
Cortical Organoids to Model Microcephaly

 ,
, 
{kind=link}
Abstract
:1. Introduction
2. Generation of Cerebral Organoids
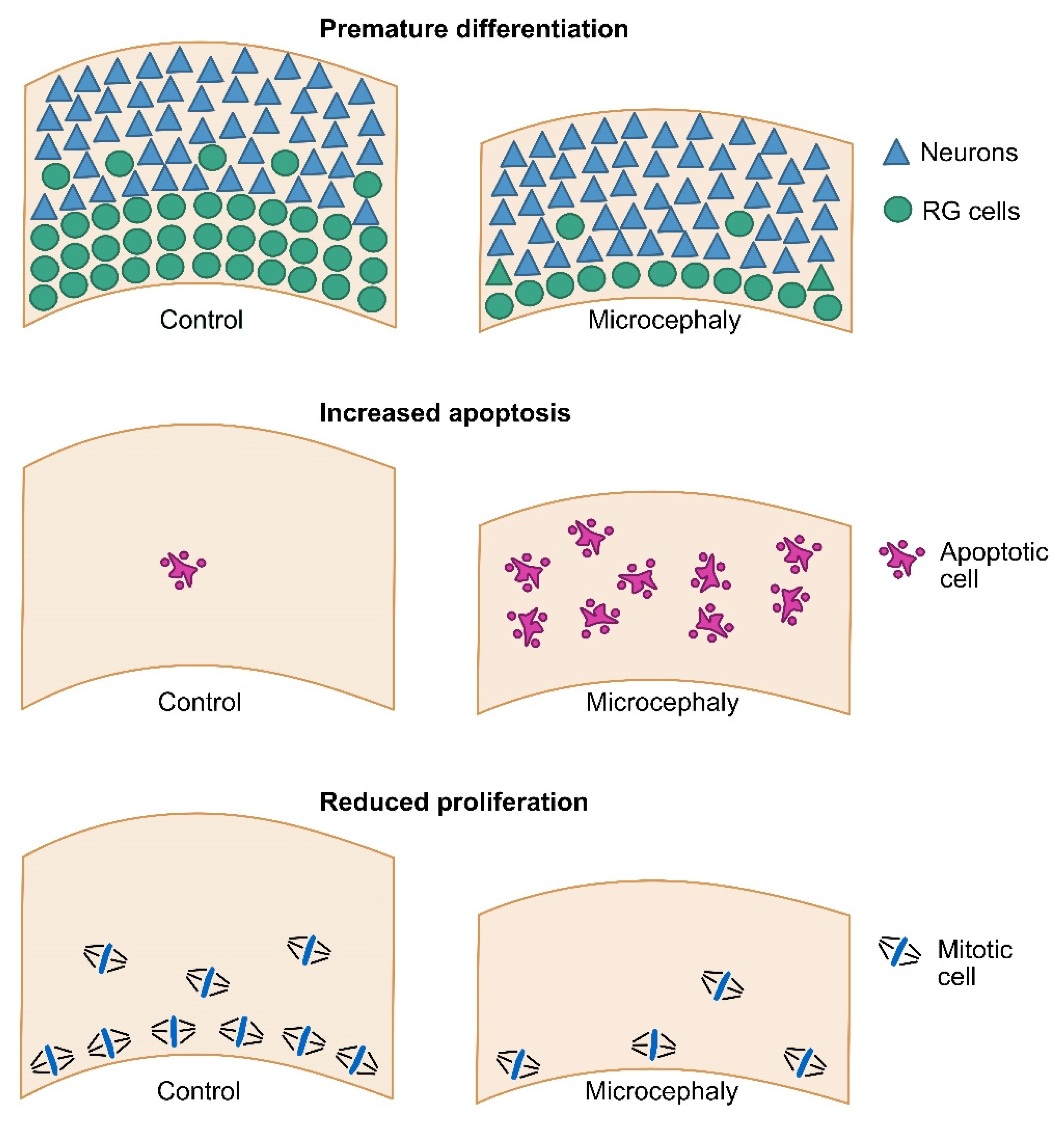
3. Modeling Primary Microcephaly Using Cerebral Organoids
4. Modeling Secondary Microcephaly Using Cerebral Organoids
5. Current Limitations and Challenges
5.1. Tissue Heterogeneity and Reproducibility Issues
5.2. Lack or Paucity of Certain Neural and Non-Neural Derivatives
5.3. Cell Death at the Core of the Organoid
5.4. Cellular Stress Pathways in Organoids
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kalebic, N.; Huttner, W.B. Basal Progenitor Morphology and Neocortex Evolution. Trends Neurosci. 2020, 43, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Fernández, V.; Llinares-Benadero, C.; Borrell, V. Cerebral cortex expansion and folding: What have we learned? EMBO J. 2016, 35, 1021–1044. [Google Scholar] [CrossRef] [PubMed]
- Penisson, M.; Ladewig, J.; Belvindrah, R.; Francis, F. Genes and Mechanisms Involved in the Generation and Amplification of Basal Radial Glial Cells. Front. Cell. Neurosci. 2019, 13, 381. [Google Scholar] [CrossRef] [PubMed]
- Heide, M.; Huttner, W.B. Human-Specific Genes, Cortical Progenitor Cells, and Microcephaly. Cells 2021, 10, 1209. [Google Scholar] [CrossRef] [PubMed]
- Dehay, C.; Kennedy, H. Cell-cycle control and cortical development. Nat. Rev. Neurosci. 2007, 8, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.L.; Skove, S.L.; Rouanet, J.P.; Pilaz, L.-J.; Bepler, T.; Gordân, R.; Wray, G.A.; Silver, D.L. Human-Chimpanzee Differences in a FZD8 Enhancer Alter Cell-Cycle Dynamics in the Developing Neocortex. Curr. Biol. 2015, 25, 772–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Souza, N. Organoids. Nat. Methods 2018, 15, 23. [Google Scholar] [CrossRef]
- Trujillo, C.A.; Muotri, A.R. Brain Organoids and the Study of Neurodevelopment. Trends Mol. Med. 2018, 24, 982–990. [Google Scholar] [CrossRef]
- Sidhaye, J.; Knoblich, J.A. Brain organoids: An ensemble of bioassays to investigate human neurodevelopment and disease. Cell Death Differ. 2021, 28, 52–67. [Google Scholar] [CrossRef]
- Kyrousi, C.; Cappello, S. Using brain organoids to study human neurodevelopment, evolution and disease. Wiley Interdiscip. Rev. Dev. Biol. 2020, 9, e347. [Google Scholar] [CrossRef]
- Zaqout, S.; Kaindl, A.M. Autosomal Recessive Primary Microcephaly: Not Just a Small Brain. Front. Cell Dev. Biol. 2021, 9, 784700. [Google Scholar] [CrossRef] [PubMed]
- Jalink, P.; Caiazzo, M. Brain Organoids: Filling the Need for a Human Model of Neurological Disorder. Biology 2021, 10, 740. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Kamiya, D.; Nishiyama, A.; Katayama, T.; Nozaki, S.; Kawasaki, H.; Watanabe, Y.; Mizuseki, K.; Sasai, Y. Directed differentiation of telencephalic precursors from embryonic stem cells. Nat. Neurosci. 2005, 8, 288–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eiraku, M.; Watanabe, K.; Matsuo-Takasaki, M.; Kawada, M.; Yonemura, S.; Matsumura, M.; Wataya, T.; Nishiyama, A.; Muguruma, K.; Sasai, Y. Self-Organized Formation of Polarized Cortical Tissues from ESCs and Its Active Manipulation by Extrinsic Signals. Cell Stem Cell 2008, 3, 519–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- Wang, H. Modeling Neurological Diseases With Human Brain Organoids. Front. Synaptic Neurosci. 2018, 10, 15. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Corsini, N.S.; Wolfinger, S.; Gustafson, E.H.; Phillips, A.W.; Burkard, T.R.; Otani, T.; Livesey, F.J.; Knoblich, J.A. Guided self-organization and cortical plate formation in human brain organoids. Nat. Biotechnol. 2017, 35, 659–666. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Wu, Q.; Wang, X. Modeling brain development and diseases with human cerebral organoids. Curr. Opin. Neurobiol. 2021, 66, 103–115. [Google Scholar] [CrossRef]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef] [Green Version]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef] [Green Version]
- Paşca, A.M.; Sloan, S.A.; Clarke, L.E.; Tian, Y.; Makinson, C.D.; Huber, N.; Kim, C.H.; Park, J.-Y.; O’Rourke, N.A.; Nguyen, K.D.; et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 2015, 12, 671–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Dosso, A.; Urenda, J.-P.; Nguyen, T.; Quadrato, G. Upgrading the Physiological Relevance of Human Brain Organoids. Neuron 2020, 107, 1014–1028. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Jacob, F.; Song, M.M.; Nguyen, H.N.; Song, H.; Ming, G.-L. Generation of human brain region-specific organoids using a miniaturized spinning bioreactor. Nat. Protoc. 2018, 13, 565–580. [Google Scholar] [CrossRef]
- Kadoshima, T.; Sakaguchi, H.; Nakano, T.; Soen, M.; Ando, S.; Eiraku, M.; Sasai, Y. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc. Natl. Acad. Sci. USA 2013, 110, 20284–20289. [Google Scholar] [CrossRef] [Green Version]
- Velasco, S.; Kedaigle, A.J.; Simmons, S.K.; Nash, A.; Rocha, M.; Quadrato, G.; Paulsen, B.; Nguyen, L.; Adiconis, X.; Regev, A.; et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 2019, 570, 523–527. [Google Scholar] [CrossRef]
- Sloan, S.A.; Andersen, J.; Pașca, A.M.; Birey, F.; Pașca, S.P. Generation and assembly of human brain region-specific three-dimensional cultures. Nat. Protoc. 2018, 13, 2062–2085. [Google Scholar] [CrossRef] [PubMed]
- Bagley, J.A.; Reumann, D.; Bian, S.; Lévi-Strauss, J.; Knoblich, J.A. Fused cerebral organoids model interactions between brain regions. Nat. Methods 2017, 14, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Birey, F.; Andersen, J.; Makinson, C.D.; Islam, S.; Wei, W.; Huber, N.; Fan, H.C.; Metzler, K.R.C.; Panagiotakos, G.; Thom, N.; et al. Assembly of functionally integrated human forebrain spheroids. Nature 2017, 545, 54–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Y.; Tanaka, Y.; Patterson, B.; Kang, Y.-J.; Govindaiah, G.; Roselaar, N.; Cakir, B.; Kim, K.-Y.; Lombroso, A.P.; Hwang, S.-M.; et al. Fusion of Regionally Specified hPSC-Derived Organoids Models Human Brain Development and Interneuron Migration. Cell Stem Cell 2017, 21, 383–398.e7. [Google Scholar] [CrossRef] [Green Version]
- Barrera, J.A.; Kao, L.R.; Hammer, R.E.; Seemann, J.; Fuchs, J.L.; Megraw, T.L. CDK5RAP2 regulates centriole engagement and cohesion in mice. Dev. Cell 2010, 18, 913–926. [Google Scholar] [CrossRef] [Green Version]
- Buchman, J.J.; Tseng, H.C.; Zhou, Y.; Frank, C.L.; Xie, Z.; Tsai, L.H. Cdk5rap2 interacts with pericentrin to maintain the neural progenitor pool in the developing neocortex. Neuron 2010, 66, 386–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lizarraga, S.B.; Margossian, S.P.; Harris, M.H.; Campagna, D.R.; Han, A.P.; Blevins, S.; Mudbhary, R.; Barker, J.E.; Walsh, C.A.; Fleming, M.D. Cdk5rap2 regulates centrosome function and chromosome segregation in neuronal progenitors. Development 2010, 137, 1907–1917. [Google Scholar] [CrossRef] [PubMed]
- Pulvers, J.N.; Bryk, J.; Fish, J.L.; Wilsch-Brauninger, M.; Arai, Y.; Schreier, D.; Naumann, R.; Helppi, J.; Habermann, B.; Vogt, J.; et al. Mutations in mouse Aspm (abnormal spindle-like microcephaly associated) cause not only microcephaly but also major defects in the germline. Proc. Natl. Acad. Sci. USA 2010, 107, 16595–16600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowakowski, T.J.; Pollen, A.A.; Sandoval-Espinosa, C.; Kriegstein, A.R. Transformation of the Radial Glia Scaffold Demarcates Two Stages of Human Cerebral Cortex Development. Neuron 2016, 91, 1219–1227. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Tsai, J.-W.; LaMonica, B.; Kriegstein, A.R. A new subtype of progenitor cell in the mouse embryonic neocortex. Nat. Neurosci. 2011, 14, 555–561. [Google Scholar] [CrossRef] [Green Version]
- Vaid, S.; Camp, J.G.; Hersemann, L.; Eugster Oegema, C.; Heninger, A.-K.; Winkler, S.; Brandl, H.; Sarov, M.; Treutlein, B.; Huttner, W.B.; et al. A novel population of Hopx-dependent basal radial glial cells in the developing mouse neocortex. Dev. Camb. Engl. 2018, 145, dev169276. [Google Scholar] [CrossRef] [Green Version]
- Qian, X.; Su, Y.; Adam, C.D.; Deutschmann, A.U.; Pather, S.R.; Goldberg, E.M.; Su, K.; Li, S.; Lu, L.; Jacob, F.; et al. Sliced Human Cortical Organoids for Modeling Distinct Cortical Layer Formation. Cell Stem Cell 2020, 26, 766–781.e9. [Google Scholar] [CrossRef]
- Iefremova, V.; Manikakis, G.; Krefft, O.; Jabali, A.; Weynans, K.; Wilkens, R.; Marsoner, F.; Brändl, B.; Müller, F.-J.; Koch, P.; et al. An Organoid-Based Model of Cortical Development Identifies Non-Cell-Autonomous Defects in Wnt Signaling Contributing to Miller-Dieker Syndrome. Cell Rep. 2017, 19, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Bershteyn, M.; Nowakowski, T.J.; Pollen, A.A.; Di Lullo, E.; Nene, A.; Wynshaw-Boris, A.; Kriegstein, A.R. Human iPSC-Derived Cerebral Organoids Model Cellular Features of Lissencephaly and Reveal Prolonged Mitosis of Outer Radial Glia. Cell Stem Cell 2017, 20, 435–449.e4. [Google Scholar] [CrossRef] [Green Version]
- Conduit, P.T.; Wainman, A.; Raff, J.W. Centrosome function and assembly in animal cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 611–624. [Google Scholar] [CrossRef]
- LaMonica, B.E.; Lui, J.H.; Hansen, D.V.; Kriegstein, A.R. Mitotic spindle orientation predicts outer radial glial cell generation in human neocortex. Nat. Commun. 2013, 4, 1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, I.; Shitamukai, A.; Kusumoto, F.; Mase, S.; Suetsugu, T.; Omori, A.; Kato, K.; Abe, T.; Shioi, G.; Konno, D.; et al. Endfoot regeneration restricts radial glial state and prevents translocation into the outer subventricular zone in early mammalian brain development. Nat. Cell Biol. 2020, 22, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Thornton, G.K.; Woods, C.G. Primary microcephaly: Do all roads lead to Rome? Trends Genet. 2009, 25, 501–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verloes, A.; Drunat, S.; Passemard, S. ASPM Primary Microcephaly. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Letard, P.; Drunat, S.; Vial, Y.; Duerinckx, S.; Ernault, A.; Amram, D.; Arpin, S.; Bertoli, M.; Busa, T.; Ceulemans, B.; et al. Autosomal recessive primary microcephaly due to ASPM mutations: An update. Hum. Mutat. 2018, 39, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.B.; Sun, X.; Kodani, A.; Borges-Monroy, R.; Girskis, K.M.; Ryu, S.C.; Wang, P.P.; Patel, K.; Gonzalez, D.M.; Woo, Y.M.; et al. Aspm knockout ferret reveals an evolutionary mechanism governing cerebral cortical size. Nature 2018, 556, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Gilardi, C.; Kalebic, N. The Ferret as a Model System for Neocortex Development and Evolution. Front. Cell Dev. Biol. 2021, 9, 661759. [Google Scholar] [CrossRef]
- Li, R.; Sun, L.; Fang, A.; Li, P.; Wu, Q.; Wang, X. Recapitulating cortical development with organoid culture in vitro and modeling abnormal spindle-like (ASPM related primary) microcephaly disease. Protein Cell 2017, 8, 823–833. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 2014, 9, 2329–2340. [Google Scholar] [CrossRef] [Green Version]
- Gabriel, E.; Wason, A.; Ramani, A.; Gooi, L.M.; Keller, P.; Pozniakovsky, A.; Poser, I.; Noack, F.; Telugu, N.S.; Calegari, F.; et al. CPAP promotes timely cilium disassembly to maintain neural progenitor pool. EMBO J. 2016, 35, 803–819. [Google Scholar] [CrossRef]
- Lange, C.; Huttner, W.B.; Calegari, F. Cdk4/cyclinD1 overexpression in neural stem cells shortens G1, delays neurogenesis, and promotes the generation and expansion of basal progenitors. Cell Stem Cell 2009, 5, 320–331. [Google Scholar] [CrossRef] [Green Version]
- Pilaz, L.-J.; Patti, D.; Marcy, G.; Ollier, E.; Pfister, S.; Douglas, R.J.; Betizeau, M.; Gautier, E.; Cortay, V.; Doerflinger, N.; et al. Forced G1-phase reduction alters mode of division, neuron number, and laminar phenotype in the cerebral cortex. Proc. Natl. Acad. Sci. USA 2009, 106, 21924–21929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Yang, S.-L.; Yang, M.; Herrlinger, S.; Shao, Q.; Collar, J.L.; Fierro, E.; Shi, Y.; Liu, A.; Lu, H.; et al. Modeling microcephaly with cerebral organoids reveals a WDR62-CEP170-KIF2A pathway promoting cilium disassembly in neural progenitors. Nat. Commun. 2019, 10, 2612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Li, Z.; Sievert, D.; Smith, D.E.C.; Mendes, M.I.; Chen, D.Y.; Stanley, V.; Ghosh, S.; Wang, Y.; Kara, M.; et al. Loss of NARS1 impairs progenitor proliferation in cortical brain organoids and leads to microcephaly. Nat. Commun. 2020, 11, 4038. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, E.; Ramani, A.; Altinisik, N.; Gopalakrishnan, J. Human Brain Organoids to Decode Mechanisms of Microcephaly. Front. Cell. Neurosci. 2020, 14, 115. [Google Scholar] [CrossRef]
- Heymann, D.L.; Hodgson, A.; Sall, A.A.; Freedman, D.O.; Staples, J.E.; Althabe, F.; Baruah, K.; Mahmud, G.; Kandun, N.; Vasconcelos, P.F.C.; et al. Zika virus and microcephaly: Why is this situation a PHEIC? Lancet 2016, 387, 719–721. [Google Scholar] [CrossRef] [Green Version]
- Cugola, F.R.; Fernandes, I.R.; Russo, F.B.; Freitas, B.C.; Dias, J.L.M.; Guimarães, K.P.; Benazzato, C.; Almeida, N.; Pignatari, G.C.; Romero, S.; et al. The Brazilian Zika virus strain causes birth defects in experimental models. Nature 2016, 534, 267–271. [Google Scholar] [CrossRef] [Green Version]
- Dang, J.; Tiwari, S.K.; Lichinchi, G.; Qin, Y.; Patil, V.S.; Eroshkin, A.M.; Rana, T.M. Zika Virus Depletes Neural Progenitors in Human Cerebral Organoids through Activation of the Innate Immune Receptor TLR3. Cell Stem Cell 2016, 19, 258–265. [Google Scholar] [CrossRef] [Green Version]
- Gabriel, E.; Ramani, A.; Karow, U.; Gottardo, M.; Natarajan, K.; Gooi, L.M.; Goranci-Buzhala, G.; Krut, O.; Peters, F.; Nikolic, M.; et al. Recent Zika Virus Isolates Induce Premature Differentiation of Neural Progenitors in Human Brain Organoids. Cell Stem Cell 2017, 20, 397–406.e5. [Google Scholar] [CrossRef] [Green Version]
- Chahrour, M.; Zoghbi, H.Y. The Story of Rett Syndrome: From Clinic to Neurobiology. Neuron 2007, 56, 422–437. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.R.; Fernandes, T.G.; Vaz, S.H.; Silva, T.P.; Bekman, E.P.; Xapelli, S.; Duarte, S.; Ghazvini, M.; Gribnau, J.; Muotri, A.R.; et al. Modeling Rett Syndrome With Human Patient-Specific Forebrain Organoids. Front. Cell Dev. Biol. 2020, 8, 610427. [Google Scholar] [CrossRef]
- McTague, A.; Howell, K.B.; Cross, J.H.; Kurian, M.A.; Scheffer, I.E. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2016, 15, 304–316. [Google Scholar] [CrossRef]
- Rasika, S.; Passemard, S.; Verloes, A.; Gressens, P.; El Ghouzzi, V. Golgipathies in Neurodevelopment: A New View of Old Defects. Dev. Neurosci. 2019, 40, 396–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberg, D.J.; Repudi, S.; Saleem, A.; Kustanovich, I.; Viukov, S.; Abudiab, B.; Banne, E.; Mahajnah, M.; Hanna, J.H.; Stern, S.; et al. Modeling genetic epileptic encephalopathies using brain organoids. EMBO Mol. Med. 2021, 13, e13610. [Google Scholar] [CrossRef] [PubMed]
- Passemard, S.; Perez, F.; Colin-Lemesre, E.; Rasika, S.; Gressens, P.; El Ghouzzi, V. Golgi trafficking defects in postnatal microcephaly: The evidence for “Golgipathies”. Prog. Neurobiol. 2017, 153, 46–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seifert, W.; Kuhnisch, J.; Maritzen, T.; Lommatzsch, S.; Hennies, H.C.; Bachmann, S.; Horn, D.; Haucke, V. Cohen syndrome-associated protein COH1 physically and functionally interacts with the small GTPase RAB6 at the Golgi complex and directs neurite outgrowth. J. Biol. Chem. 2015, 290, 3349–3358. [Google Scholar] [CrossRef] [Green Version]
- Koike, S.; Jahn, R. SNAREs define targeting specificity of trafficking vesicles by combinatorial interaction with tethering factors. Nat. Commun. 2019, 10, 1608. [Google Scholar] [CrossRef]
- Lee, Y.-K.; Hwang, S.-K.; Lee, S.-K.; Yang, J.; Kwak, J.-H.; Seo, H.; Ahn, H.; Lee, Y.-S.; Kim, J.; Lim, C.-S.; et al. Cohen Syndrome Patient iPSC-Derived Neurospheres and Forebrain-Like Glutamatergic Neurons Reveal Reduced Proliferation of Neural Progenitor Cells and Altered Expression of Synapse Genes. J. Clin. Med. 2020, 9, 1886. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Guhr, A.; Kobold, S.; Seltmann, S.; Seiler Wulczyn, A.E.M.; Kurtz, A.; Löser, P. Recent Trends in Research with Human Pluripotent Stem Cells: Impact of Research and Use of Cell Lines in Experimental Research and Clinical Trials. Stem Cell Rep. 2018, 11, 485–496. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.R.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Bar-Nur, O.; Russ, H.A.; Efrat, S.; Benvenisty, N. Epigenetic memory and preferential lineage-specific differentiation in induced pluripotent stem cells derived from human pancreatic islet beta cells. Cell Stem Cell 2011, 9, 17–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathieu, J.; Ruohola-Baker, H. Metabolic remodeling during the loss and acquisition of pluripotency. Development 2017, 144, 541–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strano, A.; Tuck, E.; Stubbs, V.E.; Livesey, F.J. Variable Outcomes in Neural Differentiation of Human PSCs Arise from Intrinsic Differences in Developmental Signaling Pathways. Cell Rep. 2020, 31, 107732. [Google Scholar] [CrossRef] [PubMed]
- Bauwens, C.L.; Peerani, R.; Niebruegge, S.; Woodhouse, K.A.; Kumacheva, E.; Husain, M.; Zandstra, P.W. Control of human embryonic stem cell colony and aggregate size heterogeneity influences differentiation trajectories. Stem Cells 2008, 26, 2300–2310. [Google Scholar] [CrossRef] [PubMed]
- Krefft, O.; Jabali, A.; Iefremova, V.; Koch, P.; Ladewig, J. Generation of Standardized and Reproducible Forebrain-type Cerebral Organoids from Human Induced Pluripotent Stem Cells. J. Vis. Exp. JoVE 2018, 131, e56768. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.-J.; Elahi, L.S.; Pașca, A.M.; Marton, R.M.; Gordon, A.; Revah, O.; Miura, Y.; Walczak, E.M.; Holdgate, G.M.; Fan, H.C.; et al. Reliability of human cortical organoid generation. Nat. Methods 2019, 16, 75–78. [Google Scholar] [CrossRef]
- Bassett, A.R. Editing the genome of hiPSC with CRISPR/Cas9: Disease models. Mamm. Genome Off. J. Int. Mamm. Genome Soc. 2017, 28, 348–364. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Bielas, S.L.; Zaki, M.S.; Ismail, S.; Farfara, D.; Um, K.; Rosti, R.O.; Scott, E.C.; Tu, S.; Chi, N.C.; et al. Biallelic Mutations in Citron Kinase Link Mitotic Cytokinesis to Human Primary Microcephaly. Am. J. Hum. Genet. 2016, 99, 501–510. [Google Scholar] [CrossRef] [Green Version]
- Ichisima, J.; Suzuki, N.M.; Samata, B.; Awaya, T.; Takahashi, J.; Hagiwara, M.; Nakahata, T.; Saito, M.K. Verification and rectification of cell type-specific splicing of a Seckel syndrome-associated ATR mutation using iPS cell model. J. Hum. Genet. 2019, 64, 445–458. [Google Scholar] [CrossRef] [Green Version]
- Lizarraga, S.B.; Ma, L.; Maguire, A.M.; van Dyck, L.I.; Wu, Q.; Ouyang, Q.; Kavanaugh, B.C.; Nagda, D.; Livi, L.L.; Pescosolido, M.F.; et al. Human neurons from Christianson syndrome iPSCs reveal mutation-specific responses to rescue strategies. Sci. Transl. Med. 2021, 13, eaaw0682. [Google Scholar] [CrossRef]
- Renner, M.; Lancaster, M.A.; Bian, S.; Choi, H.; Ku, T.; Peer, A.; Chung, K.; Knoblich, J.A. Self-organized developmental patterning and differentiation in cerebral organoids. EMBO J. 2017, 36, 1316–1329. [Google Scholar] [CrossRef] [PubMed]
- Wilpert, N.-M.; Marguet, F.; Maillard, C.; Guimiot, F.; Martinovic, J.; Drunat, S.; Attié-Bitach, T.; Razavi, F.; Tessier, A.; Capri, Y.; et al. Human neuropathology confirms projection neuron and interneuron defects and delayed oligodendrocyte production and maturation in FOXG1 syndrome. Eur. J. Med. Genet. 2021, 64, 104282. [Google Scholar] [CrossRef] [PubMed]
- Paciorkowski, A.R.; McDaniel, S.S.; Jansen, L.A.; Tully, H.; Tuttle, E.; Ghoneim, D.H.; Tupal, S.; Gunter, S.A.; Vasta, V.; Zhang, Q.; et al. Novel mutations in ATP1A3 associated with catastrophic early life epilepsy, episodic prolonged apnea, and postnatal microcephaly. Epilepsia 2015, 56, 422–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupuis, N.; Fafouri, A.; Bayot, A.; Kumar, M.; Lecharpentier, T.; Ball, G.; Edwards, D.; Bernard, V.; Dournaud, P.; Drunat, S.; et al. Dymeclin deficiency causes postnatal microcephaly, hypomyelination and reticulum-to-Golgi trafficking defects in mice and humans. Hum. Mol. Genet. 2015, 24, 2771–2783. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.L.; Martínez-Cerdeño, V.; Noctor, S.C. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 4216–4233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, A.; Wake, H.; Ishikawa, A.W.; Eto, K.; Shibata, K.; Murakoshi, H.; Koizumi, S.; Moorhouse, A.J.; Yoshimura, Y.; Nabekura, J. Microglia contact induces synapse formation in developing somatosensory cortex. Nat. Commun. 2016, 7, 12540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [Green Version]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [Green Version]
- Madhavan, M.; Nevin, Z.S.; Shick, H.E.; Garrison, E.; Clarkson-Paredes, C.; Karl, M.; Clayton, B.L.L.; Factor, D.C.; Allan, K.C.; Barbar, L.; et al. Induction of myelinating oligodendrocytes in human cortical spheroids. Nat. Methods 2018, 15, 700–706. [Google Scholar] [CrossRef]
- Marton, R.M.; Miura, Y.; Sloan, S.A.; Li, Q.; Revah, O.; Levy, R.J.; Huguenard, J.R.; Pașca, S.P. Differentiation and maturation of oligodendrocytes in human three-dimensional neural cultures. Nat. Neurosci. 2019, 22, 484–491. [Google Scholar] [CrossRef]
- Ormel, P.R.; Vieira de Sá, R.; van Bodegraven, E.J.; Karst, H.; Harschnitz, O.; Sneeboer, M.A.M.; Johansen, L.E.; van Dijk, R.E.; Scheefhals, N.; Berdenis van Berlekom, A.; et al. Microglia innately develop within cerebral organoids. Nat. Commun. 2018, 9, 4167. [Google Scholar] [CrossRef] [PubMed]
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.M.; Nguyen, C.H.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.M.; Marsh, S.E.; Fimbres, C.; et al. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 2017, 94, 278–293.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagerlund, I.; Dougalis, A.; Shakirzyanova, A.; Gomez-Budia, M.; Konttinen, H.; Ohtonen, S.; Fazaludeen, F.; Koskuvi, M.; Kuusisto, J.; Hernandez, D.; et al. Microglia Orchestrate Neuronal Activity in Brain Organoids. SSRN Electron. J. 2021. [Google Scholar] [CrossRef]
- Qian, X.; Song, H.; Ming, G. Brain organoids: Advances, applications and challenges. Development 2019, 146, dev166074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Sun, L.; Wang, M.; Liu, J.; Zhong, S.; Li, R.; Li, P.; Guo, L.; Fang, A.; Chen, R.; et al. Vascularized human cortical organoids (vOrganoids) model cortical development in vivo. PLoS Biol. 2020, 18, e3000705. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.A.; Gonçalves, J.T.; Bloyd, C.W.; Li, H.; Fernandes, S.; Quang, D.; Johnston, S.; Parylak, S.L.; Jin, X.; Gage, F.H. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 2018, 36, 432–441. [Google Scholar] [CrossRef]
- Giandomenico, S.L.; Mierau, S.B.; Gibbons, G.M.; Wenger, L.M.D.; Masullo, L.; Sit, T.; Sutcliffe, M.; Boulanger, J.; Tripodi, M.; Derivery, E.; et al. Cerebral organoids at the air-liquid interface generate diverse nerve tracts with functional output. Nat. Neurosci. 2019, 22, 669–679. [Google Scholar] [CrossRef]
- Giandomenico, S.L.; Sutcliffe, M.; Lancaster, M.A. Generation and long-term culture of advanced cerebral organoids for studying later stages of neural development. Nat. Protoc. 2021, 16, 579–602. [Google Scholar] [CrossRef]
- Pollen, A.A.; Bhaduri, A.; Andrews, M.G.; Nowakowski, T.J.; Meyerson, O.S.; Mostajo-Radji, M.A.; Di Lullo, E.; Alvarado, B.; Bedolli, M.; Dougherty, M.L.; et al. Establishing Cerebral Organoids as Models of Human-Specific Brain Evolution. Cell 2019, 176, 743–756.e17. [Google Scholar] [CrossRef] [Green Version]
- Gordon, A.; Yoon, S.-J.; Tran, S.S.; Makinson, C.D.; Park, J.Y.; Andersen, J.; Valencia, A.M.; Horvath, S.; Xiao, X.; Huguenard, J.R.; et al. Long-term maturation of human cortical organoids matches key early postnatal transitions. Nat. Neurosci. 2021, 24, 331–342. [Google Scholar] [CrossRef]
- Bhaduri, A.; Andrews, M.G.; Mancia Leon, W.; Jung, D.; Shin, D.; Allen, D.; Jung, D.; Schmunk, G.; Haeussler, M.; Salma, J.; et al. Cell stress in cortical organoids impairs molecular subtype specification. Nature 2020, 578, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Solaesa, V.; Serrano-Lorenzo, P.; Ramos-Arroyo, M.A.; Blázquez, A.; Pagola-Lorz, I.; Artigas-López, M.; Arenas, J.; Martín, M.A.; Jericó-Pascual, I. A Novel Missense Variant Associated with A Splicing Defect in A Myopathic Form of PGK1 Deficiency in The Spanish Population. Genes 2019, 10, 785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izumi, K.; Brett, M.; Nishi, E.; Drunat, S.; Tan, E.S.; Fujiki, K.; Lebon, S.; Cham, B.; Masuda, K.; Arakawa, M.; et al. ARCN1 Mutations Cause a Recognizable Craniofacial Syndrome Due to COPI-Mediated Transport Defects. Am. J. Hum. Genet. 2016, 99, 451–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passemard, S.; Perez, F.; Gressens, P.; El Ghouzzi, V. Endoplasmic reticulum and Golgi stress in microcephaly. Cell Stress 2019, 3, 369–384. [Google Scholar] [CrossRef]
- Cho, A.-N.; Jin, Y.; An, Y.; Kim, J.; Choi, Y.S.; Lee, J.S.; Kim, J.; Choi, W.-Y.; Koo, D.-J.; Yu, W.; et al. Microfluidic device with brain extracellular matrix promotes structural and functional maturation of human brain organoids. Nat. Commun. 2021, 12, 4730. [Google Scholar] [CrossRef]
- Berger, E.; Magliaro, C.; Paczia, N.; Monzel, A.S.; Antony, P.; Linster, C.L.; Bolognin, S.; Ahluwalia, A.; Schwamborn, J.C. Millifluidic culture improves human midbrain organoid vitality and differentiation. Lab. Chip 2018, 18, 3172–3183. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farcy, S.; Albert, A.; Gressens, P.; Baffet, A.D.; El Ghouzzi, V. Cortical Organoids to Model Microcephaly. Cells 2022, 11, 2135. https://doi.org/10.3390/cells11142135
Farcy S, Albert A, Gressens P, Baffet AD, El Ghouzzi V. Cortical Organoids to Model Microcephaly. Cells. 2022; 11(14):2135. https://doi.org/10.3390/cells11142135
Chicago/Turabian StyleFarcy, Sarah, Alexandra Albert, Pierre Gressens, Alexandre D. Baffet, and Vincent El Ghouzzi. 2022. "Cortical Organoids to Model Microcephaly" Cells 11, no. 14: 2135. https://doi.org/10.3390/cells11142135
APA StyleFarcy, S., Albert, A., Gressens, P., Baffet, A. D., & El Ghouzzi, V. (2022). Cortical Organoids to Model Microcephaly. Cells, 11(14), 2135. https://doi.org/10.3390/cells11142135