channel Network and Endocytic Function in Nephrocytes
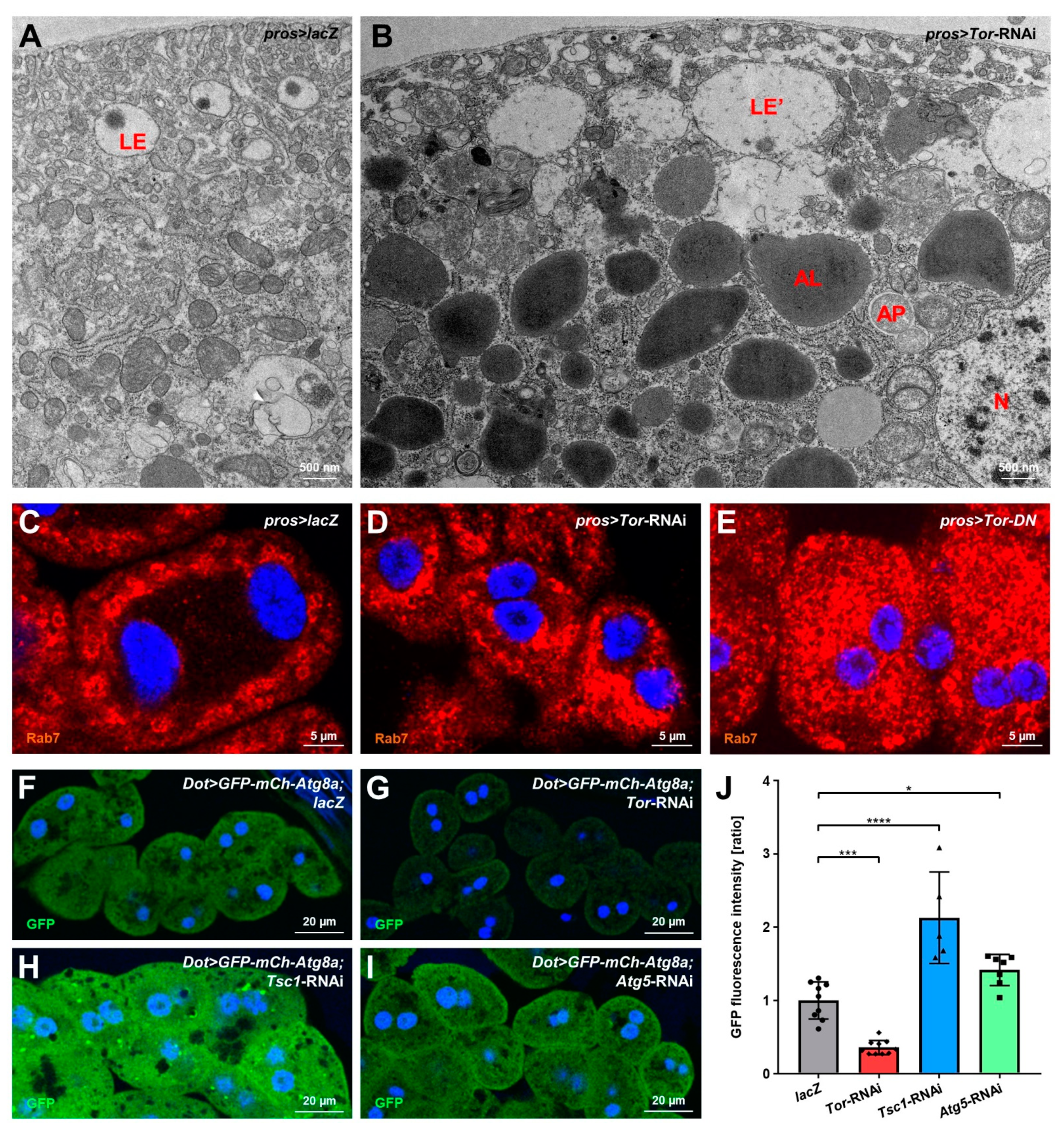
3.4. mTOR Inhibition Causes Elevated Autophagy in Nephrocytes
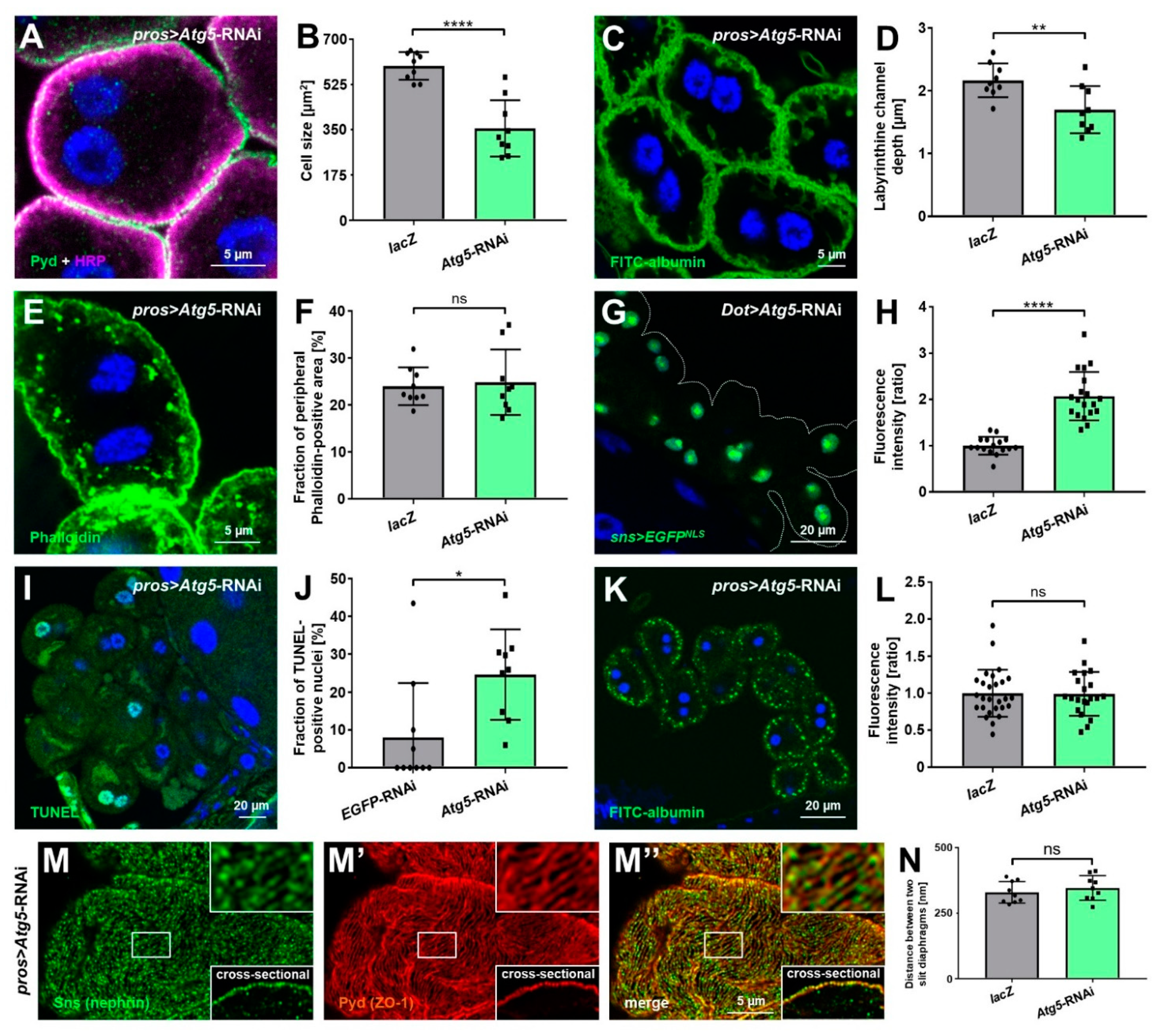
3.5. Autophagy Promotes Nephrocyte Survival but Is Dispensable for Slit Diaphragm Formation and Nephrocyte Function
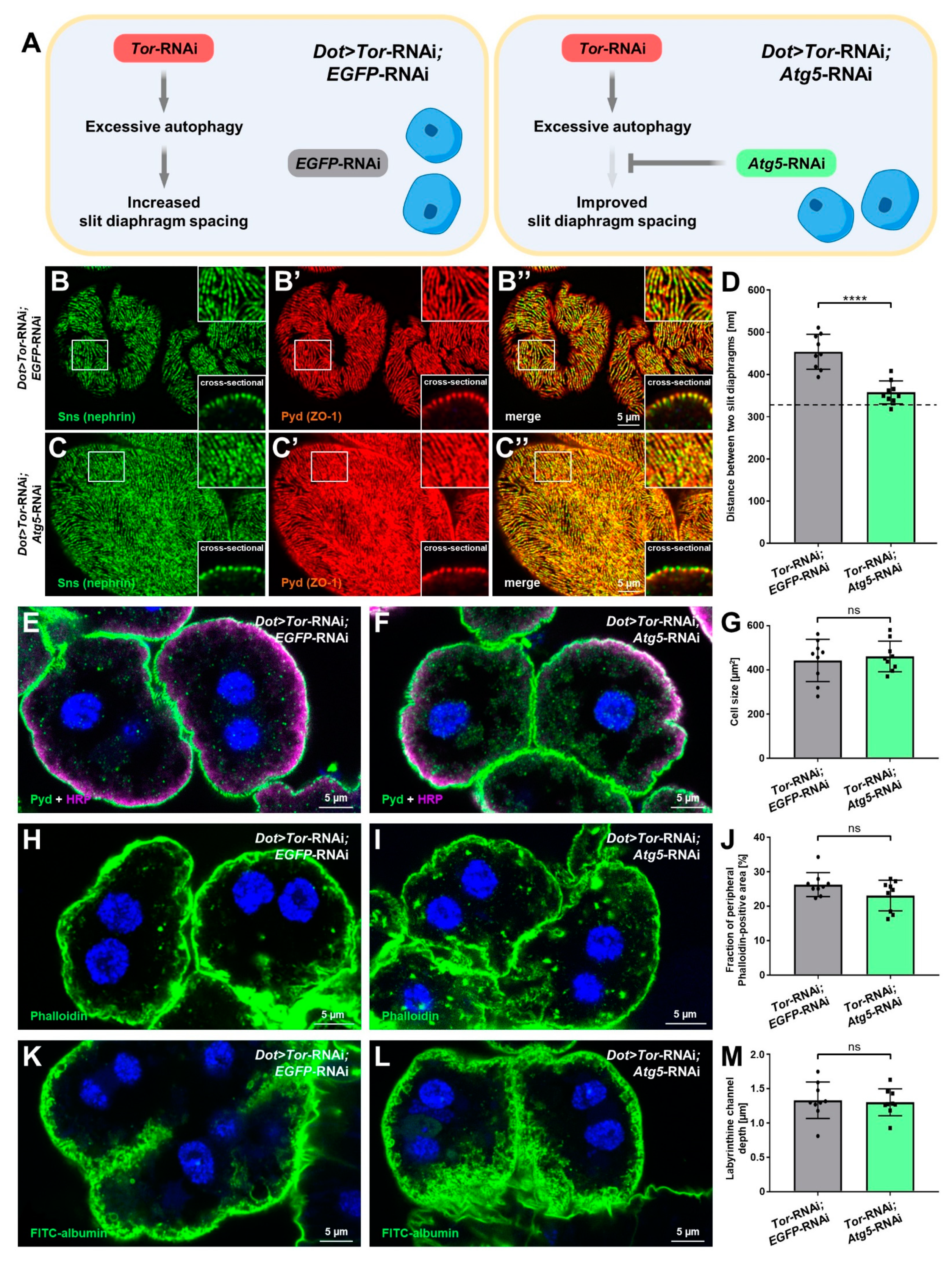
3.6. Inhibition of Autophagy Selectively Rescues Slit Diaphragm Misspacing Associated with Attenuation of mTOR Signaling
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chun, Y.; Kim, J. Autophagy: An Essential Degradation Program for Cellular Homeostasis and Life. Cells 2018, 7, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.C.; Guan, K.-L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Invest. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shemi, A.; Ben-Dor, S.; Vardi, A. Elucidating the composition and conservation of the autophagy pathway in photosynthetic eukaryotes. Autophagy 2015, 11, 701–715. [Google Scholar] [CrossRef] [Green Version]
- Deleyto-Seldas, N.; Efeyan, A. The mTOR–Autophagy Axis and the Control of Metabolism. Front. Cell Dev. Biol. 2021, 9, 1519. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Gong, L.; Pan, Q.; Yang, N. Autophagy and Inflammation Regulation in Acute Kidney Injury. Front. Physiol. 2020, 11, 576463. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Livingston, M.J.; Liu, Z.; Dong, Z. Autophagy in kidney homeostasis and disease. Nat. Rev. Nephrol. 2020, 16, 489–508. [Google Scholar] [CrossRef]
- Pollak, M.R.; Quaggin, S.E.; Hoenig, M.P.; Dworkin, L.D. The Glomerulus: The Sphere of Influence. Clin. J. Am. Soc. Nephrol. 2014, 9, 1461. [Google Scholar] [CrossRef] [Green Version]
- Benzing, T. Signaling at the Slit Diaphragm. J. Am. Soc. Nephrol. 2004, 15, 1382–1391. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.M.-W.; Nissaisorakarn, P.; Husain, I.; Jim, B. Proteinuric Kidney Diseases: A Podocyte’s Slit Diaphragm and Cytoskeleton Approach. Front. Med. 2018, 5, 221. [Google Scholar] [CrossRef]
- Holthöfer, H. Molecular architecture of the glomerular slit diaphragm: Lessons learnt for a better understanding of disease pathogenesis. Nephrol. Dial. Transplant. 2007, 22, 2124–2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieberthal, W.; Levine, J.S. The Role of the Mammalian Target Of Rapamycin (mTOR) in Renal Disease. J. Am. Soc. Nephrol. 2009, 20, 2493–2502. [Google Scholar] [CrossRef] [PubMed]
- Gödel, M.; Hartleben, B.; Herbach, N.; Liu, S.; Zschiedrich, S.; Lu, S.; Debreczeni-Mór, A.; Lindenmeyer, M.T.; Rastaldi, M.-P.; Hartleben, G.; et al. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J. Clin. Invest. 2011, 121, 2197–2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grahammer, F.; Haenisch, N.; Steinhardt, F.; Sandner, L.; Roerden, M.; Arnold, F.; Cordts, T.; Wanner, N.; Reichardt, W.; Kerjaschki, D.; et al. mTORC1 maintains renal tubular homeostasis and is essential in response to ischemic stress. Proc. Natl. Acad. Sci. USA 2014, 111, E2817–E2826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canaud, G.; Bienaimé, F.; Viau, A.; Treins, C.; Baron, W.; Nguyen, C.; Burtin, M.; Berissi, S.; Giannakakis, K.; Muda, A.O.; et al. AKT2 is essential to maintain podocyte viability and function during chronic kidney disease. Nat. Med. 2013, 19, 1288–1296. [Google Scholar] [CrossRef]
- Hartleben, B.; Gödel, M.; Meyer-Schwesinger, C.; Liu, S.; Ulrich, T.; Köbler, S.; Wiech, T.; Grahammer, F.; Arnold, S.J.; Lindenmeyer, M.T.; et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J. Clin. Invest. 2010, 120, 1084–1096. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Liu, K.; Luo, J.; Dong, Z. Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. Am. J. Pathol. 2010, 176, 1181–1192. [Google Scholar] [CrossRef] [Green Version]
- Kume, S.; Uzu, T.; Horiike, K.; Chin-Kanasaki, M.; Isshiki, K.; Araki, S.-I.; Sugimoto, T.; Haneda, M.; Kashiwagi, A.; Koya, D. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J. Clin. Invest. 2010, 120, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Teh, Y.M.; Mualif, S.A.; Lim, S.K. A comprehensive insight into autophagy and its potential signaling pathways as a therapeutic target in podocyte injury. Int. J. Biochem. Cell Biol. 2022, 143, 106153. [Google Scholar] [CrossRef]
- Kaushal, G.P.; Chandrashekar, K.; Juncos, L.A.; Shah, S.V. Autophagy Function and Regulation in Kidney Disease. Biomolecules 2020, 10, 100. [Google Scholar] [CrossRef] [Green Version]
- Stallone, G.; Infante, B.; Pontrelli, P.; Gigante, M.; Montemurno, E.; Loverre, A.; Rossini, M.; Schena, F.P.; Grandaliano, G.; Gesualdo, L. Sirolimus and Proteinuria in Renal Transplant Patients: Evidence for a Dose-Dependent Effect on Slit Diaphragm-Associated Proteins. Transplantation 2011, 91, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Biancone, L.; Bussolati, B.; Mazzucco, G.; Barreca, A.; Gallo, E.; Rossetti, M.; Messina, M.; Nuschak, B.; Fop, F.; Medica, D.; et al. Loss of Nephrin Expression in Glomeruli of Kidney-Transplanted Patients Under m-TOR Inhibitor Therapy. Am. J. Transplant. 2010, 10, 2270–2278. [Google Scholar] [CrossRef] [PubMed]
- Vollenbröker, B.; George, B.; Wolfgart, M.; Saleem, M.A.; Pavenstädt, H.; Weide, T. mTOR regulates expression of slit diaphragm proteins and cytoskeleton structure in podocytes. Am. J. Physiol. -Ren. Physiol. 2009, 296, F418–F426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.S.; Lee, J.G.; Cho, Y.; Song, S.H.; Huh, K.H.; Kim, M.S.; Kim, Y.S. Reduction of Slit Diaphragm-associated Molecules by Sirolimus: Is it Enough to Induce Proteinuria? Transplant. Proc. 2017, 49, 1165–1169. [Google Scholar] [CrossRef]
- Huber, T.B.; Walz, G.; Kuehn, E.W. mTOR and rapamycin in the kidney: Signaling and therapeutic implications beyond immunosuppression. Kidney Int. 2011, 79, 502–511. [Google Scholar] [CrossRef] [Green Version]
- Lenoir, O.; Jasiek, M.; Hénique, C.; Guyonnet, L.; Hartleben, B.; Bork, T.; Chipont, A.; Flosseau, K.; Bensaada, I.; Schmitt, A.; et al. Endothelial cell and podocyte autophagy synergistically protect from diabetes-induced glomerulosclerosis. Autophagy 2015, 11, 1130–1145. [Google Scholar] [CrossRef]
- Helmstadter, M.; Huber, T.B.; Hermle, T. Using the Drosophila Nephrocyte to Model Podocyte Function and Disease. Front. Pediatr. 2017, 5, 262. [Google Scholar] [CrossRef] [Green Version]
- Hermle, T.; Braun, D.A.; Helmstadter, M.; Huber, T.B.; Hildebrandt, F. Modeling Monogenic Human Nephrotic Syndrome in the Drosophila Garland Cell Nephrocyte. J. Am. Soc. Nephrol. 2017, 28, 1521–1533. [Google Scholar] [CrossRef] [Green Version]
- Weavers, H.; Prieto-Sanchez, S.; Grawe, F.; Garcia-Lopez, A.; Artero, R.; Wilsch-Brauninger, M.; Ruiz-Gomez, M.; Skaer, H.; Denholm, B. The insect nephrocyte is a podocyte-like cell with a filtration slit diaphragm. Nature 2009, 457, 322–326. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, S.; Shao, H.; Guo, F.; Trimble, R.; Pearce, E.; Abmayr, S.M. Sns and Kirre, the Drosophila orthologs of Nephrin and Neph1, direct adhesion, fusion and formation of a slit diaphragm-like structure in insect nephrocytes. Development 2009, 136, 2335–2344. [Google Scholar] [CrossRef] [Green Version]
- Denton, D.; O’Keefe, L.; Kumar, S. Drosophila as a model to understand autophagy deregulation in human disorders. Prog. Mol. Biol. Transl. Sci. 2020, 172, 375–409. [Google Scholar] [CrossRef] [PubMed]
- Khezri, R.; Rusten, T.E. Autophagy and Tumorigenesis in Drosophila. In The Drosophila Model in Cancer; Deng, W.-M., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 113–127. [Google Scholar]
- Maruzs, T.; Simon-Vecsei, Z.; Kiss, V.; Csizmadia, T.; Juhász, G. On the Fly: Recent Progress on Autophagy and Aging in Drosophila. Front. Cell Dev. Biol. 2019, 7, 140. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Kumar, S.; Denton, D. Chapter Twenty-Three—Characterization of Autophagic Responses in Drosophila melanogaster. In Methods in Enzymology; Galluzzi, L., Bravo-San Pedro, J.M., Kroemer, G., Eds.; Academic Press: Cambridge, MA, USA, 2017; Volume 588, pp. 445–465. [Google Scholar]
- Lőrincz, P.; Mauvezin, C.; Juhász, G. Exploring Autophagy in Drosophila. Cells 2017, 6, 22. [Google Scholar] [CrossRef] [Green Version]
- Rigon, L.; De Filippis, C.; Napoli, B.; Tomanin, R.; Orso, G. Exploiting the Potential of Drosophila Models in Lysosomal Storage Disorders: Pathological Mechanisms and Drug Discovery. Biomedicines 2021, 9, 268. [Google Scholar] [CrossRef]
- Puelles, V.G.; van der Wolde, J.W.; Wanner, N.; Scheppach, M.W.; Cullen-McEwen, L.A.; Bork, T.; Lindenmeyer, M.T.; Gernhold, L.; Wong, M.N.; Braun, F.; et al. mTOR-mediated podocyte hypertrophy regulates glomerular integrity in mice and humans. JCI insight 2019, 4, e99271. [Google Scholar] [CrossRef]
- Lin, Q.; Banu, K.; Ni, Z.; Leventhal, J.S.; Menon, M.C. Podocyte Autophagy in Homeostasis and Disease. J. Clin. Med. 2021, 10, 1184. [Google Scholar] [CrossRef]
- Gerstner, L.; Chen, M.; Kampf, L.L.; Milosavljevic, J.; Lang, K.; Schneider, R.; Hildebrandt, F.; Helmstädter, M.; Walz, G.; Hermle, T. Inhibition of endoplasmic reticulum stress signaling rescues cytotoxicity of human apolipoprotein-L1 risk variants in Drosophila. Kidney Int. 2022. [Google Scholar] [CrossRef] [PubMed]
- Ivy, J.R.; Drechsler, M.; Catterson, J.H.; Bodmer, R.; Ocorr, K.; Paululat, A.; Hartley, P.S. Klf15 Is Critical for the Development and Differentiation of Drosophila Nephrocytes. PLoS ONE 2015, 10, e0134620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nezis, I.P.; Lamark, T.; Velentzas, A.D.; Rusten, T.E.; Bjørkøy, G.; Johansen, T.; Papassideri, I.S.; Stravopodis, D.J.; Margaritis, L.H.; Stenmark, H.; et al. Cell death during Drosophila melanogaster early oogenesis is mediated through autophagy. Autophagy 2009, 5, 298–302. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.S.; Vautier, M.; Allenbach, Y.; Zahr, N.; Benveniste, O.; Funck-Brentano, C.; Salem, J.-E. Sirolimus and mTOR Inhibitors: A Review of Side Effects and Specific Management in Solid Organ Transplantation. Drug Safety 2019, 42, 813–825. [Google Scholar] [CrossRef]
- Bork, T.; Liang, W.; Yamahara, K.; Lee, P.; Tian, Z.; Liu, S.; Schell, C.; Thedieck, K.; Hartleben, B.; Patel, K.; et al. Podocytes maintain high basal levels of autophagy independent of mtor signaling. Autophagy 2020, 16, 1932–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spitz, D.; Comas, M.; Gerstner, L.; Kayser, S.; Helmstädter, M.; Walz, G.; Hermle, T. mTOR-Dependent Autophagy Regulates Slit Diaphragm Density in Podocyte-like Drosophila Nephrocytes. Cells 2022, 11, 2103. https://doi.org/10.3390/cells11132103
Spitz D, Comas M, Gerstner L, Kayser S, Helmstädter M, Walz G, Hermle T. mTOR-Dependent Autophagy Regulates Slit Diaphragm Density in Podocyte-like Drosophila Nephrocytes. Cells. 2022; 11(13):2103. https://doi.org/10.3390/cells11132103
Chicago/Turabian StyleSpitz, Dominik, Maria Comas, Lea Gerstner, Séverine Kayser, Martin Helmstädter, Gerd Walz, and Tobias Hermle. 2022. "mTOR-Dependent Autophagy Regulates Slit Diaphragm Density in Podocyte-like Drosophila Nephrocytes" Cells 11, no. 13: 2103. https://doi.org/10.3390/cells11132103
APA StyleSpitz, D., Comas, M., Gerstner, L., Kayser, S., Helmstädter, M., Walz, G., & Hermle, T. (2022). mTOR-Dependent Autophagy Regulates Slit Diaphragm Density in Podocyte-like Drosophila Nephrocytes. Cells, 11(13), 2103. https://doi.org/10.3390/cells11132103