
Integrin Signaling Shaping BTK-Inhibitor Resistance

 ,
, 
Abstract
:1. Introduction
2. Cell Adhesion-Mediated Drug Resistance
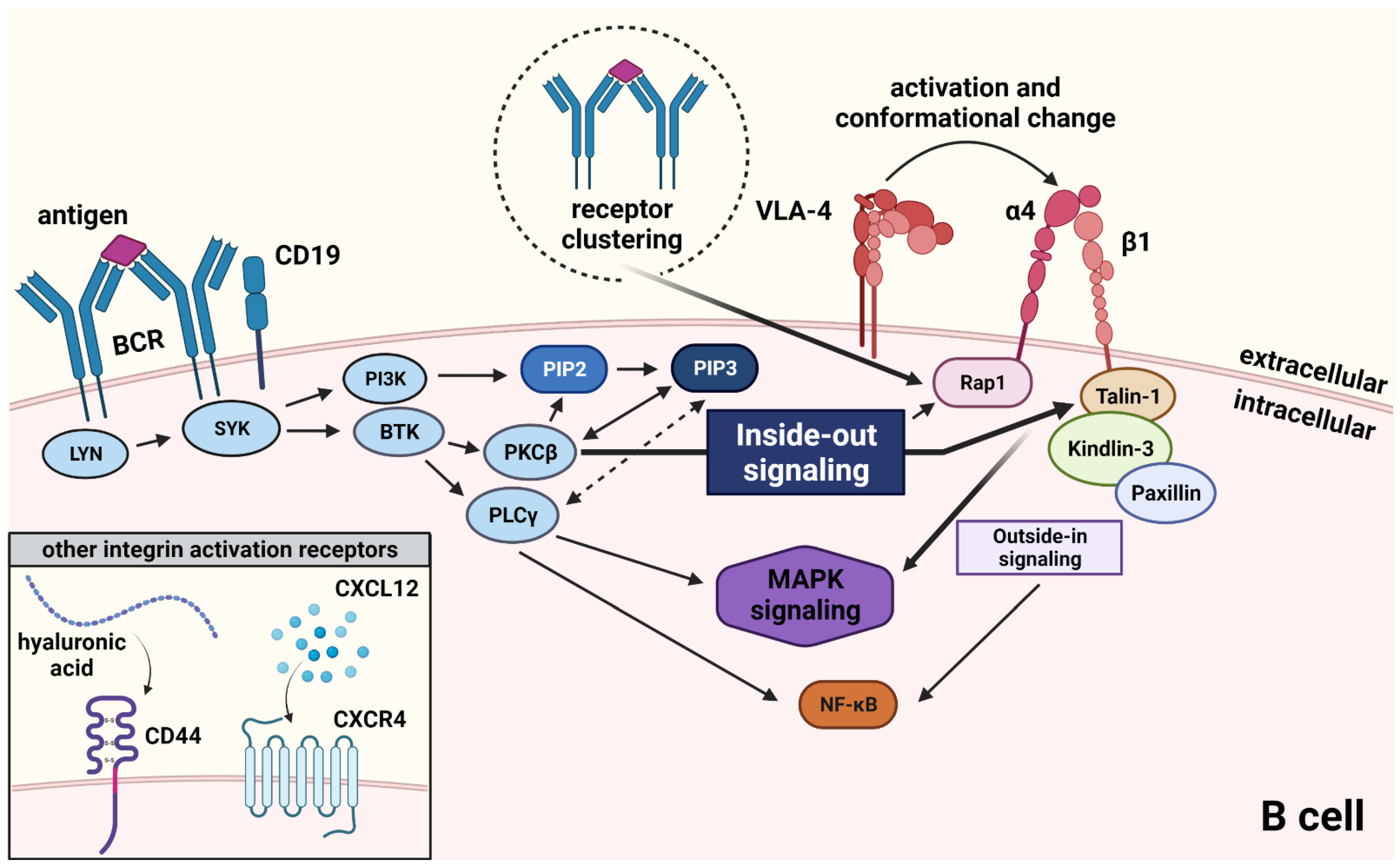
3. VLA-4 Signaling—The Inside-Out Cascade
4. BTK Inhibitor Therapy in Chronic Lymphocytic Leukemia
5. Next-Generation BTK Inhibitors
6. Prognostic and Predictive Markers in the Era of BTK-Inhibitor Therapy
7. Potential Bypass of BTK in the VLA-4 Signaling Activation Cascade
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gaballa, S.; Pinilla-Ibarz, J. BTK inhibitors in chronic Lymphocytic Leukemia. Curr. Hematol. Malig. Rep. 2021, 16, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Malavasi, F. Chronic lymphocytic leukemia microenvironment: Shifting the balance from apoptosis to proliferation. Haematologica 2009, 94, 752–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harzschel, A.; Zucchetto, A.; Gattei, V.; Hartmann, T.N. VLA-4 expression and activation in B cell malignancies: Functional and clinical aspects. Int. J. Mol. Sci. 2020, 21, 2206. [Google Scholar] [CrossRef] [Green Version]
- Rip, J.; de Bruijn, M.J.W.; Appelman, M.K.; Pal Singh, S.; Hendriks, R.W.; Corneth, O.B.J. Toll-like receptor signaling drives btk-mediated autoimmune disease. Front. Immunol. 2019, 10, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendriks, R.W.; Yuvaraj, S.; Kil, L.P. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat. Rev. Cancer 2014, 14, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Ponader, S.; Chen, S.S.; Buggy, J.J.; Balakrishnan, K.; Gandhi, V.; Wierda, W.G.; Keating, M.J.; O’Brien, S.; Chiorazzi, N.; Burger, J.A. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood 2012, 119, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Tissino, E.; Benedetti, D.; Herman, S.E.M.; Ten Hacken, E.; Ahn, I.E.; Chaffee, K.G.; Rossi, F.M.; Dal Bo, M.; Bulian, P.; Bomben, R.; et al. Functional and clinical relevance of VLA-4 (CD49d/CD29) in ibrutinib-treated chronic lymphocytic leukemia. J. Exp. Med. 2018, 215, 681–697. [Google Scholar] [CrossRef] [Green Version]
- Damiano, J.S.; Cress, A.E.; Hazlehurst, L.A.; Shtil, A.A.; Dalton, W.S. Cell adhesion mediated drug resistance (CAM-DR): Role of integrins and resistance to apoptosis in human myeloma cell lines. Blood 1999, 93, 1658–1667. [Google Scholar] [CrossRef] [Green Version]
- Cervantes-Villagrana, R.D.; Albores-Garcia, D.; Cervantes-Villagrana, A.R.; Garcia-Acevez, S.J. Tumor-induced neurogenesis and immune evasion as targets of innovative anti-cancer therapies. Signal Transduct. Target. Ther. 2020, 5, 99. [Google Scholar] [CrossRef]
- Ata, R.; Antonescu, C.N. Integrins and cell metabolism: An intimate relationship impacting cancer. Int. J. Mol. Sci. 2017, 18, 189. [Google Scholar] [CrossRef] [Green Version]
- Blandin, A.F.; Renner, G.; Lehmann, M.; Lelong-Rebel, I.; Martin, S.; Dontenwill, M. Beta1 integrins as therapeutic targets to disrupt hallmarks of cancer. Front. Pharmacol. 2015, 6, 279. [Google Scholar] [CrossRef] [PubMed]
- Byron, A.; Frame, M.C. Adhesion protein networks reveal functions proximal and distal to cell-matrix contacts. Curr. Opin. Cell Biol. 2016, 39, 93–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Windmoller, B.A.; Beshay, M.; Helweg, L.P.; Flottmann, C.; Beermann, M.; Forster, C.; Wilkens, L.; Greiner, J.F.W.; Kaltschmidt, C.; Kaltschmidt, B. Novel primary human cancer stem-like cell populations from non-small cell lung cancer: Inhibition of cell survival by targeting NF-kappaB and MYC signaling. Cells 2021, 10, 1024. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta 2011, 1813, 1619–1633. [Google Scholar] [CrossRef] [Green Version]
- Afasizheva, A.; Devine, A.; Tillman, H.; Fung, K.L.; Vieira, W.D.; Blehm, B.H.; Kotobuki, Y.; Busby, B.; Chen, E.I.; Tanner, K. Mitogen-activated protein kinase signaling causes malignant melanoma cells to differentially alter extracellular matrix biosynthesis to promote cell survival. BMC Cancer 2016, 16, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, H.; Quearry, B.; Ruiz-Vela, A.; Korsmeyer, S.J. Survival factor-induced extracellular signal-regulated kinase phosphorylates BIM, inhibiting its association with BAX and proapoptotic activity. Proc. Natl. Acad. Sci. USA 2004, 101, 15313–15317. [Google Scholar] [CrossRef] [Green Version]
- Hazlehurst, L.A.; Argilagos, R.F.; Dalton, W.S. Beta1 integrin mediated adhesion increases Bim protein degradation and contributes to drug resistance in leukaemia cells. Br. J. Haematol. 2007, 136, 269–275. [Google Scholar] [CrossRef]
- Weston, C.R.; Balmanno, K.; Chalmers, C.; Hadfield, K.; Molton, S.A.; Ley, R.; Wagner, E.F.; Cook, S.J. Activation of ERK1/2 by deltaRaf-1:ER* represses Bim expression independently of the JNK or PI3K pathways. Oncogene 2003, 22, 1281–1293. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Bhattacharyya, J.; Jaganathan, B.G. Adhesion to stromal cells mediates imatinib resistance in chronic myeloid leukemia through ERK and BMP signaling pathways. Sci. Rep. 2017, 7, 9535. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Blenis, J.; Yuan, J. Activation of PI3K/Akt and MAPK pathways regulates Myc-mediated transcription by phosphorylating and promoting the degradation of Mad1. Proc. Natl. Acad. Sci. USA 2008, 105, 6584–6589. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Piao, C.; Zhang, Z.; Jiang, Y.; Kong, C. The potential role of c-MYC and polyamine metabolism in multiple drug resistance in bladder cancer investigated by metabonomics. Genomics 2022, 114, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Fang, L.; Kwantwi, L.B.; He, G.; Luo, W.; Yang, L.; Huang, Y.; Yin, S.; Cai, Y.; Ma, W.; et al. N-Myc promotes angiogenesis and therapeutic resistance of prostate cancer by TEM8. Med. Oncol. 2021, 38, 127. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Verma, R.; Nakamoto-Matsubara, R.; Siu, K.T.; Panaroni, C.; Fulzele, K.S.; Mukaihara, K.; Onyewadume, C.; Maebius, A.; Kato, H.; et al. Low NCOR2 levels in multiple myeloma patients drive multidrug resistance via MYC upregulation. Blood Cancer J. 2021, 11, 194. [Google Scholar] [CrossRef] [PubMed]
- Sodir, N.M.; Kortlever, R.M.; Barthet, V.J.A.; Campos, T.; Pellegrinet, L.; Kupczak, S.; Anastasiou, P.; Swigart, L.B.; Soucek, L.; Arends, M.J.; et al. MYC Instructs and Maintains Pancreatic Adenocarcinoma Phenotype. Cancer Discov. 2020, 10, 588–607. [Google Scholar] [CrossRef] [Green Version]
- Benaud, C.M.; Dickson, R.B. Regulation of the expression of c-Myc by beta1 integrins in epithelial cells. Oncogene 2001, 20, 759–768. [Google Scholar] [CrossRef] [Green Version]
- van Golen, C.M.; Soules, M.E.; Grauman, A.R.; Feldman, E.L. N-Myc overexpression leads to decreased beta1 integrin expression and increased apoptosis in human neuroblastoma cells. Oncogene 2003, 22, 2664–2673. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.M.; Giltnane, J.M.; Balko, J.M.; Schwarz, L.J.; Guerrero-Zotano, A.L.; Hutchinson, K.E.; Nixon, M.J.; Estrada, M.V.; Sanchez, V.; Sanders, M.E.; et al. MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab. 2017, 26, 633–647.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dharmaraja, A.T. Role of Reactive Oxygen Species (ROS) in Therapeutics and Drug Resistance in Cancer and Bacteria. J. Med. Chem. 2017, 60, 3221–3240. [Google Scholar] [CrossRef]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Fouani, L.; Kovacevic, Z.; Richardson, D.R. Targeting Oncogenic Nuclear Factor Kappa B Signaling with Redox-Active Agents for Cancer Treatment. Antioxid. Redox. Signal. 2019, 30, 1096–1123. [Google Scholar] [CrossRef]
- Bentires-Alj, M.; Barbu, V.; Fillet, M.; Chariot, A.; Relic, B.; Jacobs, N.; Gielen, J.; Merville, M.P.; Bours, V. NF-kappaB transcription factor induces drug resistance through MDR1 expression in cancer cells. Oncogene 2003, 22, 90–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Q.; Xiao, W.; Sun, S.; Sohrabi, A.; Liang, J.; Seidlits, S.K. Extracellular Matrix Proteins Confer Cell Adhesion-Mediated Drug Resistance Through Integrin alpha v in Glioblastoma Cells. Front. Cell Dev. Biol. 2021, 9, 616580. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Shi, Y.; Zhao, S.; Shi, T.; Zhang, G. NF-kappaB signaling and integrin-beta1 inhibition attenuates osteosarcoma metastasis via increased cell apoptosis. Int. J. Biol. Macromol. 2019, 123, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.M.; Zhang, H.; Park, C.C. NF-kappaB regulates radioresistance mediated by beta1-integrin in three-dimensional culture of breast cancer cells. Cancer Res. 2013, 73, 3737–3748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arroyo, A.G.; Sanchez-Mateos, P.; Campanero, M.R.; Martin-Padura, I.; Dejana, E.; Sanchez-Madrid, F. Regulation of the VLA integrin-ligand interactions through the beta 1 subunit. J. Cell Biol. 1992, 117, 659–670. [Google Scholar] [CrossRef] [Green Version]
- Ryan, D.H.; Nuccie, B.L.; Abboud, C.N.; Winslow, J.M. Vascular cell adhesion molecule-1 and the integrin VLA-4 mediate adhesion of human B cell precursors to cultured bone marrow adherent cells. J. Clin. Invest. 1991, 88, 995–1004. [Google Scholar] [CrossRef]
- Baiula, M.; Spampinato, S.; Gentilucci, L.; Tolomelli, A. Novel ligands targeting alpha4beta1 integrin: Therapeutic applications and perspectives. Front. Chem. 2019, 7, 489. [Google Scholar] [CrossRef]
- Tissino, E.; Pivetta, E.; Capuano, A.; Capasso, G.; Bomben, R.; Caldana, C.; Rossi, F.M.; Pozzo, F.; Benedetti, D.; Boldorini, R.; et al. Elastin MIcrofibriL INterfacer1 (EMILIN-1) is an alternative prosurvival VLA-4 ligand in chronic lymphocytic leukemia. Hematol. Oncol. 2022, 40, 181–190. [Google Scholar] [CrossRef]
- Bayless, K.J.; Meininger, G.A.; Scholtz, J.M.; Davis, G.E. Osteopontin is a ligand for the alpha4beta1 integrin. J. Cell Sci. 1998, 111 Pt 9, 1165–1174. [Google Scholar] [CrossRef]
- Scalici, J.M.; Harrer, C.; Allen, A.; Jazaeri, A.; Atkins, K.A.; McLachlan, K.R.; Slack-Davis, J.K. Inhibition of alpha4beta1 integrin increases ovarian cancer response to carboplatin. Gynecol. Oncol. 2014, 132, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Klemke, M.; Weschenfelder, T.; Konstandin, M.H.; Samstag, Y. High affinity interaction of integrin alpha4beta1 (VLA-4) and vascular cell adhesion molecule 1 (VCAM-1) enhances migration of human melanoma cells across activated endothelial cell layers. J. Cell Physiol. 2007, 212, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Soto, M.S.; Serres, S.; Anthony, D.C.; Sibson, N.R. Functional role of endothelial adhesion molecules in the early stages of brain metastasis. Neuro. Oncol. 2014, 16, 540–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.; Sharma, R.; Khaket, T.P.; Dutta, C.; Chakraborty, B.; Mukherjee, T.K. Breast cancer metastasis: Putative therapeutic role of vascular cell adhesion molecule-1. Cell Oncol. 2017, 40, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Rettig, M.P.; Ansstas, G.; DiPersio, J.F. Mobilization of hematopoietic stem and progenitor cells using inhibitors of CXCR4 and VLA-4. Leukemia 2012, 26, 34–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldschmidt, J.M.; Simon, A.; Wider, D.; Muller, S.J.; Follo, M.; Ihorst, G.; Decker, S.; Lorenz, J.; Chatterjee, M.; Azab, A.K.; et al. CXCL12 and CXCR7 are relevant targets to reverse cell adhesion-mediated drug resistance in multiple myeloma. Br. J. Haematol. 2017, 179, 36–49. [Google Scholar] [CrossRef]
- Fontana, F.; Scott, M.J.; Allen, J.S.; Yang, X.; Cui, G.; Pan, D.; Yanaba, N.; Fiala, M.A.; O’Neal, J.; Schmieder-Atteberry, A.H.; et al. VLA4-Targeted Nanoparticles Hijack Cell Adhesion-Mediated Drug Resistance to Target Refractory Myeloma Cells and Prolong Survival. Clin. Cancer Res. 2021, 27, 1974–1986. [Google Scholar] [CrossRef]
- Takeda, T.; Tsubak, M.; Genno, S.; Matsuda, T.; Yamamoto, Y.; Ueda, E.; Imano, M.; Satou, T.; Nishida, S. CD49d and CD49e induce cell adhesion-mediated drug resistance through the nuclear factor-kappaB pathway in Burkitt lymphoma. J. Physiol. Pharmacol. 2020, 71, 467–471. [Google Scholar] [CrossRef]
- Gutjahr, J.C.; Bayer, E.; Yu, X.; Laufer, J.M.; Hopner, J.P.; Tesanovic, S.; Harzschel, A.; Auer, G.; Riess, T.; Salmhofer, A.; et al. CD44 engagement enhances acute myeloid leukemia cell adhesion to the bone marrow microenvironment by increasing VLA-4 avidity. Haematologica 2021, 106, 2102–2113. [Google Scholar] [CrossRef]
- Jung, O.; Beauvais, D.M.; Adams, K.M.; Rapraeger, A.C. VLA-4 phosphorylation during tumor and immune cell migration relies on its coupling to VEGFR2 and CXCR4 by syndecan-1. J. Cell Sci. 2019, 132, jcs232645. [Google Scholar] [CrossRef]
- Gauld, S.B.; Cambier, J.C. Src-family kinases in B-cell development and signaling. Oncogene 2004, 23, 8001–8006. [Google Scholar] [CrossRef] [Green Version]
- Tse, K.W.; Dang-Lawson, M.; Lee, R.L.; Vong, D.; Bulic, A.; Buckbinder, L.; Gold, M.R. B cell receptor-induced phosphorylation of Pyk2 and focal adhesion kinase involves integrins and the Rap GTPases and is required for B cell spreading. J. Biol. Chem. 2009, 284, 22865–22877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reedquist, K.A.; Ross, E.; Koop, E.A.; Wolthuis, R.M.; Zwartkruis, F.J.; van Kooyk, Y.; Salmon, M.; Buckley, C.D.; Bos, J.L. The small GTPase, Rap1, mediates CD31-induced integrin adhesion. J. Cell Biol. 2000, 148, 1151–1158. [Google Scholar] [CrossRef]
- Lagarrigue, F.; Paul, D.S.; Gingras, A.R.; Valadez, A.J.; Sun, H.; Lin, J.; Cuevas, M.N.; Ablack, J.N.; Lopez-Ramirez, M.A.; Bergmeier, W.; et al. Talin-1 is the principal platelet Rap1 effector of integrin activation. Blood 2020, 136, 1180–1190. [Google Scholar] [CrossRef]
- Han, J.; Lim, C.J.; Watanabe, N.; Soriani, A.; Ratnikov, B.; Calderwood, D.A.; Puzon-McLaughlin, W.; Lafuente, E.M.; Boussiotis, V.A.; Shattil, S.J.; et al. Reconstructing and deconstructing agonist-induced activation of integrin alphaIIbbeta3. Curr. Biol. 2006, 16, 1796–1806. [Google Scholar] [CrossRef] [Green Version]
- Hyduk, S.J.; Rullo, J.; Cano, A.P.; Xiao, H.; Chen, M.; Moser, M.; Cybulsky, M.I. Talin-1 and kindlin-3 regulate alpha4beta1 integrin-mediated adhesion stabilization, but not G protein-coupled receptor-induced affinity upregulation. J. Immunol. 2011, 187, 4360–4368. [Google Scholar] [CrossRef] [Green Version]
- Moser, M.; Nieswandt, B.; Ussar, S.; Pozgajova, M.; Fassler, R. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat. Med. 2008, 14, 325–330. [Google Scholar] [CrossRef]
- Harzschel, A.; Li, L.; Krenn, P.W.; Szenes-Nagy, E.; Andrieux, G.; Bayer, E.; Pfeifer, D.; Polcik, L.; Denk, U.; Hopner, J.P.; et al. Kindlin-3 maintains marginal zone B cells but confines follicular B cell activation and differentiation. J. Leukoc. Biol. 2022, 111, 745–758. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.T.; Xu, Z.; Shi, X.; Liu, S.; Schulte, M.L.; White, G.C.; Ma, Y.Q. Paxillin binding to the PH domain of kindlin-3 in platelets is required to support integrin alphaIIbbeta3 outside-in signaling. J. Thromb. Haemost. 2021, 19, 3126–3138. [Google Scholar] [CrossRef] [PubMed]
- Alon, R.; Feigelson, S.W.; Manevich, E.; Rose, D.M.; Schmitz, J.; Overby, D.R.; Winter, E.; Grabovsky, V.; Shinder, V.; Matthews, B.D.; et al. Alpha4beta1-dependent adhesion strengthening under mechanical strain is regulated by paxillin association with the alpha4-cytoplasmic domain. J. Cell Biol. 2005, 171, 1073–1084. [Google Scholar] [CrossRef]
- Ross, S.H.; Spanjaard, E.; Post, A.; Vliem, M.J.; Kristyanto, H.; Bos, J.L.; de Rooij, J. Rap1 can bypass the FAK-Src-Paxillin cascade to induce cell spreading and focal adhesion formation. PLoS ONE 2012, 7, e50072. [Google Scholar] [CrossRef] [Green Version]
- Chigaev, A.; Waller, A.; Zwartz, G.J.; Buranda, T.; Sklar, L.A. Regulation of cell adhesion by affinity and conformational unbending of alpha4beta1 integrin. J. Immunol. 2007, 178, 6828–6839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartfield, P.J.; Greaves, M.W.; Camp, R.D. Beta 1 integrin-mediated T cell adhesion is regulated by calcium ionophores and endoplasmic reticulum Ca(2+)-ATPase inhibitors. Biochem. Biophys. Res. Commun. 1993, 196, 1183–1187. [Google Scholar] [CrossRef] [PubMed]
- Sjaastad, M.D.; Nelson, W.J. Integrin-mediated calcium signaling and regulation of cell adhesion by intracellular calcium. Bioessays 1997, 19, 47–55. [Google Scholar] [CrossRef]
- Roman-Garcia, S.; Merino-Cortes, S.V.; Gardeta, S.R.; de Bruijn, M.J.W.; Hendriks, R.W.; Carrasco, Y.R. Distinct roles for bruton’s tyrosine kinase in B cell immune synapse formation. Front. Immunol. 2018, 9, 2027. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Wahl, M.I.; Afar, D.E.; Turck, C.W.; Rawlings, D.J.; Tam, C.; Scharenberg, A.M.; Kinet, J.P.; Witte, O.N. Regulation of Btk function by a major autophosphorylation site within the SH3 domain. Immunity 1996, 4, 515–525. [Google Scholar] [CrossRef] [Green Version]
- Pal Singh, S.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 2018, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Wahl, M.I.; Fluckiger, A.C.; Kato, R.M.; Park, H.; Witte, O.N.; Rawlings, D.J. Phosphorylation of two regulatory tyrosine residues in the activation of Bruton’s tyrosine kinase via alternative receptors. Proc. Natl. Acad. Sci. USA 1997, 94, 11526–11533. [Google Scholar] [CrossRef] [Green Version]
- Scharenberg, A.M.; Kinet, J.P. PtdIns-3,4,5-P3: A regulatory nexus between tyrosine kinases and sustained calcium signals. Cell 1998, 94, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef] [Green Version]
- Eichhorst, B.; Robak, T.; Montserrat, E.; Ghia, P.; Niemann, C.U.; Kater, A.P.; Gregor, M.; Cymbalista, F.; Buske, C.; Hillmen, P.; et al. Chronic lymphocytic leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2021, 32, 23–33. [Google Scholar] [CrossRef]
- Ghia, P.; Ferreri, A.M.; Caligaris-Cappio, F. Chronic lymphocytic leukemia. Crit. Rev. Oncol. Hematol. 2007, 64, 234–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer, G.; Montserrat, E. Critical molecular pathways in CLL therapy. Mol. Med. 2018, 24, 9. [Google Scholar] [CrossRef] [PubMed]
- Furstenau, M.; Bahlo, J.; Fink, A.M.; Lange, E.; Dreger, P.; Dreyling, M.; Hess, G.; Ritgen, M.; Kneba, M.; Dohner, H.; et al. Residual abdominal lymphadenopathy after intensive frontline chemoimmunotherapy is associated with inferior outcome independently of minimal residual disease status in chronic lymphocytic leukemia. Leukemia 2020, 34, 924–928. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Zhou, X.; Yang, J.; Shi, H.; Li, H.; Zhao, X.; Ma, X. The Role of Tumor-Stroma Interactions in Drug Resistance Within Tumor Microenvironment. Front. Cell Dev. Biol. 2021, 9, 637675. [Google Scholar] [CrossRef] [PubMed]
- Abrams, S.L.; Steelman, L.S.; Shelton, J.G.; Wong, E.W.; Chappell, W.H.; Basecke, J.; Stivala, F.; Donia, M.; Nicoletti, F.; Libra, M.; et al. The Raf/MEK/ERK pathway can govern drug resistance, apoptosis and sensitivity to targeted therapy. Cell Cycle 2010, 9, 1781–1791. [Google Scholar] [CrossRef] [Green Version]
- Tambaro, F.P.; De Novellis, D.; Wierda, W.G. The Role of BTK Inhibition in the Treatment of Chronic Lymphocytic Leukemia: A Clinical View. J. Exp. Pharmacol. 2021, 13, 923–935. [Google Scholar] [CrossRef]
- Woyach, J.A.; Smucker, K.; Smith, L.L.; Lozanski, A.; Zhong, Y.; Ruppert, A.S.; Lucas, D.; Williams, K.; Zhao, W.; Rassenti, L.; et al. Prolonged lymphocytosis during ibrutinib therapy is associated with distinct molecular characteristics and does not indicate a suboptimal response to therapy. Blood 2014, 123, 1810–1817. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, S.; Jones, J.A.; Coutre, S.E.; Mato, A.R.; Hillmen, P.; Tam, C.; Osterborg, A.; Siddiqi, T.; Thirman, M.J.; Furman, R.R.; et al. Ibrutinib for patients with relapsed or refractory chronic lymphocytic leukaemia with 17p deletion (RESONATE-17): A phase 2, open-label, multicentre study. Lancet Oncol. 2016, 17, 1409–1418. [Google Scholar] [CrossRef]
- Burger, J.A.; Barr, P.M.; Robak, T.; Owen, C.; Ghia, P.; Tedeschi, A.; Bairey, O.; Hillmen, P.; Coutre, S.E.; Devereux, S.; et al. Long-term efficacy and safety of first-line ibrutinib treatment for patients with CLL/SLL: 5 years of follow-up from the phase 3 RESONATE-2 study. Leukemia 2020, 34, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Burger, J.A.; Tedeschi, A.; Barr, P.M.; Robak, T.; Owen, C.; Ghia, P.; Bairey, O.; Hillmen, P.; Bartlett, N.L.; Li, J.; et al. Ibrutinib as Initial Therapy for Patients with Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2015, 373, 2425–2437. [Google Scholar] [CrossRef]
- Pula, B.; Golos, A.; Gorniak, P.; Jamroziak, K. Overcoming Ibrutinib Resistance in Chronic Lymphocytic Leukemia. Cancers 2019, 11, 1834. [Google Scholar] [CrossRef] [Green Version]
- Paydas, S. Management of adverse effects/toxicity of ibrutinib. Crit. Rev. Oncol. Hematol. 2019, 136, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Mato, A.R.; Nabhan, C.; Thompson, M.C.; Lamanna, N.; Brander, D.M.; Hill, B.; Howlett, C.; Skarbnik, A.; Cheson, B.D.; Zent, C.; et al. Toxicities and outcomes of 616 ibrutinib-treated patients in the United States: A real-world analysis. Haematologica 2018, 103, 874–879. [Google Scholar] [CrossRef] [PubMed]
- Byrd, J.C.; Furman, R.R.; Coutre, S.E.; Flinn, I.W.; Burger, J.A.; Blum, K.A.; Grant, B.; Sharman, J.P.; Coleman, M.; Wierda, W.G.; et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2013, 369, 32–42. [Google Scholar] [CrossRef]
- Levy, V.; Delmer, A.; Cymbalista, F. Frontline treatment in CLL: The case for time-limited treatment. Hematol. Am. Soc. Hematol. Educ. Program 2021, 2021, 59–67. [Google Scholar] [CrossRef]
- Woyach, J.A.; Johnson, A.J. Targeted therapies in CLL: Mechanisms of resistance and strategies for management. Blood 2015, 126, 471–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddocks, K.J.; Ruppert, A.S.; Lozanski, G.; Heerema, N.A.; Zhao, W.; Abruzzo, L.; Lozanski, A.; Davis, M.; Gordon, A.; Smith, L.L.; et al. Etiology of ibrutinib therapy discontinuation and outcomes in patients with chronic lymphocytic leukemia. JAMA Oncol. 2015, 1, 80–87. [Google Scholar] [CrossRef]
- Sedlarikova, L.; Petrackova, A.; Papajik, T.; Turcsanyi, P.; Kriegova, E. Resistance-associated mutations in chronic lymphocytic leukemia patients treated with novel agents. Front. Oncol. 2020, 10, 894. [Google Scholar] [CrossRef]
- Byrd, J.C.; Harrington, B.; O’Brien, S.; Jones, J.A.; Schuh, A.; Devereux, S.; Chaves, J.; Wierda, W.G.; Awan, F.T.; Brown, J.R.; et al. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2016, 374, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Miao, Y.; Xu, W.; Li, J. Assessing the pharmacokinetics of acalabrutinib in the treatment of chronic lymphocytic leukemia. Expert Opin. Drug Metab. Toxicol. 2021, 17, 1023–1030. [Google Scholar] [CrossRef]
- Byrd, J.C.; Hillmen, P.; Ghia, P.; Kater, A.P.; Chanan-Khan, A.; Furman, R.R.; O’Brien, S.; Yenerel, M.N.; Illes, A.; Kay, N.; et al. Acalabrutinib versus ibrutinib in previously treated chronic lymphocytic leukemia: Results of the first randomized phase III trial. J. Clin. Oncol. 2021, 39, 3441–3452. [Google Scholar] [CrossRef] [PubMed]
- Woyach, J.; Huang, Y.; Rogers, K.; Bhat, S.A.; Grever, M.R.; Lozanski, A.; Doong, T.-J.; Blachly, J.S.; Lozanski, G.; Jones, D.; et al. Resistance to acalabrutinib in CLL is mediated primarily by BTK mutations. Blood 2019, 134, 504. [Google Scholar] [CrossRef]
- Hillmen, P.; Brown, J.R.; Eichhorst, B.F.; Lamanna, N.; O’Brien, S.M.; Qiu, L.; Salmi, T.; Hilger, J.; Wu, K.; Cohen, A.; et al. ALPINE: Zanubrutinib versus ibrutinib in relapsed/refractory chronic lymphocytic leukemia/small lymphocytic lymphoma. Future Oncol. 2020, 16, 517–523. [Google Scholar] [CrossRef] [Green Version]
- Soumerai, J.D.; Mato, A.R.; Dogan, A.; Seshan, V.E.; Joffe, E.; Flaherty, K.; Carter, J.; Hochberg, E.; Barnes, J.A.; Hamilton, A.M.; et al. Zanubrutinib, obinutuzumab, and venetoclax with minimal residual disease-driven discontinuation in previously untreated patients with chronic lymphocytic leukaemia or small lymphocytic lymphoma: A multicentre, single-arm, phase 2 trial. Lancet Haematol. 2021, 8, e879–e890. [Google Scholar] [CrossRef]
- Tam, C.S.; Robak, T.; Ghia, P.; Kahl, B.S.; Walker, P.; Janowski, W.; Simpson, D.; Shadman, M.; Ganly, P.S.; Laurenti, L.; et al. Zanubrutinib monotherapy for patients with treatment naive chronic lymphocytic leukemia and 17p deletion. Haematologica 2020, 106, 2354–2363. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.S.; Trotman, J.; Opat, S.; Burger, J.A.; Cull, G.; Gottlieb, D.; Harrup, R.; Johnston, P.B.; Marlton, P.; Munoz, J.; et al. Phase 1 study of the selective BTK inhibitor zanubrutinib in B-cell malignancies and safety and efficacy evaluation in CLL. Blood 2019, 134, 851–859. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, S.; Zhou, K.; Pan, L.; Li, Z.; Zhou, J.; Gao, S.; Zhou, D.; Hu, J.; Feng, R.; et al. Treatment of relapsed/refractory chronic lymphocytic leukemia/small lymphocytic lymphoma with the BTK inhibitor zanubrutinib: Phase 2, single-arm, multicenter study. J. Hematol. Oncol. 2020, 13, 48. [Google Scholar] [CrossRef] [PubMed]
- Byrd, J.C.; Brown, J.R.; O’Brien, S.; Barrientos, J.C.; Kay, N.E.; Reddy, N.M.; Coutre, S.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N. Engl. J. Med. 2014, 371, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Shadman, M.; Flinn, I.W.; Levy, M.Y.; Porter, R.; Burke, J.M.; Cultrera, J.L.; Misleh, J.; Zafar, S.F.; Freeman, B.; Rao, S.S.; et al. Phase 2 Study of Zanubrutinib in BTK Inhibitor-Intolerant Patients (Pts) with Relapsed/Refractory B-Cell Malignancies. Blood 2021, 138, 1410. [Google Scholar] [CrossRef]
- Estupinan, H.Y.; Wang, Q.; Berglof, A.; Schaafsma, G.C.P.; Shi, Y.; Zhou, L.; Mohammad, D.K.; Yu, L.; Vihinen, M.; Zain, R.; et al. BTK gatekeeper residue variation combined with cysteine 481 substitution causes super-resistance to irreversible inhibitors acalabrutinib, ibrutinib and zanubrutinib. Leukemia 2021, 35, 1317–1329. [Google Scholar] [CrossRef]
- Wang, M.; Shah, N.N.; Alencar, A.J.; Gerson, J.N.; Patel, M.R.; Fakhri, B.; Jurczak, W.; Tan, X.N.; Lewis, K.; Fenske, T.S.; et al. Pirtobrutinib, A Next Generation, Highly Selective, Non-Covalent BTK Inhibitor in Previously Treated Mantle Cell Lymphoma: Updated Results from the Phase 1/2 BRUIN Study. Blood 2021, 138, 381. [Google Scholar] [CrossRef]
- Jain, P.; O’Brien, S. Richter’s transformation in chronic lymphocytic leukemia. Oncology 2012, 26, 1146–1152. [Google Scholar] [PubMed]
- Parikh, S.A.; Shanafelt, T.D. Risk factors for Richter syndrome in chronic lymphocytic leukemia. Curr. Hematol. Malig. Rep. 2014, 9, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Brieghel, C.; Aarup, K.; Torp, M.H.; Andersen, M.A.; Yde, C.W.; Tian, X.; Wiestner, A.; Ahn, I.E.; Niemann, C.U. Clinical Outcomes in Patients with Multi-Hit TP53 Chronic Lymphocytic Leukemia Treated with Ibrutinib. Clin. Cancer Res. 2021, 27, 4531–4538. [Google Scholar] [CrossRef]
- Jain, P.; Keating, M.; Wierda, W.; Estrov, Z.; Ferrajoli, A.; Jain, N.; George, B.; James, D.; Kantarjian, H.; Burger, J.; et al. Outcomes of patients with chronic lymphocytic leukemia after discontinuing ibrutinib. Blood 2015, 125, 2062–2067. [Google Scholar] [CrossRef] [Green Version]
- Kipps, T.J.; Fraser, G.; Coutre, S.E.; Brown, J.R.; Barrientos, J.C.; Barr, P.M.; Byrd, J.C.; O’Brien, S.M.; Dilhuydy, M.S.; Hillmen, P.; et al. Long-Term studies assessing outcomes of ibrutinib therapy in patients with Del(11q) chronic lymphocytic leukemia. Clin. Lymphoma Myeloma Leuk. 2019, 19, 715–722.e716. [Google Scholar] [CrossRef] [Green Version]
- Dal-Bo, M.; Bertoni, F.; Forconi, F.; Zucchetto, A.; Bomben, R.; Marasca, R.; Deaglio, S.; Laurenti, L.; Efremov, D.G.; Gaidano, G.; et al. Intrinsic and extrinsic factors influencing the clinical course of B-cell chronic lymphocytic leukemia: Prognostic markers with pathogenetic relevance. J. Transl. Med. 2009, 7, 76. [Google Scholar] [CrossRef] [Green Version]
- Landau, D.A.; Tausch, E.; Taylor-Weiner, A.N.; Stewart, C.; Reiter, J.G.; Bahlo, J.; Kluth, S.; Bozic, I.; Lawrence, M.; Bottcher, S.; et al. Mutations driving CLL and their evolution in progression and relapse. Nature 2015, 526, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Malcikova, J.; Tausch, E.; Rossi, D.; Sutton, L.A.; Soussi, T.; Zenz, T.; Kater, A.P.; Niemann, C.U.; Gonzalez, D.; Davi, F.; et al. ERIC recommendations for TP53 mutation analysis in chronic lymphocytic leukemia-update on methodological approaches and results interpretation. Leukemia 2018, 32, 1070–1080. [Google Scholar] [CrossRef] [Green Version]
- Dufour, A.; Palermo, G.; Zellmeier, E.; Mellert, G.; Duchateau-Nguyen, G.; Schneider, S.; Benthaus, T.; Kakadia, P.M.; Spiekermann, K.; Hiddemann, W.; et al. Inactivation of TP53 correlates with disease progression and low miR-34a expression in previously treated chronic lymphocytic leukemia patients. Blood 2013, 121, 3650–3657. [Google Scholar] [CrossRef] [Green Version]
- Hallek, M.; Al-Sawaf, O. Chronic lymphocytic leukemia: 2022 update on diagnostic and therapeutic procedures. Am. J. Hematol. 2021, 96, 1679–1705. [Google Scholar] [CrossRef] [PubMed]
- Rotbain, E.C.; Frederiksen, H.; Hjalgrim, H.; Rostgaard, K.; Egholm, G.J.; Zahedi, B.; Poulsen, C.B.; Enggard, L.; da Cunha-Bang, C.; Niemann, C.U. IGHV mutational status and outcome for patients with chronic lymphocytic leukemia upon treatment: A Danish nationwide population-based study. Haematologica 2020, 105, 1621–1629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, K.; Bahlo, J.; Fink, A.M.; Goede, V.; Herling, C.D.; Cramer, P.; Langerbeins, P.; von Tresckow, J.; Engelke, A.; Maurer, C.; et al. Long-term remissions after FCR chemoimmunotherapy in previously untreated patients with CLL: Updated results of the CLL8 trial. Blood 2016, 127, 208–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai-Adisaksopha, C.; Brown, J.R. FCR achieves long-term durable remissions in patients with IGHV-mutated CLL. Blood 2017, 130, 2278–2282. [Google Scholar] [CrossRef]
- Shanafelt, T.D.; Wang, X.V.; Hanson, C.A.; Paietta, E.M.; O’Brien, S.; Barrientos, J.C.; Jelinek, D.F.; Braggio, E.; Leis, J.F.; Zhang, C.C.; et al. Long-term Outcomes for Ibrutinib-Rituximab and Chemoimmunotherapy in CLL: Updated Results of the E1912 Trial. Blood 2022, 140, 112–120. [Google Scholar] [CrossRef]
- Shanafelt, T.D.; Wang, X.V.; Kay, N.E.; Hanson, C.A.; O’Brien, S.; Barrientos, J.; Jelinek, D.F.; Braggio, E.; Leis, J.F.; Zhang, C.C.; et al. Ibrutinib-Rituximab or Chemoimmunotherapy for Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2019, 381, 432–443. [Google Scholar] [CrossRef]
- Gattei, V.; Bulian, P.; Del Principe, M.I.; Zucchetto, A.; Maurillo, L.; Buccisano, F.; Bomben, R.; Dal-Bo, M.; Luciano, F.; Rossi, F.M.; et al. Relevance of CD49d protein expression as overall survival and progressive disease prognosticator in chronic lymphocytic leukemia. Blood 2008, 111, 865–873. [Google Scholar] [CrossRef] [Green Version]
- Shanafelt, T.D.; Geyer, S.M.; Bone, N.D.; Tschumper, R.C.; Witzig, T.E.; Nowakowski, G.S.; Zent, C.S.; Call, T.G.; Laplant, B.; Dewald, G.W.; et al. CD49d expression is an independent predictor of overall survival in patients with chronic lymphocytic leukaemia: A prognostic parameter with therapeutic potential. Br. J. Haematol. 2008, 140, 537–546. [Google Scholar] [CrossRef]
- Rossi, D.; Zucchetto, A.; Rossi, F.M.; Capello, D.; Cerri, M.; Deambrogi, C.; Cresta, S.; Rasi, S.; De Paoli, L.; Bodoni, C.L.; et al. CD49d expression is an independent risk factor of progressive disease in early stage chronic lymphocytic leukemia. Haematologica 2008, 93, 1575–1579. [Google Scholar] [CrossRef] [Green Version]
- Baumann, T.; Delgado, J.; Santacruz, R.; Martinez-Trillos, A.; Rozman, M.; Aymerich, M.; Lopez, C.; Costa, D.; Carrio, A.; Villamor, N.; et al. CD49d (ITGA4) expression is a predictor of time to first treatment in patients with chronic lymphocytic leukaemia and mutated IGHV status. Br. J. Haematol. 2016, 172, 48–55. [Google Scholar] [CrossRef] [Green Version]
- Rossi, D.; Bodoni, C.L.; Zucchetto, A.; Rasi, S.; De Paoli, L.; Fangazio, M.; Rossi, F.M.; Ladetto, M.; Gattei, V.; Gaidano, G. Low CD49d expression and long telomere identify a chronic lymphocytic leukemia subset with highly favourable outcome. Am. J. Hematol. 2010, 85, 619–622. [Google Scholar] [CrossRef]
- Tissino, E.; Pozzo, F.; Benedetti, D.; Caldana, C.; Bittolo, T.; Rossi, F.M.; Bomben, R.; Nanni, P.; Chivilo, H.; Cattarossi, I.; et al. CD49d promotes disease progression in chronic lymphocytic leukemia: New insights from CD49d bimodal expression. Blood 2020, 135, 1244–1254. [Google Scholar] [CrossRef] [PubMed]
- Zucchetto, A.; Caldana, C.; Benedetti, D.; Tissino, E.; Rossi, F.M.; Hutterer, E.; Pozzo, F.; Bomben, R.; Dal Bo, M.; D’Arena, G.; et al. CD49d is overexpressed by trisomy 12 chronic lymphocytic leukemia cells: Evidence for a methylation-dependent regulation mechanism. Blood 2013, 122, 3317–3321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsimberidou, A.M.; Keating, M.J. Richter syndrome: Biology, incidence, and therapeutic strategies. Cancer 2005, 103, 216–228. [Google Scholar] [CrossRef]
- Bulian, P.; Shanafelt, T.D.; Fegan, C.; Zucchetto, A.; Cro, L.; Nuckel, H.; Baldini, L.; Kurtova, A.V.; Ferrajoli, A.; Burger, J.A.; et al. CD49d is the strongest flow cytometry-based predictor of overall survival in chronic lymphocytic leukemia. J. Clin. Oncol. 2014, 32, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Dal Bo, M.; Bulian, P.; Bomben, R.; Zucchetto, A.; Rossi, F.M.; Pozzo, F.; Tissino, E.; Benedetti, D.; Bittolo, T.; Nanni, P.; et al. CD49d prevails over the novel recurrent mutations as independent prognosticator of overall survival in chronic lymphocytic leukemia. Leukemia 2016, 30, 2011–2018. [Google Scholar] [CrossRef]
- Balatti, V.; Bottoni, A.; Palamarchuk, A.; Alder, H.; Rassenti, L.Z.; Kipps, T.J.; Pekarsky, Y.; Croce, C.M. NOTCH1 mutations in CLL associated with trisomy 12. Blood 2012, 119, 329–331. [Google Scholar] [CrossRef]
- Benedetti, D.; Tissino, E.; Pozzo, F.; Bittolo, T.; Caldana, C.; Perini, C.; Martorelli, D.; Bravin, V.; D’Agaro, T.; Rossi, F.M.; et al. NOTCH1 mutations are associated with high CD49d expression in chronic lymphocytic leukemia: Link between the NOTCH1 and the NF-kappaB pathways. Leukemia 2018, 32, 654–662. [Google Scholar] [CrossRef]
- Carrasco, Y.R.; Batista, F.D. B cell recognition of membrane-bound antigen: An exquisite way of sensing ligands. Curr. Opin. Immunol. 2006, 18, 286–291. [Google Scholar] [CrossRef]
- Herman, S.E.; Mustafa, R.Z.; Jones, J.; Wong, D.H.; Farooqui, M.; Wiestner, A. Treatment with ibrutinib inhibits BTK- and VLA-4-dependent adhesion of chronic lymphocytic leukemia cells in vivo. Clin. Cancer Res. 2015, 21, 4642–4651. [Google Scholar] [CrossRef] [Green Version]
- de Rooij, M.F.; Kuil, A.; Geest, C.R.; Eldering, E.; Chang, B.Y.; Buggy, J.J.; Pals, S.T.; Spaargaren, M. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood 2012, 119, 2590–2594. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R. Phosphatidylinositol 3 kinase δ inhibitors: Present and future. Cancer J. 2019, 25, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Davids, M.S.; Kim, H.T.; Nicotra, A.; Savell, A.; Francoeur, K.; Hellman, J.M.; Bazemore, J.; Miskin, H.P.; Sportelli, P.; Stampleman, L.; et al. Umbralisib in combination with ibrutinib in patients with relapsed or refractory chronic lymphocytic leukaemia or mantle cell lymphoma: A multicentre phase 1-1b study. Lancet Haematol. 2019, 6, e38–e47. [Google Scholar] [CrossRef]
- Nastoupil, L.J.; Lunning, M.A.; Vose, J.M.; Schreeder, M.T.; Siddiqi, T.; Flowers, C.R.; Cohen, J.B.; Burger, J.A.; Wierda, W.G.; O’Brien, S.; et al. Tolerability and activity of ublituximab, umbralisib, and ibrutinib in patients with chronic lymphocytic leukaemia and non-Hodgkin lymphoma: A phase 1 dose escalation and expansion trial. Lancet Haematol. 2019, 6, e100–e109. [Google Scholar] [CrossRef]
- Zelenetz, A.D.; Reddy, N.; Jagadeesh, D.; Stathis, A.; Salman, H.S.; Soumerai, J.D.; Kenkre, V.P.; Asch, A.S.; Llorin-Sangalang, J.; Li, J.; et al. Tolerability and durable respones of the PI3Kδ inhibitor ME-401 administered on an intermittent schedule in relapsed/refractory (R/R) follicular lymphoma (FL) and other B-cell malignancies. J. Clin. Oncol. 2020, 38, 8016. [Google Scholar] [CrossRef]
- Ghia, P.; Pluta, A.; Wach, M.; Lysak, D.; Kozak, T.; Simkovic, M.; Kaplan, P.; Kraychok, I.; Illes, A.; de la Serna, J.; et al. ASCEND: Phase III, randomized trial of acalabrutinib versus idelalisib plus rituximab or bendamustine plus rituximab in relapsed or refractory chronic lymphocytic leukemia. J. Clin. Oncol. 2020, 38, 2849–2861. [Google Scholar] [CrossRef]
- Barr, P.M.; Saylors, G.B.; Spurgeon, S.E.; Cheson, B.D.; Greenwald, D.R.; O’Brien, S.M.; Liem, A.K.; McLntyre, R.E.; Joshi, A.; Abella-Dominicis, E.; et al. Phase 2 study of idelalisib and entospletinib: Pneumonitis limits combination therapy in relapsed refractory CLL and NHL. Blood 2016, 127, 2411–2415. [Google Scholar] [CrossRef] [Green Version]
- Cheah, C.Y.; Nastoupil, L.J.; Neelapu, S.S.; Forbes, S.G.; Oki, Y.; Fowler, N.H. Lenalidomide, idelalisib, and rituximab are unacceptably toxic in patients with relapsed/refractory indolent lymphoma. Blood 2015, 125, 3357–3359. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.M.; Pitcher, B.N.; Jung, S.H.; Bartlett, N.L.; Wagner-Johnston, N.; Park, S.I.; Richards, K.L.; Cashen, A.F.; Jaslowski, A.; Smith, S.E.; et al. Safety and tolerability of idelalisib, lenalidomide, and rituximab in relapsed and refractory lymphoma: The alliance for clinical trials in oncology A051201 and A051202 phase 1 trials. Lancet Haematol. 2017, 4, e176–e182. [Google Scholar] [CrossRef] [Green Version]
- Flinn, I.W.; O’Brien, S.; Kahl, B.; Patel, M.; Oki, Y.; Foss, F.F.; Porcu, P.; Jones, J.; Burger, J.A.; Jain, N.; et al. Duvelisib, a novel oral dual inhibitor of PI3K-delta, gamma, is clinically active in advanced hematologic malignancies. Blood 2018, 131, 877–887. [Google Scholar] [CrossRef] [Green Version]
- Murali, I.; Kasar, S.; Naeem, A.; Tyekucheva, S.; Khalsa, J.K.; Thrash, E.M.; Itchaki, G.; Livitz, D.; Leshchiner, I.; Dong, S.; et al. Activation of the MAPK pathway mediates resistance to PI3K inhibitors in chronic lymphocytic leukemia. Blood 2021, 138, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Scheffold, A.; Jebaraj, B.M.C.; Tausch, E.; Bloehdorn, J.; Ghia, P.; Yahiaoui, A.; Dolnik, A.; Blatte, T.J.; Bullinger, L.; Dheenadayalan, R.P.; et al. IGF1R as druggable target mediating PI3K-delta inhibitor resistance in a murine model of chronic lymphocytic leukemia. Blood 2019, 134, 534–547. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Elias, E.E.; Sarapura Martinez, V.J.; Amondarain, M.; Colado, A.; Cordini, G.; Bezares, R.F.; Fernandez Grecco, H.; Custidiano, M.D.R.; Sanchez Avalos, J.C.; Garate, G.; et al. Venetoclax-resistant CLL cells show a highly activated and proliferative phenotype. Cancer Immunol. Immunother. 2022, 71, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Lin, V.S.; Lew, T.E.; Handunnetti, S.M.; Blombery, P.; Nguyen, T.; Westerman, D.A.; Kuss, B.J.; Tam, C.S.; Roberts, A.W.; Seymour, J.F.; et al. BTK inhibitor therapy is effective in patients with CLL resistant to venetoclax. Blood 2020, 135, 2266–2270. [Google Scholar] [CrossRef]
- Chen, S.-S.; Kutok, J.L.; Ferrer, G.; Ravichandran, P.; Ibrahim, M.; Kieso, Y.; Barrientos, J.C.; Weaver, D.T.; Pachter, J.A.; Rai, K.R.; et al. Dual inhibition of PI3K-δ and PI3K-γ by duvelisib eliminates CLL B cells, impairs CLL-supporting cells, and overcomes ibrutinib resistance in a patient-derived xenograft model. Blood 2018, 132, 4420. [Google Scholar] [CrossRef]
- Bichi, R.; Shinton, S.A.; Martin, E.S.; Koval, A.; Calin, G.A.; Cesari, R.; Russo, G.; Hardy, R.R.; Croce, C.M. Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc. Natl. Acad. Sci. USA 2002, 99, 6955–6960. [Google Scholar] [CrossRef] [Green Version]
- Simonetti, G.; Bertilaccio, M.T.; Ghia, P.; Klein, U. Mouse models in the study of chronic lymphocytic leukemia pathogenesis and therapy. Blood 2014, 124, 1010–1019. [Google Scholar] [CrossRef] [Green Version]
- Hofbauer, J.P.; Heyder, C.; Denk, U.; Kocher, T.; Holler, C.; Trapin, D.; Asslaber, D.; Tinhofer, I.; Greil, R.; Egle, A. Development of CLL in the TCL1 transgenic mouse model is associated with severe skewing of the T-cell compartment homologous to human CLL. Leukemia 2011, 25, 1452–1458. [Google Scholar] [CrossRef]
- Szenes, E.; Harzschel, A.; Decker, S.; Tissino, E.; Pischeli, J.; Gutjahr, J.C.; Kissel, S.; Pennisi, S.; Hopner, J.P.; Egle, A.; et al. TCL1 transgenic mice as a model for CD49d-high chronic lymphocytic leukemia. Leukemia 2020, 34, 2498–2502. [Google Scholar] [CrossRef] [Green Version]
- Estupinan, H.Y.; Bouderlique, T.; He, C.; Berglof, A.; Gupta, D.; Saher, O.; Daza Cruz, M.A.; Pena-Perez, L.; Yu, L.; Zain, R.; et al. Novel mouse model resistant to irreversible BTK inhibitors: A tool identifying new therapeutic targets and side effects. Blood Adv. 2020, 4, 2439–2450. [Google Scholar] [CrossRef] [PubMed]
- Aslan, B.; Kismali, G.; Chen, L.S.; Iles, L.R.; Mahendra, M.; Peoples, M.; Gagea, M.; Fowlkes, N.W.; Zheng, X.; Wang, J.; et al. Development and characterization of prototypes for in vitro and in vivo mouse models of ibrutinib-resistant CLL. Blood Adv. 2021, 5, 3134–3146. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Ibrutinib | Acalabrutinib | Zanubrutinib | Pirtobrutinib | |
|---|---|---|---|---|
| Mechanism of action | Covalent | Covalent | Covalent | Non-covalent |
| Approval | 02/2014, CLL | 11/2019, CLL | 11/2019, MCL | Phase I/II clinical trial (BRUIN) |
| Most prevalent adverse events | Infection Diarrhea Bleeding/Bruising | Infection Bleeding/Bruising Headache | Infection Neutropenia | Fatigue Diarrhea Contusion |
| Most prevalent resistance mutations | C481S/F/C/R, T474, L528, T316A, PLCγ2 mutations | Patient data not available yet | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Polcik, L.; Dannewitz Prosseda, S.; Pozzo, F.; Zucchetto, A.; Gattei, V.; Hartmann, T.N. Integrin Signaling Shaping BTK-Inhibitor Resistance. Cells 2022, 11, 2235. https://doi.org/10.3390/cells11142235
Polcik L, Dannewitz Prosseda S, Pozzo F, Zucchetto A, Gattei V, Hartmann TN. Integrin Signaling Shaping BTK-Inhibitor Resistance. Cells. 2022; 11(14):2235. https://doi.org/10.3390/cells11142235
Chicago/Turabian StylePolcik, Laura, Svenja Dannewitz Prosseda, Federico Pozzo, Antonella Zucchetto, Valter Gattei, and Tanja Nicole Hartmann. 2022. "Integrin Signaling Shaping BTK-Inhibitor Resistance" Cells 11, no. 14: 2235. https://doi.org/10.3390/cells11142235
APA StylePolcik, L., Dannewitz Prosseda, S., Pozzo, F., Zucchetto, A., Gattei, V., & Hartmann, T. N. (2022). Integrin Signaling Shaping BTK-Inhibitor Resistance. Cells, 11(14), 2235. https://doi.org/10.3390/cells11142235