
In Vitro Drug Screening Using iPSC-Derived Cardiomyocytes of a Long QT-Syndrome Patient Carrying KCNQ1 & TRPM4 Dual Mutation: An Experimental Personalized Treatment

 , , , , , , , , , , , , and
, , , , , , , , , , , , and 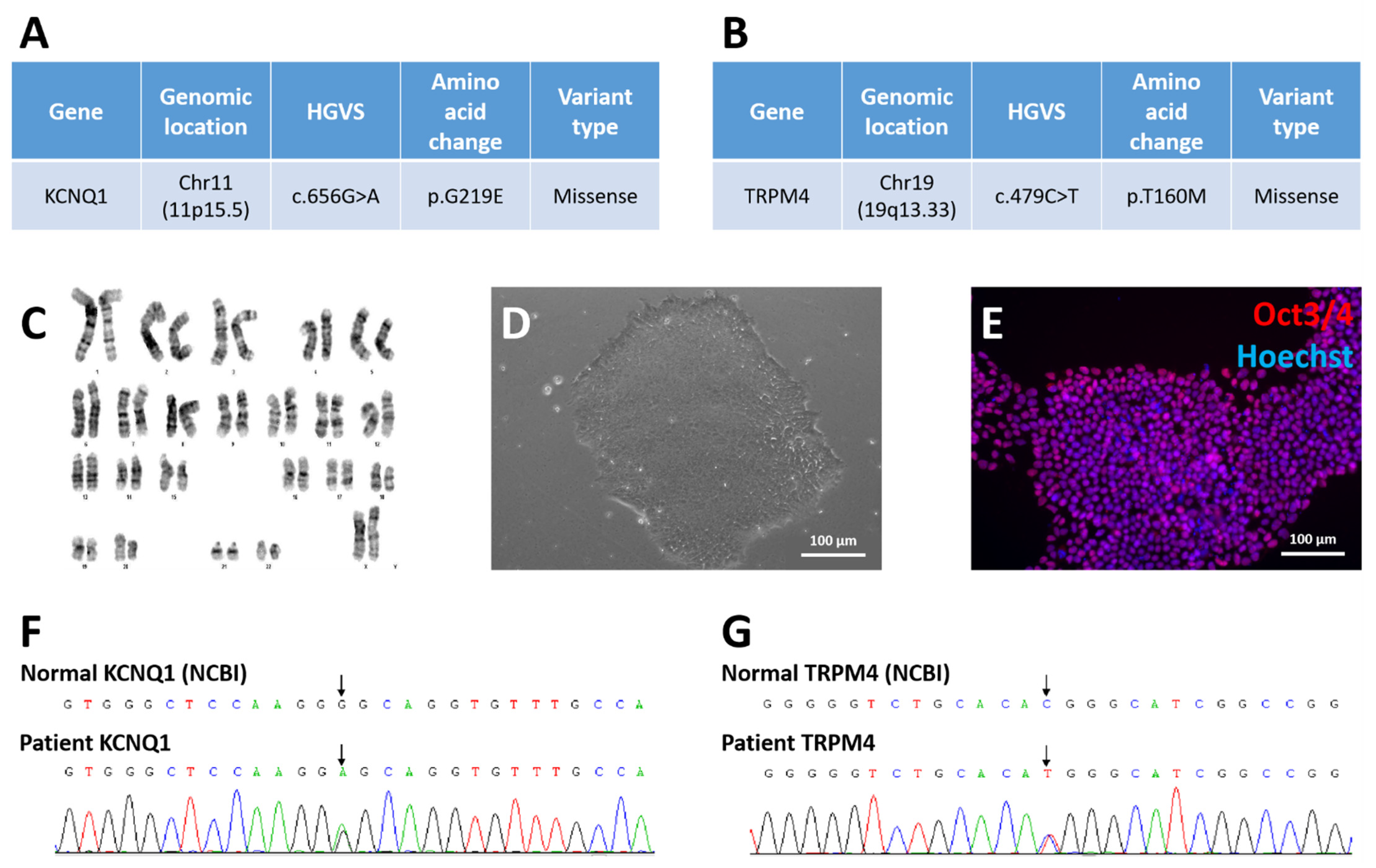{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Characteristics
2.2. PBMC Isolation and Reprogramming into iPSCs
2.3. Cardiomyocyte Differentiation
2.4. Drug Treatment and Electrophysiological Examination
2.5. Immunofluorescence
2.6. Flow Cytometry
2.7. STR Analysis
2.8. Karyotyping
2.9. Whole Exome Sequencing
2.10. Statistical Analysis
2.11. Information on Material Suppliers
3. Results
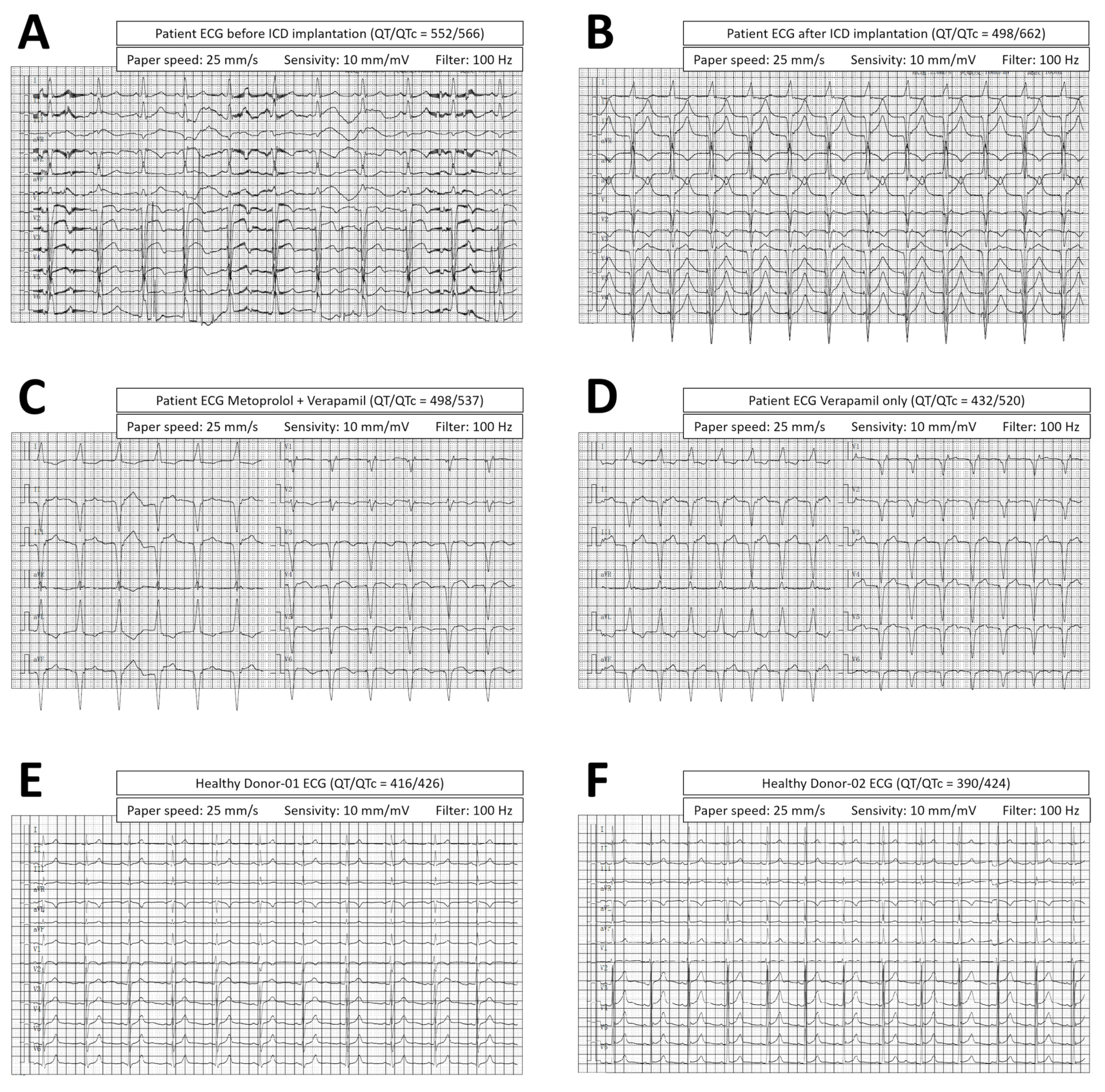
3.1. Patient Diagnosis and Treatment
3.2. Derivation of Long QT Patient-Specific iPSC Line FAHXMUi001-A
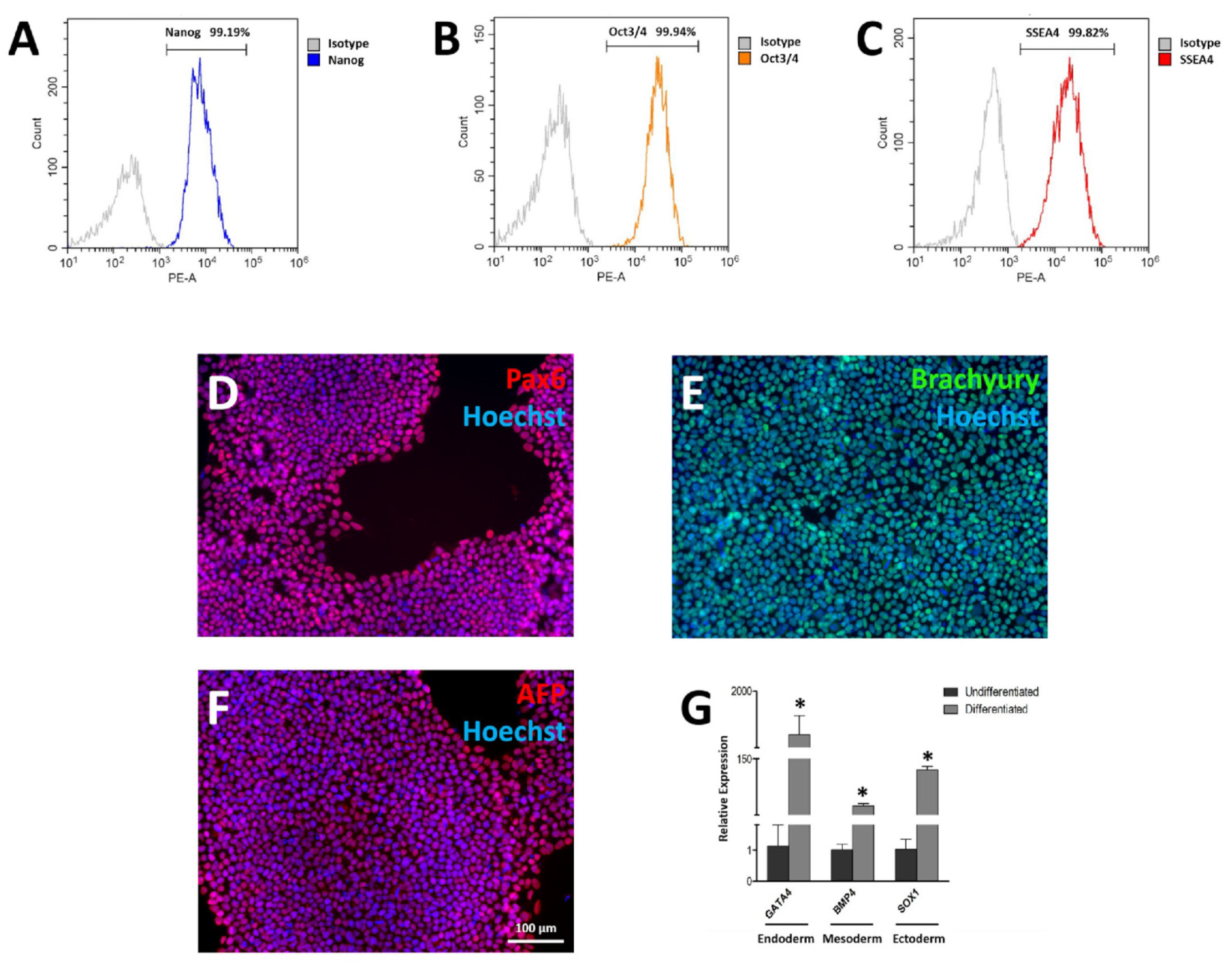
3.3. Pluripotency Characterization of FAHXMUi001-A
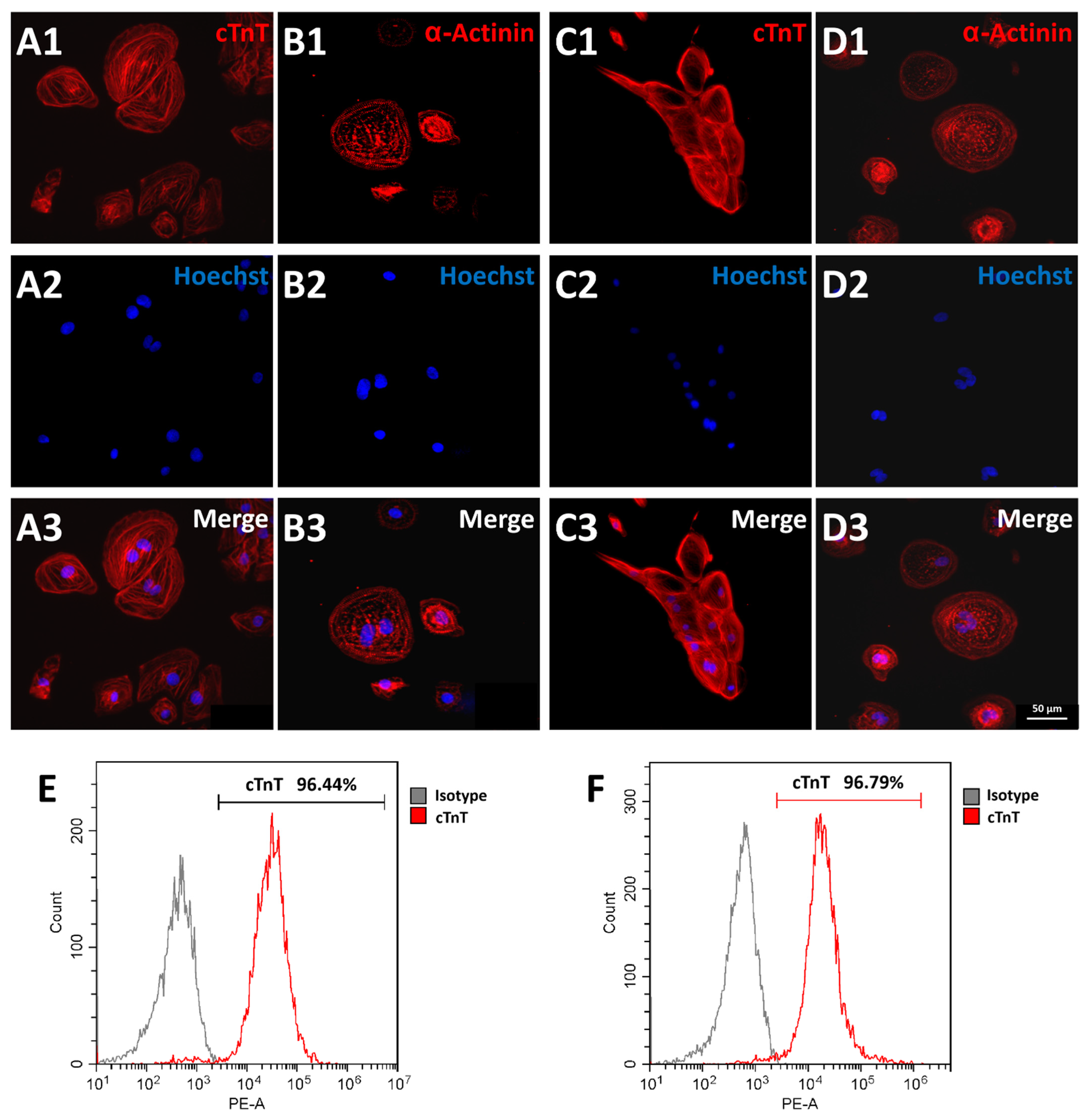
3.4. Cardiomyocyte Differentiation of FAHXMUi001-A
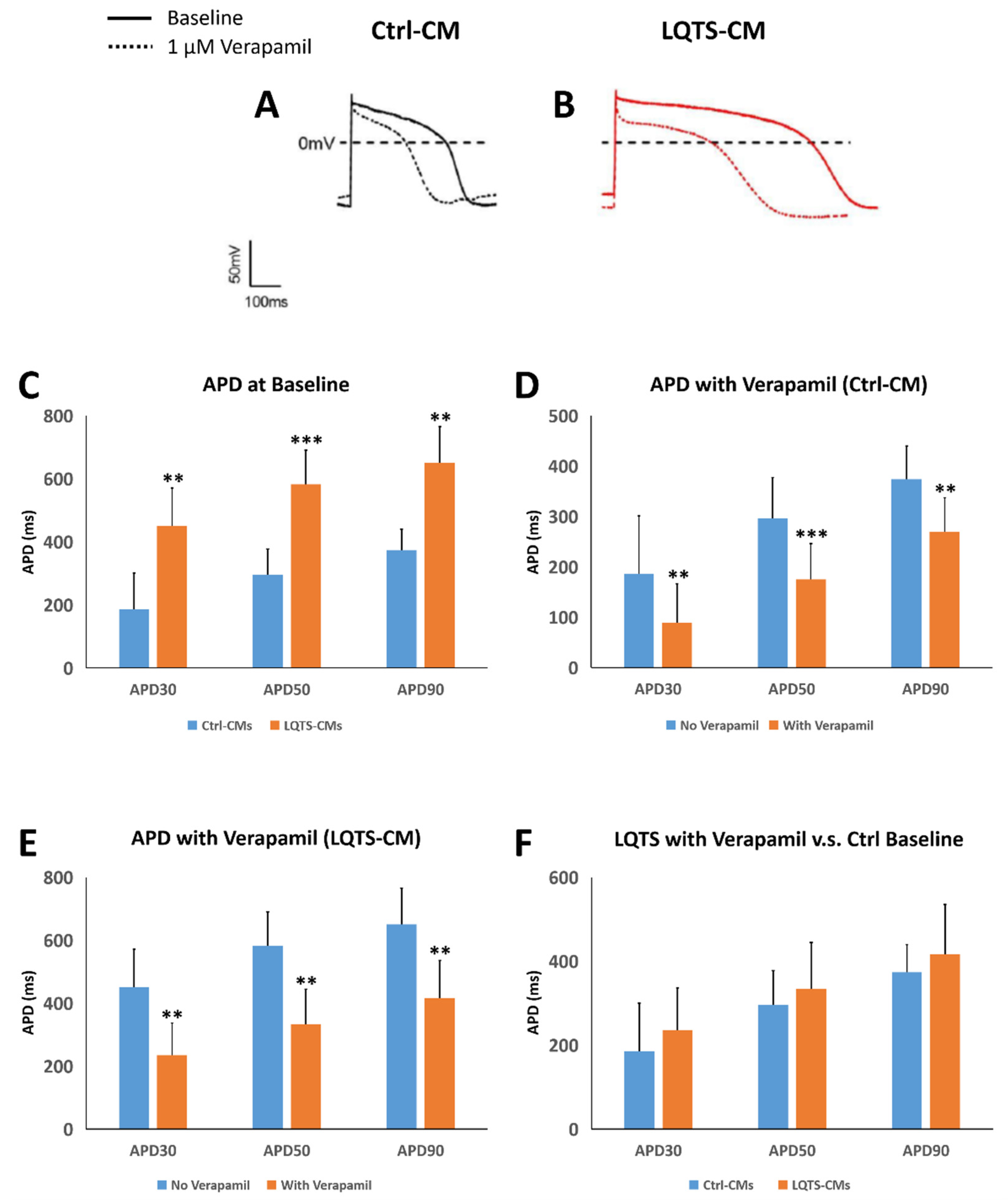
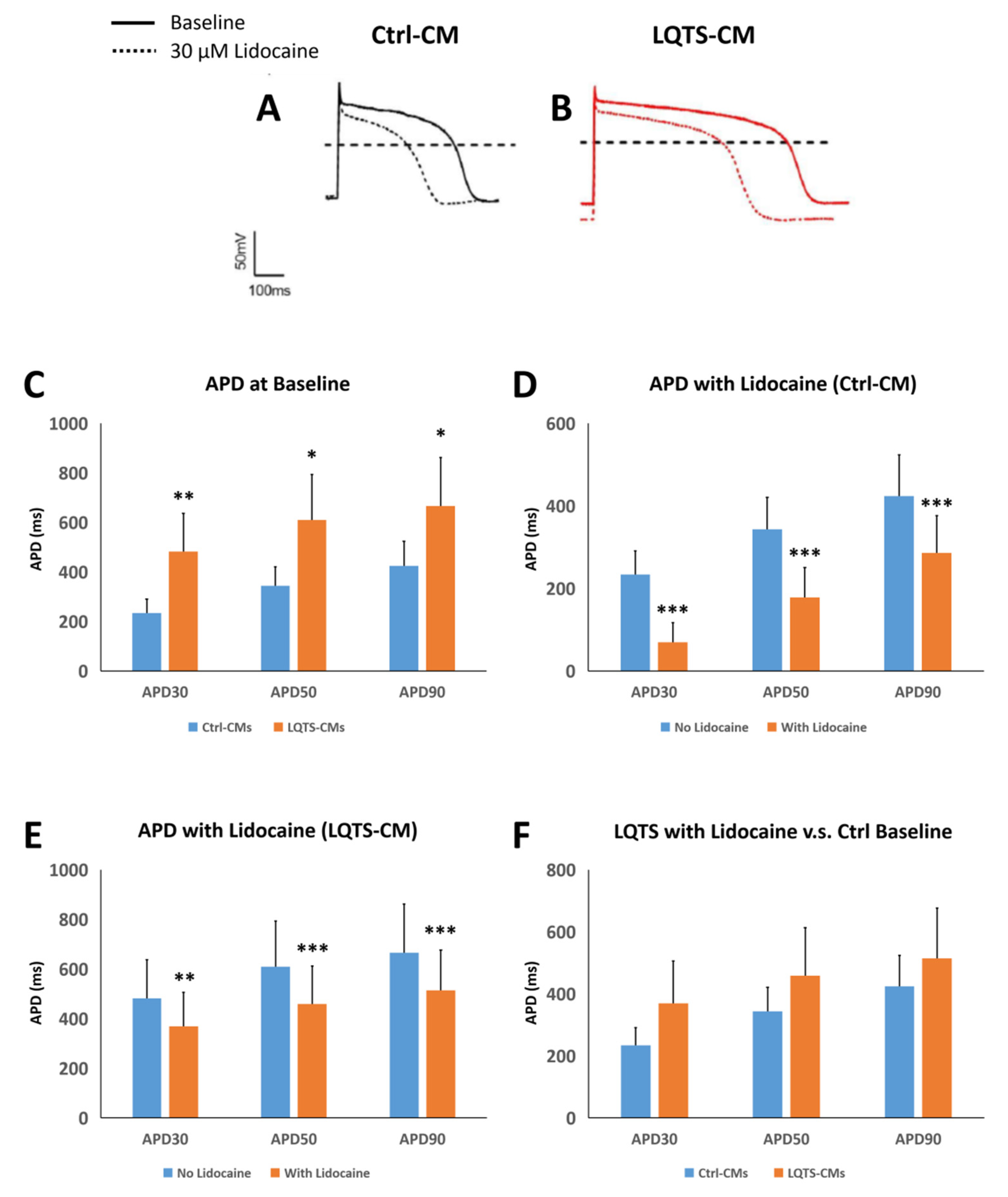
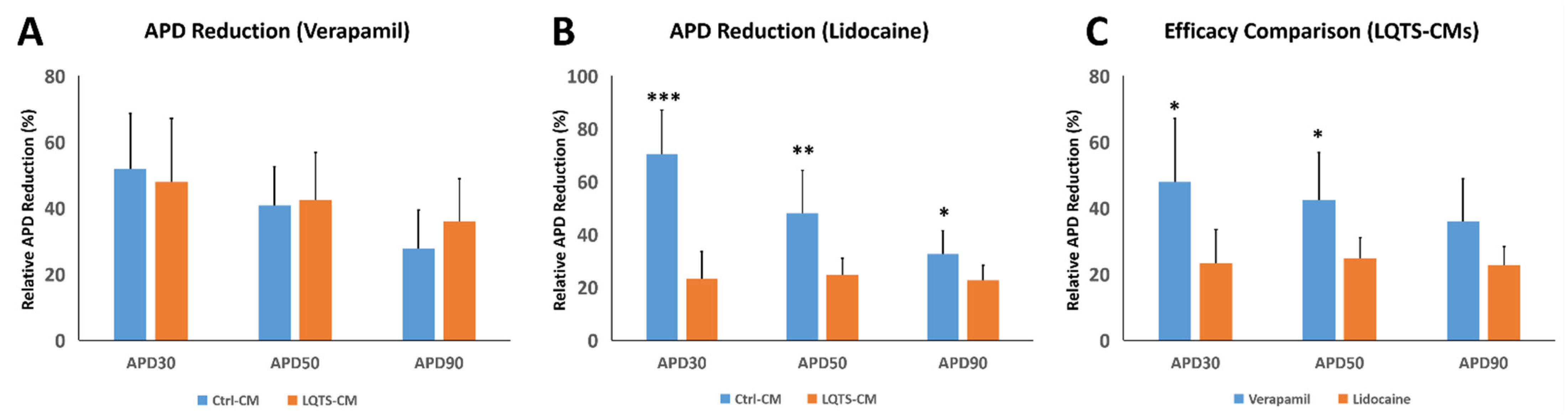
3.5. Drug Treatment and Electrophysiological Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schwartz, P.J.; Crotti, L.; Insolia, R. Long-QT syndrome from genetics to management. Circ. Arrhythmia Electrophysiol. 2012, 5, 868–877. [Google Scholar] [CrossRef]
- Shah, S.R.; Park, K.; Alweis, R. Long QT syndrome: A comprehensive review of the literature and current evidence. Curr. Probl. Cardiol. 2019, 44, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.A.M.; Amin, A.S.; Postema, P.G. Diagnosis, management and therapeutic strategies for congenital long QT syndrome. Heart 2022, 108, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Stramba-Badiale, M.; Lia Crotti Pedrazzini, M.; Besana, A.; Bosi, G.; Gabbarini, F.; Goulene, K.; Insolia, R.; Mannarino, S.; Mosca, F.; et al. Prevalence of the congenital long QT syndrome. Circulation 2009, 120, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.T.; Gula, L.J.; Klein, G.J.; Skanes, A.C.; Yee, R.; Leong-Sit, P.; Chattha, I.; Sy, R.; Jones, D.L.; Krahn, A.D. Effect of beta-blockers on QT dynamics in the long QT syndrome: Measuring the benefit. EP Eur. 2014, 6, 1847–1851. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.A.M.; Ackerman, M.J. Beta-blockers in the treatment of congenital long QT syndrome: Is one beta-blocker superior to another? J. Am. Coll. Cardiol. 2014, 64, 1359–1361. [Google Scholar] [CrossRef] [PubMed]
- Abu-Zeitone, A.; Peterson, D.R.; Polonsky, B.; McNitt, S.; Moss, A.J. Efficacy of different beta-blockers in the treatment of long QT syndrome. J. Am. Coll. Cardiol. 2014, 64, 1352–1358. [Google Scholar] [CrossRef]
- Moss, A.J.; Zareba, W.; Hall, W.J.; Schwartz, P.J.; Crampton, R.S.; Benhorin, J.; Vincent, G.M.; Locati, E.H.; Priori, S.G.; Napolitano, C.; et al. Effectiveness and limitations of β-blocker therapy in congenital long-QT syndrome. Circulation 2000, 101, 616–623. [Google Scholar] [CrossRef]
- Carroll, S.L.; Strachan, P.H.; Laat, S.; Schwartz, L.; Arthur, H.M. Patients’ decision making to accept or decline an implantable cardioverter defibrillator for primary prevention of sudden cardiac death. Health Expect 2013, 16, 69–79. [Google Scholar] [CrossRef]
- Persson, R.; Earley, A.; Garlitski, A.C.; Balk, E.M.; Uhlig, K. Adverse events following implantable cardioverter defibrillator implantation: A systematic review. J. Interv. Card. Electrophysiol. 2014, 40, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Tomzik, J.; Koltermann, K.C.; Zabel, M.; Willich, S.N.; Reinhold, T. Quality of life in patients with an implantable cardioverter defibrillator: A systematic review. Front. Cardiovasc. Med. 2015, 2, 34. [Google Scholar] [CrossRef]
- Giudicessi, J.R.; Ackerman, M.J. Genotype- and phenotype-guided management of congenital long QT syndrome. Curr. Probl. Cardiol. 2013, 38, 417–455. [Google Scholar] [CrossRef]
- Han, L.; Liu, F.; Li, Q.; Qing, T.; Zhai, Z.; Xia, Z.; Li, J. The efficacy of beta-blockers in patients with long QT syndrome 1-3 according to individuals’ gender, age, and QTc intervals: A network meta-analysis. Front. Pharmacol. 2020, 11, 579525. [Google Scholar] [CrossRef]
- Ahn, J.; Kim, H.J.; Choi, J.I.; Lee, K.N.; Shim, J.; Ahn, H.S.; Kim, Y.H. Effectiveness of beta-blockers depending on the genotype of congenital long-QT syndrome: A meta-analysis. PLoS ONE 2017, 12, e0185680. [Google Scholar] [CrossRef]
- Nakano, Y.; Shimizu, W. Genetics of long-QT syndrome. J. Hum. Gene 2016, 61, 51–55. [Google Scholar] [CrossRef]
- Wallace, E.; Howard, L.; Liu, M.; O’Brien, T.; Ward, D.; Shen, S.; Prendiville, T. Long QT syndrome: Genetics and future perspective. Pediatr. Cardiol. 2019, 40, 1419–1430. [Google Scholar] [CrossRef]
- Salama, G.; London, B. Mouse models of long QT syndrome. J. Physiol. 2007, 578 Pt 1, 43–53. [Google Scholar] [CrossRef]
- Mummery, C.L. Perspectives on the use of human induced pluripotent stem cell-derived cardiomyocytes in biomedical research. Stem Cell Rep. 2018, 11, 1306–1311. [Google Scholar] [CrossRef]
- Liu, H.; El Zein, L.; Kruse, M.; Guinamard, R.; Beckmann, A.; Bozio, A.; Kurtbay, G.; Mégarbané, A.; Ohmert, I.; Blaysat, G.; et al. Gain-of-function mutations in TRPM4 cause autosomal dominant isolated cardiac conduction disease. Circ. Cardiovasc. Genet. 2010, 3, 374–385. [Google Scholar] [CrossRef]
- Bianchi, B.; Ozhathil, L.C.; Medeiros-Domingo, A.; Gollob, M.H.; Abriel, H. Four TRPM4 cation channel mutations found in cardiac conduction diseases lead to altered protein stability. Front. Physiol. 2018, 9, 177. [Google Scholar] [CrossRef]
- Saito, Y.; Nakamura, K.; Nishi, N.; Igawa, O.; Yoshida, M.; Miyoshi, T.; Watanabe, A.; Morita, H.; Ito, H. TRPM4 mutation in patients with ventricular noncompaction and cardiac conduction disease. Circ. Genom. Precis. Med. 2018, 11, e002103. [Google Scholar] [CrossRef]
- Janin, A.; Bessière, F.; Georgescu, T.; Chanavat, V.; Chevalier, P.; Millat, G. TRPM4 mutations to cause autosomal recessive and not autosomal dominant Brugada type 1 syndrome. Eur. J. Med. Genet. 2019, 62, 103527. [Google Scholar] [CrossRef]
- Pepine, C.J.; Faich, G.; Makuch, R. Verapamil use in patients with cardiovascular disease: An overview of randomized trials. Clin. Cardiol. 1998, 21, 633–641. [Google Scholar] [CrossRef]
- Collinsworth, K.A.; Kalman, S.M.; Harrison, D.C. The clinical pharmacology of lidocaine as an antiarrhythymic drug. Circulation 1974, 50, 1217–1230. [Google Scholar] [CrossRef]
- Masic, D.; Liang, E.; Long, C.; Sterk, E.J.; Barbas, B.; Rech, M.A. Intravenous lidocaine for acute pain: A systematic review. Pharmacotherapy 2018, 38, 1250–1259. [Google Scholar] [CrossRef]
- Aiba, T.; Shimizu, W.; Inagaki, M.; Noda, T.; Miyoshi, S.; Ding, W.G.; Zankov, D.P.; Toyoda, F.; Matsuura, H.; Horie, M.; et al. Cellular and ionic mechanism for drug-induced long QT syndrome and effectiveness of verapamil. J. Am. Coll. Cardiol. 2005, 45, 300–307. [Google Scholar] [CrossRef]
- Owczuk, R.; Wujtewicz, M.A.; Sawicka, W.; Piankowski, A.; Polak-Krzeminska, A.; Morzuch, E.; Wujtewicz, M. The effect of intravenous lidocaine on QT changes during tracheal intubation. Anaesthesia 2008, 63, 924–931. [Google Scholar] [CrossRef]
- Tohyama, S.; Hattori, F.; Sano, M.; Hishiki, T.; Nagahata, Y.; Matsuura, T.; Hashimoto, H.; Suzuki, T.; Yamashita, H.; Satoh, Y.; et al. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell 2013, 12, 127–137. [Google Scholar] [CrossRef]
- Yokoo, N.; Baba, S.; Kaichi, S.; Niwa, A.; Mima, T.; Doi, H.; Yamanaka, S.; Nakahata, T.; Heike, T. The effects of cardioactive drugs on cardiomyocytes derived from human induced pluripotent stem cells. Biochem. Biophys. Res. Commun. 2009, 387, 482–488. [Google Scholar] [CrossRef]
- Olschewski, A.; Bräu, M.E.; Olschewski, H.; Hempelmann, G.; Vogel, W. ATP-dependent potassium channel in rat cardiomyocytes is blocked by lidocaine. Possible impact on the antiarrhythmic action of lidocaine. Circulation 1996, 93, 656–659. [Google Scholar] [CrossRef]
- Balcells, J.; Rodríguez, M.; Pujol, M.; Iglesias, J. Successful treatment of long QT syndrome-induced ventricular tachycardia with esmolol. Pediatr. Cardiol. 2004, 25, 160–162. [Google Scholar] [CrossRef]
- Chockalingam, P.; Crotti, L.; Girardengo, G.; Johnson, J.N.; Harris, K.M.; van der Heijden, J.F.; Hauer, R.N.; Beckmann, B.M.; Spazzolini, C.; Rordorf, R.; et al. Not all beta-blockers are equal in the management of long QT syndrome types 1 and 2: Higher recurrence of events under metoprolol. J. Am. Coll. Cardiol. 2012, 60, 2092–2099. [Google Scholar] [CrossRef]
- Auer, J.; Berent, R.; Eber, B. Amiodarone in the prevention and treatment of arrhythmia. Curr. Opin. Investig. Drugs 2002, 3, 1037–1044. [Google Scholar]
- Aronow, W.S.; Mercando, A.D.; Epstein, S. Effect of benazepril on complex ventricular arrhythmias in older patients with congestive heart failure, prior myocardial infarction, and normal left ventricular ejection fraction. Am. J. Cardiol. 1998, 81, 1368–1370. [Google Scholar] [CrossRef]
- Taylor, K.C.; Kang, P.W.; Hou, P.; Yang, N.D.; Kuenze, G.; Smith, J.A.; Shi, J.; Huang, H.; White, K.M.; Peng, D.; et al. Structure and physiological function of the human KCNQ1 channel voltage sensor intermediate state. elife 2020, 9, e53901. [Google Scholar] [CrossRef]
- Huang, H.; Kuenze, G.; Smith, J.A.; Taylor, K.C.; Duran, A.M.; Hadziselimovic, A.; Meiler, J.; Vanoye, C.G.; George, A.L., Jr.; Sanders, C.R. Mechanisms of KCNQ1 channel dysfunction in long QT syndrome involving voltage sensor domain mutations. Sci. Adv. 2018, 4, eaar2631. [Google Scholar] [CrossRef]
- Dong, Y.; Du, R.; Fan, L.L.; Jin, J.Y.; Huang, H.; Chen, Y.Q.; Bi, D.D.; Xiang, R. Whole-exome sequencing identifies a novel TRPM4 mutation in a Chinese family with atrioventricular block. Biomed. Res. Int. 2021, 2021, 9247541. [Google Scholar] [CrossRef]
- Xian, W.; Hui, X.; Tian, Q.; Wang, H.; Moretti, A.; Laugwitz, K.L.; Flockerzi, V.; Ruppenthal, S.; Lipp, P. Aberrant deactivation-induced gain of function in TRPM4 mutant is associated with human cardiac conduction block. Cell Rep. 2018, 24, 724–731. [Google Scholar] [CrossRef]
- Wang, Z.; Tristani-Firouzi, M.; Xu, Q.; Lin, M.; Keating, M.T.; Sanguinetti, M.C. Functional effects of mutations in KvLQT1 that cause long QT syndrome. J. Cardiovasc. Electrophysiol. 1999, 10, 817–826. [Google Scholar] [CrossRef]
- Leng, T.; Lin, S.; Xiong, Z.; Lin, J. Lidocaine suppresses glioma cell proliferation by inhibiting TRPM7 channels. Int. J. Physiol. Pathophysiol. Pharmacol. 2017, 9, 8–15. [Google Scholar]
- Wolff, M.; Schnöbel-Ehehalt, R.; Mühling, J.; Weigand, M.A.; Olschewski, A. Mechanisms of lidocaine’s action on subtypes of spinal dorsal horn neurons subject to the diverse roles of Na(+) and K(+) channels in action potential generation. Anesth. Analg. 2014, 119, 463–470. [Google Scholar] [CrossRef]
- Yang, X.; Wei, X.; Mu, Y.; Li, Q.; Liu, J. A review of the mechanism of the central analgesic effect of lidocaine. Medicine 2020, 99, e19898. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, F.; Han, Y.; Sang, W.; Wang, L.; Liang, X.; Wang, L.; Xing, Q.; Guo, Y.; Zhang, J.; Zhang, L.; et al. In Vitro Drug Screening Using iPSC-Derived Cardiomyocytes of a Long QT-Syndrome Patient Carrying KCNQ1 & TRPM4 Dual Mutation: An Experimental Personalized Treatment. Cells 2022, 11, 2495. https://doi.org/10.3390/cells11162495
Wang F, Han Y, Sang W, Wang L, Liang X, Wang L, Xing Q, Guo Y, Zhang J, Zhang L, et al. In Vitro Drug Screening Using iPSC-Derived Cardiomyocytes of a Long QT-Syndrome Patient Carrying KCNQ1 & TRPM4 Dual Mutation: An Experimental Personalized Treatment. Cells. 2022; 11(16):2495. https://doi.org/10.3390/cells11162495
Chicago/Turabian StyleWang, Feifei, Yafan Han, Wanyue Sang, Lu Wang, Xiaoyan Liang, Liang Wang, Qiang Xing, Yankai Guo, Jianghua Zhang, Ling Zhang, and et al. 2022. "In Vitro Drug Screening Using iPSC-Derived Cardiomyocytes of a Long QT-Syndrome Patient Carrying KCNQ1 & TRPM4 Dual Mutation: An Experimental Personalized Treatment" Cells 11, no. 16: 2495. https://doi.org/10.3390/cells11162495
APA StyleWang, F., Han, Y., Sang, W., Wang, L., Liang, X., Wang, L., Xing, Q., Guo, Y., Zhang, J., Zhang, L., Zukela, T., Xiaokereti, J., Lu, Y., Zhou, X., Tang, B., & Li, Y. (2022). In Vitro Drug Screening Using iPSC-Derived Cardiomyocytes of a Long QT-Syndrome Patient Carrying KCNQ1 & TRPM4 Dual Mutation: An Experimental Personalized Treatment. Cells, 11(16), 2495. https://doi.org/10.3390/cells11162495