
Promises and Challenges of Immunogenic Chemotherapy in Multiple Myeloma

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
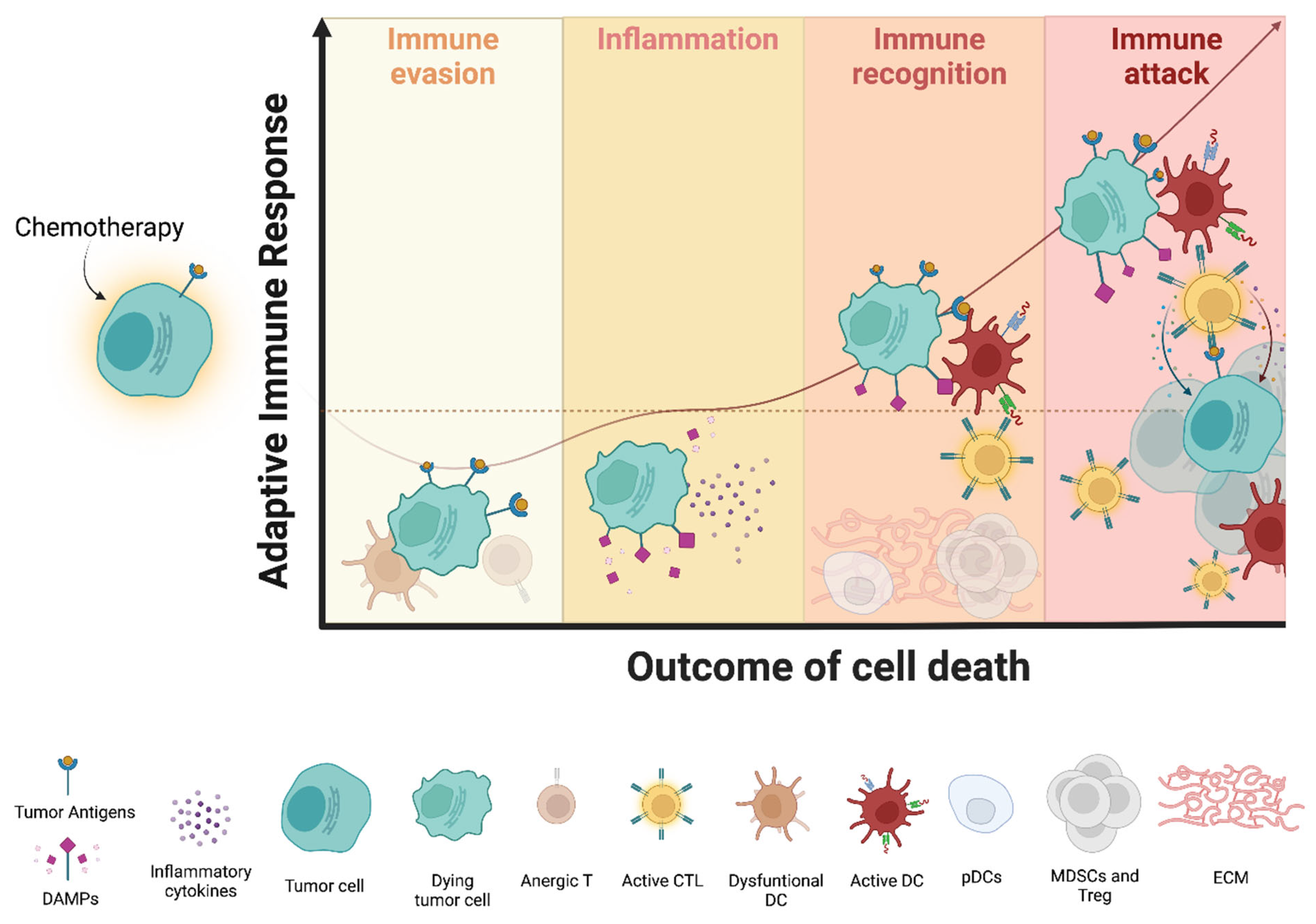
2. Immunogenic Cell Death: Using Cancer to Beat Cancer
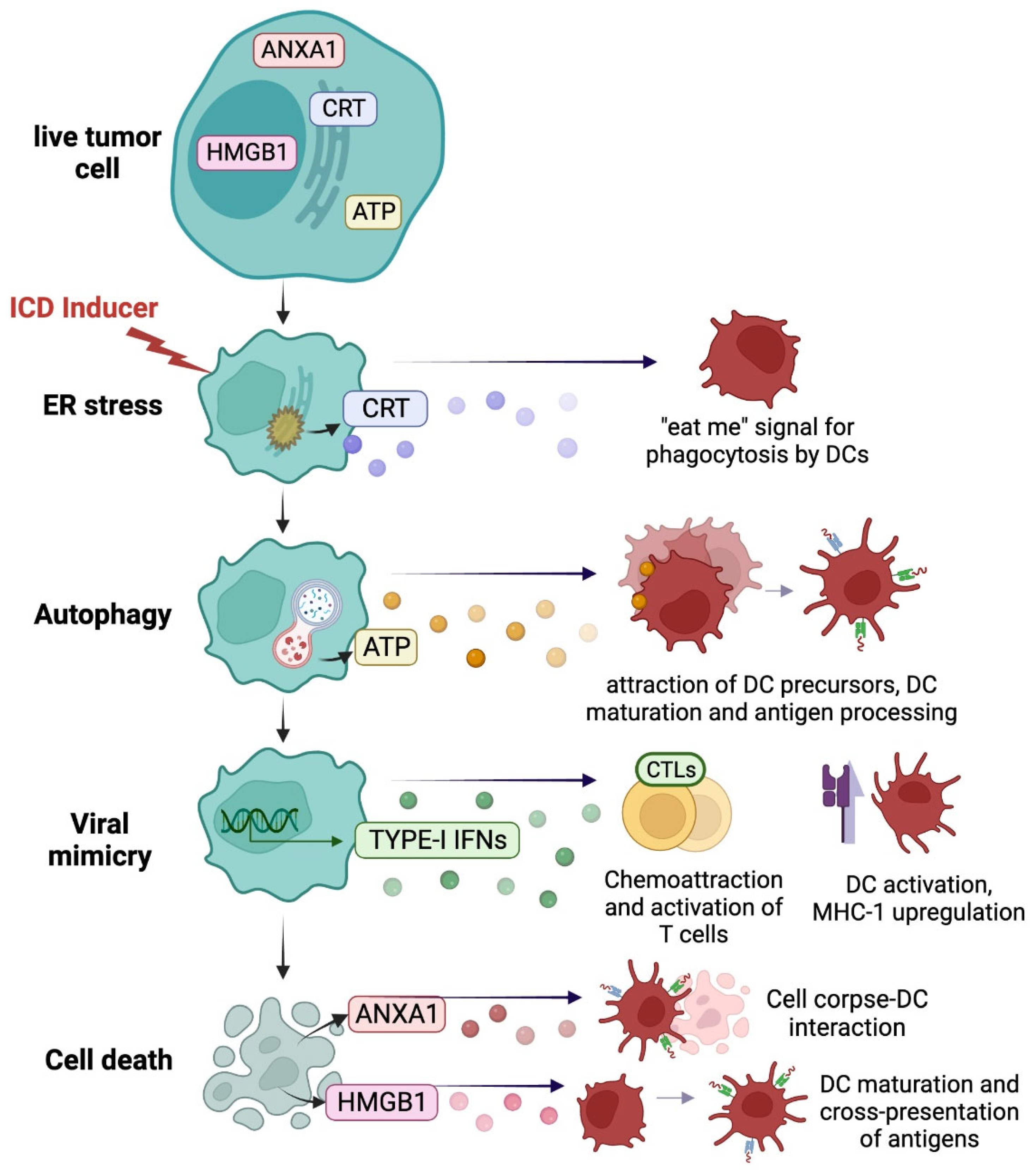
2.1. Immunogenic Danger Signals from Dying Cancer Cells
2.1.1. ER Stress Response and Calreticulin Exposure
2.1.2. Autophagy and ATP Release
2.1.3. Viral Mimicry
2.1.4. Cell Death and Loss of Plasma Membrane Integrity
2.2. Perception of Immunogenicity by Immune Cells: A Focus on MM
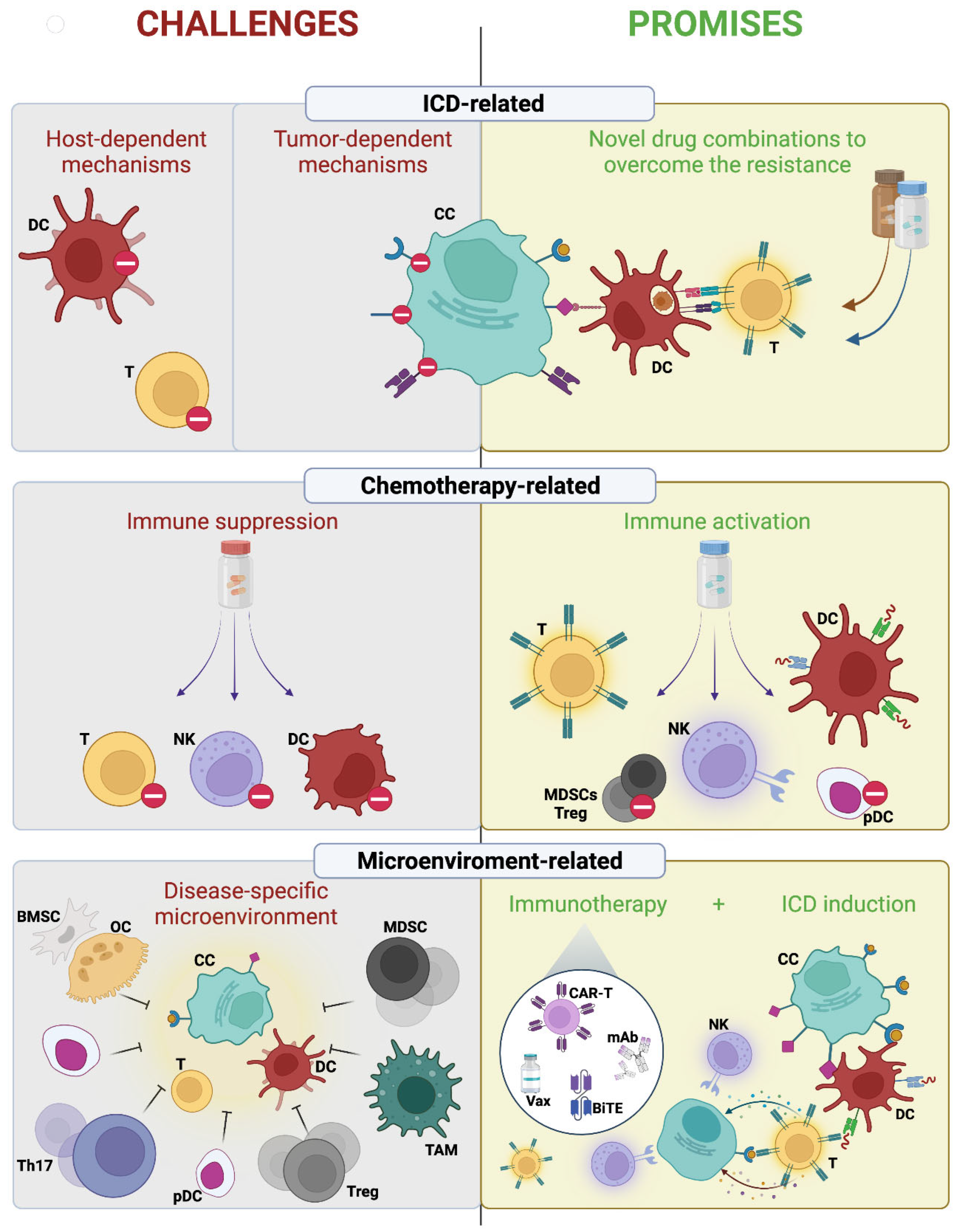
3. Challenges of Immunogenic Chemotherapy in Multiple Myeloma
3.1. Tumor-Dependent ICD Resistance Mechanisms
3.2. Host-Dependent Mechanisms
3.3. Drug-Dependent Mechanisms
4. Promises of Immunogenic Chemotherapy in Multiple Myeloma
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Gulla, A.; Anderson, K.C. Multiple myeloma: The (r)evolution of current therapy and a glance into future. Haematologica 2020, 105, 2358–2367. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, L.; Amodio, N.; Gulla, A.; Anderson, K.C. Harnessing the Immune System Against Multiple Myeloma: Challenges and Opportunities. Front. Oncol. 2020, 10, 606368. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; Kroemer, G. Immunological aspects of cancer chemotherapy. Nat. Rev. Immunol. 2008, 8, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Gulla, A.; Morelli, E.; Samur, M.K.; Botta, C.; Hideshima, T.; Bianchi, G.; Fulciniti, M.; Malvestiti, S.; Prabhala, R.H.; Talluri, S.; et al. Bortezomib induces anti-multiple myeloma immune response mediated by cGAS/STING pathway activation. Blood Cancer Discov. 2021, 2, 468–483. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galassi, C.; Zitvogel, L.; Galluzzi, L. Immunogenic cell stress and death. Nat. Immunol. 2022, 23, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Humeau, J.; Buque, A.; Zitvogel, L.; Kroemer, G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2020, 17, 725–741. [Google Scholar] [CrossRef]
- Matzinger, P. The danger model: A renewed sense of self. Science 2002, 296, 301–305. [Google Scholar] [CrossRef]
- Legrand, A.J.; Konstantinou, M.; Goode, E.F.; Meier, P. The Diversification of Cell Death and Immunity: Memento Mori. Mol. Cell 2019, 76, 232–242. [Google Scholar] [CrossRef]
- Bianchi, G.; Oliva, L.; Cascio, P.; Pengo, N.; Fontana, F.; Cerruti, F.; Orsi, A.; Pasqualetto, E.; Mezghrani, A.; Calbi, V.; et al. The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition. Blood 2009, 113, 3040–3049. [Google Scholar] [CrossRef]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed]
- Fucikova, J.; Spisek, R.; Kroemer, G.; Galluzzi, L. Calreticulin and cancer. Cell Res. 2021, 31, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Bezu, L.; Sauvat, A.; Humeau, J.; Gomes-da-Silva, L.C.; Iribarren, K.; Forveille, S.; Garcia, P.; Zhao, L.; Liu, P.; Zitvogel, L.; et al. eIF2alpha phosphorylation is pathognomonic for immunogenic cell death. Cell Death Differ. 2018, 25, 1375–1393. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Spisek, R.; Charalambous, A.; Mazumder, A.; Vesole, D.H.; Jagannath, S.; Dhodapkar, M.V. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: Therapeutic implications. Blood 2007, 109, 4839–4845. [Google Scholar] [CrossRef] [PubMed]
- Montes de Oca, R.; Alavi, A.S.; Vitali, N.; Bhattacharya, S.; Blackwell, C.; Patel, K.; Seestaller-Wehr, L.; Kaczynski, H.; Shi, H.; Dobrzynski, E.; et al. Belantamab Mafodotin (GSK2857916) Drives Immunogenic Cell Death and Immune-mediated Antitumor Responses In Vivo. Mol. Cancer Ther. 2021, 20, 1941–1955. [Google Scholar] [CrossRef] [PubMed]
- De Beck, L.; Melhaoui, S.; De Veirman, K.; Menu, E.; De Bruyne, E.; Vanderkerken, K.; Breckpot, K.; Maes, K. Epigenetic treatment of multiple myeloma mediates tumor intrinsic and extrinsic immunomodulatory effects. Oncoimmunology 2018, 7, e1484981. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 2020, 8, e000337. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef]
- Li, Y.; Wang, L.X.; Yang, G.; Hao, F.; Urba, W.J.; Hu, H.M. Efficient cross-presentation depends on autophagy in tumor cells. Cancer Res. 2008, 68, 6889–6895. [Google Scholar] [CrossRef]
- Yamazaki, T.; Bravo-San Pedro, J.M.; Galluzzi, L.; Kroemer, G.; Pietrocola, F. Autophagy in the cancer-immunity dialogue. Adv. Drug Deliv. Rev. 2021, 169, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Green, D.R.; Zou, W. Autophagy in tumour immunity and therapy. Nat. Rev. Cancer 2021, 21, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discov. 2019, 9, 1167–1181. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, G.; Chen, Y.; Wang, H.; Hua, Y.; Cai, Z. Immunogenic cell death in cancer therapy: Present and emerging inducers. J. Cell Mol. Med. 2019, 23, 4854–4865. [Google Scholar] [CrossRef] [PubMed]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Bezu, L.; Yamazaki, T.; Di Virgilio, F.; Smyth, M.J.; Kroemer, G.; Galluzzi, L. ATP and cancer immunosurveillance. EMBO J. 2021, 40, e108130. [Google Scholar] [CrossRef]
- Lazarowski, E.R.; Sesma, J.I.; Seminario-Vidal, L.; Kreda, S.M. Molecular mechanisms of purine and pyrimidine nucleotide release. Adv. Pharmacol. 2011, 61, 221–261. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef]
- Zitvogel, L.; Kepp, O.; Galluzzi, L.; Kroemer, G. Inflammasomes in carcinogenesis and anticancer immune responses. Nat. Immunol. 2012, 13, 343–351. [Google Scholar] [CrossRef]
- Ladoire, S.; Enot, D.; Senovilla, L.; Ghiringhelli, F.; Poirier-Colame, V.; Chaba, K.; Semeraro, M.; Chaix, M.; Penault-Llorca, F.; Arnould, L.; et al. The presence of LC3B puncta and HMGB1 expression in malignant cells correlate with the immune infiltrate in breast cancer. Autophagy 2016, 12, 864–875. [Google Scholar] [CrossRef]
- Pietrocola, F.; Pol, J.; Vacchelli, E.; Rao, S.; Enot, D.P.; Baracco, E.E.; Levesque, S.; Castoldi, F.; Jacquelot, N.; Yamazaki, T.; et al. Caloric Restriction Mimetics Enhance Anticancer Immunosurveillance. Cancer Cell 2016, 30, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Ferrere, G.; Tidjani Alou, M.; Liu, P.; Goubet, A.G.; Fidelle, M.; Kepp, O.; Durand, S.; Iebba, V.; Fluckiger, A.; Daillere, R.; et al. Ketogenic diet and ketone bodies enhance the anticancer effects of PD-1 blockade. JCI Insight 2021, 6, e145207. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Araki, K.; Li, S.; Han, J.H.; Ye, L.; Tan, W.G.; Konieczny, B.T.; Bruinsma, M.W.; Martinez, J.; Pearce, E.L.; et al. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat. Immunol. 2014, 15, 1152–1161. [Google Scholar] [CrossRef] [PubMed]
- Yun, Z.; Zhichao, J.; Hao, Y.; Ou, J.; Ran, Y.; Wen, D.; Qun, S. Targeting autophagy in multiple myeloma. Leuk. Res. 2017, 59, 97–104. [Google Scholar] [CrossRef]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remedios, C.; et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef]
- Yamazaki, T.; Kirchmair, A.; Sato, A.; Buque, A.; Rybstein, M.; Petroni, G.; Bloy, N.; Finotello, F.; Stafford, L.; Navarro Manzano, E.; et al. Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nat. Immunol. 2020, 21, 1160–1171. [Google Scholar] [CrossRef]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef]
- Papewalis, C.; Jacobs, B.; Wuttke, M.; Ullrich, E.; Baehring, T.; Fenk, R.; Willenberg, H.S.; Schinner, S.; Cohnen, M.; Seissler, J.; et al. IFN-alpha skews monocytes into CD56+-expressing dendritic cells with potent functional activities in vitro and in vivo. J. Immunol. 2008, 180, 1462–1470. [Google Scholar] [CrossRef]
- Guillot, B.; Portales, P.; Thanh, A.D.; Merlet, S.; Dereure, O.; Clot, J.; Corbeau, P. The expression of cytotoxic mediators is altered in mononuclear cells of patients with melanoma and increased by interferon-alpha treatment. Br. J. Dermatol. 2005, 152, 690–696. [Google Scholar] [CrossRef]
- Ilander, M.; Kreutzman, A.; Rohon, P.; Melo, T.; Faber, E.; Porkka, K.; Vakkila, J.; Mustjoki, S. Enlarged memory T-cell pool and enhanced Th1-type responses in chronic myeloid leukemia patients who have successfully discontinued IFN-alpha monotherapy. PLoS ONE 2014, 9, e87794. [Google Scholar] [CrossRef] [PubMed]
- Crouse, J.; Bedenikovic, G.; Wiesel, M.; Ibberson, M.; Xenarios, I.; Von Laer, D.; Kalinke, U.; Vivier, E.; Jonjic, S.; Oxenius, A. Type I interferons protect T cells against NK cell attack mediated by the activating receptor NCR1. Immunity 2014, 40, 961–973. [Google Scholar] [CrossRef]
- Xu, H.C.; Grusdat, M.; Pandyra, A.A.; Polz, R.; Huang, J.; Sharma, P.; Deenen, R.; Kohrer, K.; Rahbar, R.; Diefenbach, A.; et al. Type I interferon protects antiviral CD8+ T cells from NK cell cytotoxicity. Immunity 2014, 40, 949–960. [Google Scholar] [CrossRef]
- Bacher, N.; Raker, V.; Hofmann, C.; Graulich, E.; Schwenk, M.; Baumgrass, R.; Bopp, T.; Zechner, U.; Merten, L.; Becker, C.; et al. Interferon-alpha suppresses cAMP to disarm human regulatory T cells. Cancer Res. 2013, 73, 5647–5656. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tai, Y.T.; Ho, M.Z.G.; Qiu, L.; Anderson, K.C. Interferon-alpha-based immunotherapies in the treatment of B cell-derived hematologic neoplasms in today’s treat-to-target era. Exp. Hematol. Oncol. 2017, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in inflammation and immunity. Cell 2010, 140, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Chavan, S.S.; Andersson, U. High Mobility Group Box Protein 1 (HMGB1): The Prototypical Endogenous Danger Molecule. Mol. Med. 2015, 21 (Suppl. 1), S6–S12. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef]
- Vacchelli, E.; Ma, Y.; Baracco, E.E.; Sistigu, A.; Enot, D.P.; Pietrocola, F.; Yang, H.; Adjemian, S.; Chaba, K.; Semeraro, M.; et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science 2015, 350, 972–978. [Google Scholar] [CrossRef]
- Lee, C.H.; Yelensky, R.; Jooss, K.; Chan, T.A. Update on Tumor Neoantigens and Their Utility: Why It Is Good to Be Different. Trends Immunol. 2018, 39, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, H.; Vesely, M.D.; Koboldt, D.C.; Rickert, C.G.; Uppaluri, R.; Magrini, V.J.; Arthur, C.D.; White, J.M.; Chen, Y.S.; Shea, L.K.; et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012, 482, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Tureci, O.; Vormehr, M.; Diken, M.; Kreiter, S.; Huber, C.; Sahin, U. Targeting the Heterogeneity of Cancer with Individualized Neoepitope Vaccines. Clin. Cancer Res. 2016, 22, 1885–1896. [Google Scholar] [CrossRef] [PubMed]
- Perumal, D.; Imai, N.; Lagana, A.; Finnigan, J.; Melnekoff, D.; Leshchenko, V.V.; Solovyov, A.; Madduri, D.; Chari, A.; Cho, H.J.; et al. Mutation-derived Neoantigen-specific T-cell Responses in Multiple Myeloma. Clin. Cancer Res. 2020, 26, 450–464. [Google Scholar] [CrossRef] [PubMed]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef]
- Walz, S.; Stickel, J.S.; Kowalewski, D.J.; Schuster, H.; Weisel, K.; Backert, L.; Kahn, S.; Nelde, A.; Stroh, T.; Handel, M.; et al. The antigenic landscape of multiple myeloma: Mass spectrometry (re)defines targets for T-cell-based immunotherapy. Blood 2015, 126, 1203–1213. [Google Scholar] [CrossRef]
- Neri, P.; Bahlis, N.J. Genomic instability in multiple myeloma: Mechanisms and therapeutic implications. Expert Opin. Biol. Ther. 2013, 13 (Suppl. 1), S69–S82. [Google Scholar] [CrossRef]
- Shammas, M.A.; Shmookler Reis, R.J.; Koley, H.; Batchu, R.B.; Li, C.; Munshi, N.C. Dysfunctional homologous recombination mediates genomic instability and progression in myeloma. Blood 2009, 113, 2290–2297. [Google Scholar] [CrossRef]
- Samur, M.K.; Aktas Samur, A.; Fulciniti, M.; Szalat, R.; Han, T.; Shammas, M.; Richardson, P.; Magrangeas, F.; Minvielle, S.; Corre, J.; et al. Genome-Wide Somatic Alterations in Multiple Myeloma Reveal a Superior Outcome Group. J. Clin. Oncol. 2020, 38, 3107–3118. [Google Scholar] [CrossRef]
- Miller, A.; Asmann, Y.; Cattaneo, L.; Braggio, E.; Keats, J.; Auclair, D.; Lonial, S.; Network, M.C.; Russell, S.J.; Stewart, A.K. High somatic mutation and neoantigen burden are correlated with decreased progression-free survival in multiple myeloma. Blood Cancer J. 2017, 7, e612. [Google Scholar] [CrossRef]
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 2021, 21, 298–312. [Google Scholar] [CrossRef] [PubMed]
- Paludan, S.R.; Pradeu, T.; Masters, S.L.; Mogensen, T.H. Constitutive immune mechanisms: Mediators of host defence and immune regulation. Nat. Rev. Immunol. 2021, 21, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Dudek-Peric, A.M.; Ferreira, G.B.; Muchowicz, A.; Wouters, J.; Prada, N.; Martin, S.; Kiviluoto, S.; Winiarska, M.; Boon, L.; Mathieu, C.; et al. Antitumor immunity triggered by melphalan is potentiated by melanoma cell surface-associated calreticulin. Cancer Res. 2015, 75, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Ding, Z.C.; Cao, Y.; Liu, C.; Habtetsion, T.; Yu, M.; Lemos, H.; Salman, H.; Xu, H.; Mellor, A.L.; et al. Alkylating agent melphalan augments the efficacy of adoptive immunotherapy using tumor-specific CD4+ T cells. J. Immunol. 2015, 194, 2011–2021. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.D.; Raje, N.; Fowler, J.A.; Mezzi, K.; Scott, E.C.; Dhodapkar, M.V. How to Train Your T Cells: Overcoming Immune Dysfunction in Multiple Myeloma. Clin. Cancer Res. 2020, 26, 1541–1554. [Google Scholar] [CrossRef]
- Zavidij, O.; Haradhvala, N.J.; Mouhieddine, T.H.; Sklavenitis-Pistofidis, R.; Cai, S.; Reidy, M.; Rahmat, M.; Flaifel, A.; Ferland, B.; Su, N.K.; et al. Single-cell RNA sequencing reveals compromised immune microenvironment in precursor stages of multiple myeloma. Nat. Cancer 2020, 1, 493–506. [Google Scholar] [CrossRef]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259–1271.e1211. [Google Scholar] [CrossRef]
- Garrido, F. HLA Class-I Expression and Cancer Immunotherapy. Adv. Exp. Med. Biol. 2019, 1151, 79–90. [Google Scholar] [CrossRef]
- Carbone, E.; Neri, P.; Mesuraca, M.; Fulciniti, M.T.; Otsuki, T.; Pende, D.; Groh, V.; Spies, T.; Pollio, G.; Cosman, D.; et al. HLA class I, NKG2D, and natural cytotoxicity receptors regulate multiple myeloma cell recognition by natural killer cells. Blood 2005, 105, 251–258. [Google Scholar] [CrossRef]
- Samur, M.K.; Fulciniti, M.; Aktas Samur, A.; Bazarbachi, A.H.; Tai, Y.T.; Prabhala, R.; Alonso, A.; Sperling, A.S.; Campbell, T.; Petrocca, F.; et al. Biallelic loss of BCMA as a resistance mechanism to CAR T cell therapy in a patient with multiple myeloma. Nat. Commun. 2021, 12, 868. [Google Scholar] [CrossRef]
- van de Donk, N.; Usmani, S.Z. CD38 Antibodies in Multiple Myeloma: Mechanisms of Action and Modes of Resistance. Front. Immunol. 2018, 9, 2134. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Zhou, Z.; Li, H.; Xue, Y.; Lu, X.; Bahar, I.; Kepp, O.; Hung, M.C.; Kroemer, G.; Wan, Y. Pharmacologic Suppression of B7-H4 Glycosylation Restores Antitumor Immunity in Immune-Cold Breast Cancers. Cancer Discov. 2020, 10, 1872–1893. [Google Scholar] [CrossRef] [PubMed]
- Fucikova, J.; Truxova, I.; Hensler, M.; Becht, E.; Kasikova, L.; Moserova, I.; Vosahlikova, S.; Klouckova, J.; Church, S.E.; Cremer, I.; et al. Calreticulin exposure by malignant blasts correlates with robust anticancer immunity and improved clinical outcome in AML patients. Blood 2016, 128, 3113–3124. [Google Scholar] [CrossRef]
- Liu, P.; Zhao, L.; Loos, F.; Marty, C.; Xie, W.; Martins, I.; Lachkar, S.; Qu, B.; Waeckel-Enee, E.; Plo, I.; et al. Immunosuppression by Mutated Calreticulin Released from Malignant Cells. Mol. Cell 2020, 77, 748–760.e749. [Google Scholar] [CrossRef]
- Lin, H.; Kryczek, I.; Li, S.; Green, M.D.; Ali, A.; Hamasha, R.; Wei, S.; Vatan, L.; Szeliga, W.; Grove, S.; et al. Stanniocalcin 1 is a phagocytosis checkpoint driving tumor immune resistance. Cancer Cell 2021, 39, 480–493.e486. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Marjon, K.D.; Zhu, F.; Weissman-Tsukamoto, R.; Levett, A.; Sullivan, K.; Kao, K.S.; Markovic, M.; Bump, P.A.; Jackson, H.M.; et al. Programmed cell removal by calreticulin in tissue homeostasis and cancer. Nat. Commun. 2018, 9, 3194. [Google Scholar] [CrossRef]
- Sun, J.; Muz, B.; Alhallak, K.; Markovic, M.; Gurley, S.; Wang, Z.; Guenthner, N.; Wasden, K.; Fiala, M.; King, J.; et al. Targeting CD47 as a Novel Immunotherapy for Multiple Myeloma. Cancers 2020, 12, 305. [Google Scholar] [CrossRef]
- Rybstein, M.D.; Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. The autophagic network and cancer. Nat. Cell Biol. 2018, 20, 243–251. [Google Scholar] [CrossRef]
- Allard, B.; Allard, D.; Buisseret, L.; Stagg, J. The adenosine pathway in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 17, 611–629. [Google Scholar] [CrossRef]
- Moesta, A.K.; Li, X.Y.; Smyth, M.J. Targeting CD39 in cancer. Nat. Rev. Immunol. 2020, 20, 739–755. [Google Scholar] [CrossRef]
- Baracco, E.E.; Stoll, G.; Van Endert, P.; Zitvogel, L.; Vacchelli, E.; Kroemer, G. Contribution of annexin A1 to anticancer immunosurveillance. Oncoimmunology 2019, 8, e1647760. [Google Scholar] [CrossRef]
- Bailur, J.K.; McCachren, S.S.; Doxie, D.B.; Shrestha, M.; Pendleton, K.; Nooka, A.K.; Neparidze, N.; Parker, T.L.; Bar, N.; Kaufman, J.L.; et al. Early alterations in stem-like/resident T cells, innate and myeloid cells in the bone marrow in preneoplastic gammopathy. JCI Insight 2019, 5, e127807. [Google Scholar] [CrossRef] [PubMed]
- Prabhala, R.H.; Neri, P.; Bae, J.E.; Tassone, P.; Shammas, M.A.; Allam, C.K.; Daley, J.F.; Chauhan, D.; Blanchard, E.; Thatte, H.S.; et al. Dysfunctional T regulatory cells in multiple myeloma. Blood 2006, 107, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Dhodapkar, K.M.; Barbuto, S.; Matthews, P.; Kukreja, A.; Mazumder, A.; Vesole, D.; Jagannath, S.; Dhodapkar, M.V. Dendritic cells mediate the induction of polyfunctional human IL17-producing cells (Th17-1 cells) enriched in the bone marrow of patients with myeloma. Blood 2008, 112, 2878–2885. [Google Scholar] [CrossRef]
- Prabhala, R.H.; Pelluru, D.; Fulciniti, M.; Prabhala, H.K.; Nanjappa, P.; Song, W.; Pai, C.; Amin, S.; Tai, Y.T.; Richardson, P.G.; et al. Elevated IL-17 produced by TH17 cells promotes myeloma cell growth and inhibits immune function in multiple myeloma. Blood 2010, 115, 5385–5392. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; van Duin, M.; Sonneveld, P.; Mateos, M.V.; Gay, F.; Anderson, K.C. Multiple myeloma. Nat. Rev. Dis. Primers 2017, 3, 17046. [Google Scholar] [CrossRef] [PubMed]
- Burwick, N.; Sharma, S. Glucocorticoids in multiple myeloma: Past, present, and future. Ann. Hematol. 2019, 98, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xia, L.; Chen, J.; Zhang, S.; Martin, V.; Li, Q.; Lin, S.; Chen, J.; Calmette, J.; Lu, M.; et al. Stress-glucocorticoid-TSC22D3 axis compromises therapy-induced antitumor immunity. Nat. Med. 2019, 25, 1428–1441. [Google Scholar] [CrossRef]
- Rozkova, D.; Horvath, R.; Bartunkova, J.; Spisek, R. Glucocorticoids severely impair differentiation and antigen presenting function of dendritic cells despite upregulation of Toll-like receptors. Clin. Immunol. 2006, 120, 260–271. [Google Scholar] [CrossRef]
- Chiossone, L.; Vitale, C.; Cottalasso, F.; Moretti, S.; Azzarone, B.; Moretta, L.; Mingari, M.C. Molecular analysis of the methylprednisolone-mediated inhibition of NK-cell function: Evidence for different susceptibility of IL-2- versus IL-15-activated NK cells. Blood 2007, 109, 3767–3775. [Google Scholar] [CrossRef]
- Franco, L.M.; Gadkari, M.; Howe, K.N.; Sun, J.; Kardava, L.; Kumar, P.; Kumari, S.; Hu, Z.; Fraser, I.D.C.; Moir, S.; et al. Immune regulation by glucocorticoids can be linked to cell type-dependent transcriptional responses. J. Exp. Med. 2019, 216, 384–406. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Ogiya, D.; Liu, J.; Harada, T.; Kurata, K.; Bae, J.; Massefski, W.; Anderson, K.C. Immunomodulatory drugs activate NK cells via both Zap-70 and cereblon-dependent pathways. Leukemia 2021, 35, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.; Hideshima, T.; Tai, Y.T.; Song, Y.; Richardson, P.; Raje, N.; Munshi, N.C.; Anderson, K.C. Histone deacetylase (HDAC) inhibitor ACY241 enhances anti-tumor activities of antigen-specific central memory cytotoxic T lymphocytes against multiple myeloma and solid tumors. Leukemia 2018, 32, 1932–1947. [Google Scholar] [CrossRef] [PubMed]
- Gandolfi, S.; Laubach, J.P.; Hideshima, T.; Chauhan, D.; Anderson, K.C.; Richardson, P.G. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 2017, 36, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.; Kepp, O.; Schlemmer, F.; Adjemian, S.; Tailler, M.; Shen, S.; Michaud, M.; Menger, L.; Gdoura, A.; Tajeddine, N.; et al. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene 2011, 30, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- .Kepp, O.; Galluzzi, L.; Giordanetto, F.; Tesniere, A.; Vitale, I.; Martins, I.; Schlemmer, F.; Adjemian, S.; Zitvogel, L.; Kroemer, G. Disruption of the PP1/GADD34 complex induces calreticulin exposure. Cell Cycle 2009, 8, 3971–3977. [Google Scholar] [CrossRef]
- Le Naour, J.; Liu, P.; Zhao, L.; Adjemian, S.; Sztupinszki, Z.; Taieb, J.; Mulot, C.; Silvin, A.; Dutertre, C.A.; Ginhoux, F.; et al. A TLR3 Ligand Reestablishes Chemotherapeutic Responses in the Context of FPR1 Deficiency. Cancer Discov. 2021, 11, 408–423. [Google Scholar] [CrossRef]
- Amouzegar, A.; Chelvanambi, M.; Filderman, J.N.; Storkus, W.J.; Luke, J.J. STING Agonists as Cancer Therapeutics. Cancers 2021, 13, 2695. [Google Scholar] [CrossRef]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J. Clin. Oncol. 2016, 34, 2698–2704. [Google Scholar] [CrossRef]
- Costello, C. The future of checkpoint inhibition in multiple myeloma? Lancet Haematol. 2019, 6, e439–e440. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnstone, M.; Vinaixa, D.; Turi, M.; Morelli, E.; Anderson, K.C.; Gulla, A. Promises and Challenges of Immunogenic Chemotherapy in Multiple Myeloma. Cells 2022, 11, 2519. https://doi.org/10.3390/cells11162519
Johnstone M, Vinaixa D, Turi M, Morelli E, Anderson KC, Gulla A. Promises and Challenges of Immunogenic Chemotherapy in Multiple Myeloma. Cells. 2022; 11(16):2519. https://doi.org/10.3390/cells11162519
Chicago/Turabian StyleJohnstone, Megan, Delaney Vinaixa, Marcello Turi, Eugenio Morelli, Kenneth Carl Anderson, and Annamaria Gulla. 2022. "Promises and Challenges of Immunogenic Chemotherapy in Multiple Myeloma" Cells 11, no. 16: 2519. https://doi.org/10.3390/cells11162519
APA StyleJohnstone, M., Vinaixa, D., Turi, M., Morelli, E., Anderson, K. C., & Gulla, A. (2022). Promises and Challenges of Immunogenic Chemotherapy in Multiple Myeloma. Cells, 11(16), 2519. https://doi.org/10.3390/cells11162519