
Integrated Multi-Omics Signature Predicts Survival in Head and Neck Cancer

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Sample
2.2. Statistical Analysis
2.2.1. Multi-Omics HNSCC Data Reduction
2.2.2. Principal Components Analysis and Survival Analysis
2.2.3. Classification Using the First Ten PCs as Predictors for Survival
2.2.4. Selection of Genes for the Multi-Omics Signature
2.2.5. Evaluation of the Proposed Multi-Omics Signature
2.2.6. Association of the Obtained Clusters with the Metastatic Status of the Patients
3. Results
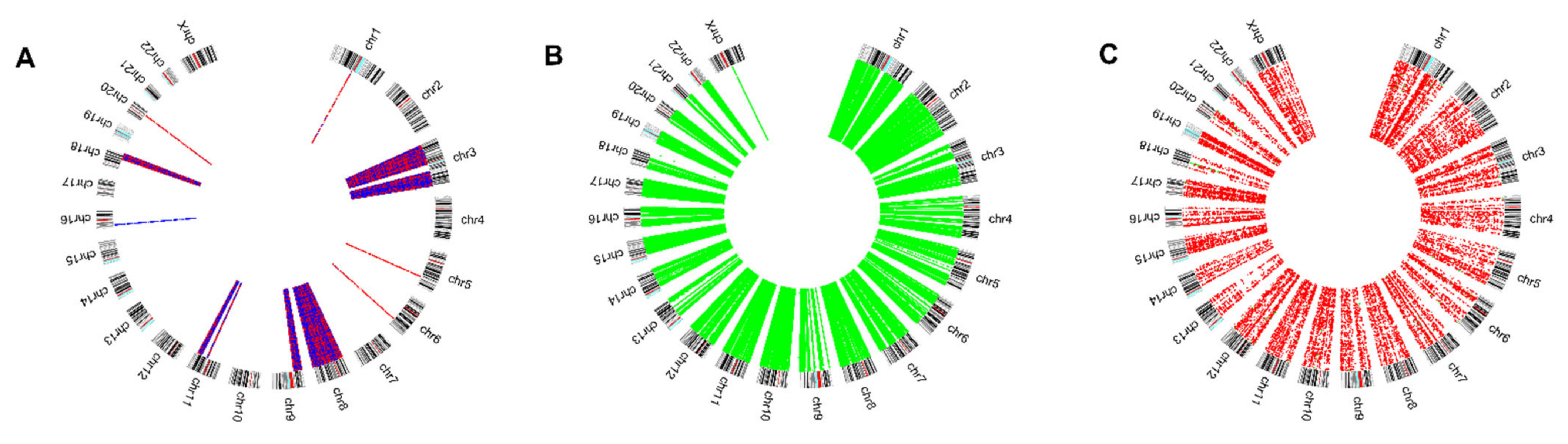
3.1. Multi-Omics HNSCC Data Reduction
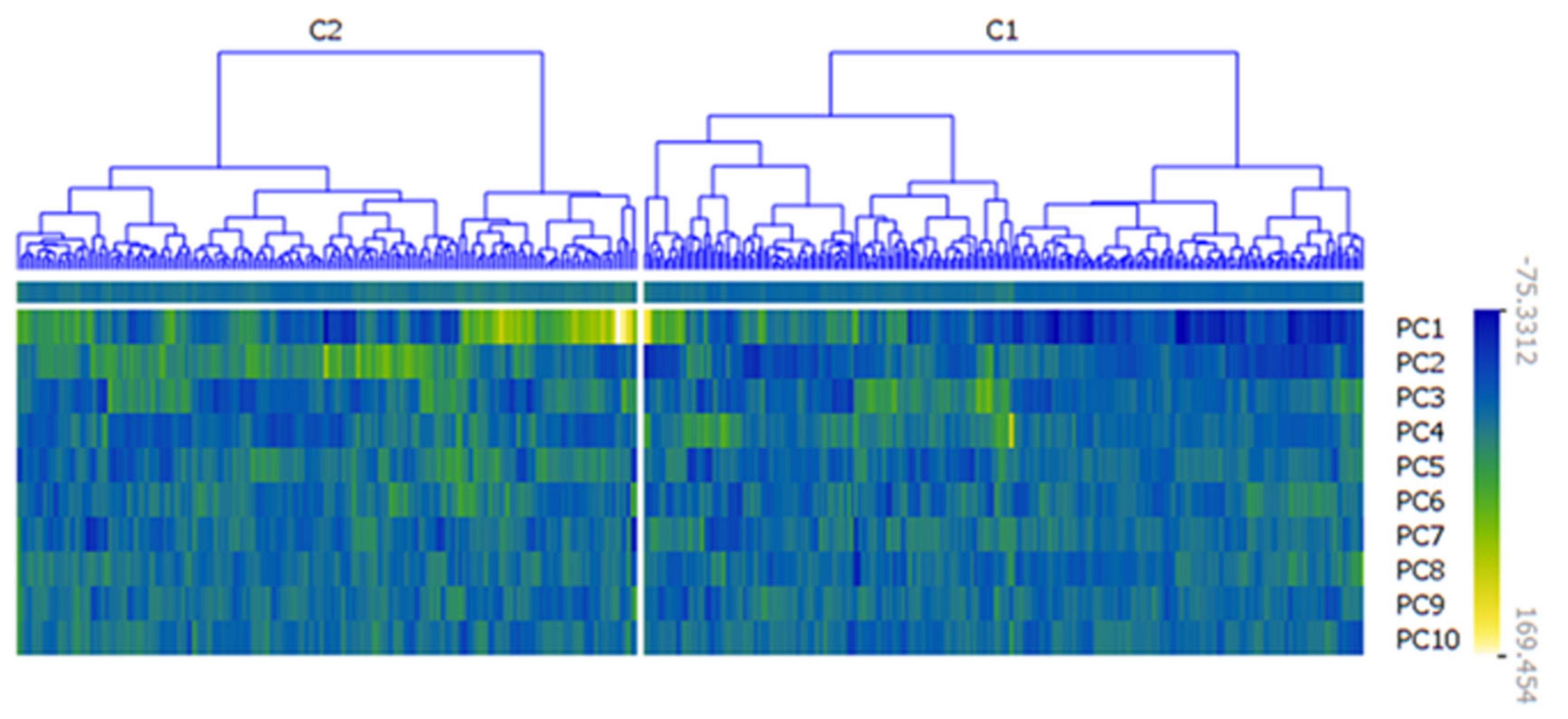
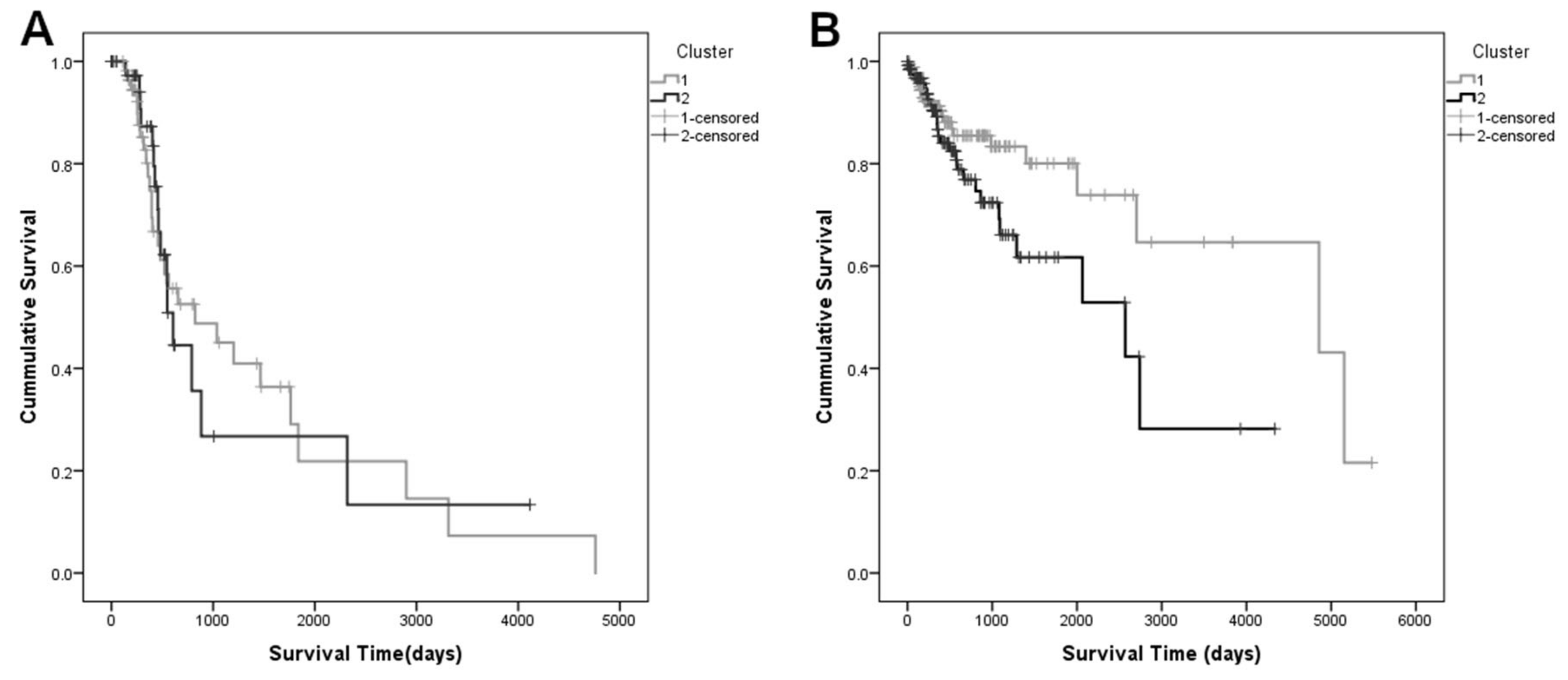
3.2. Principal Components Analysis and Survival Analysis
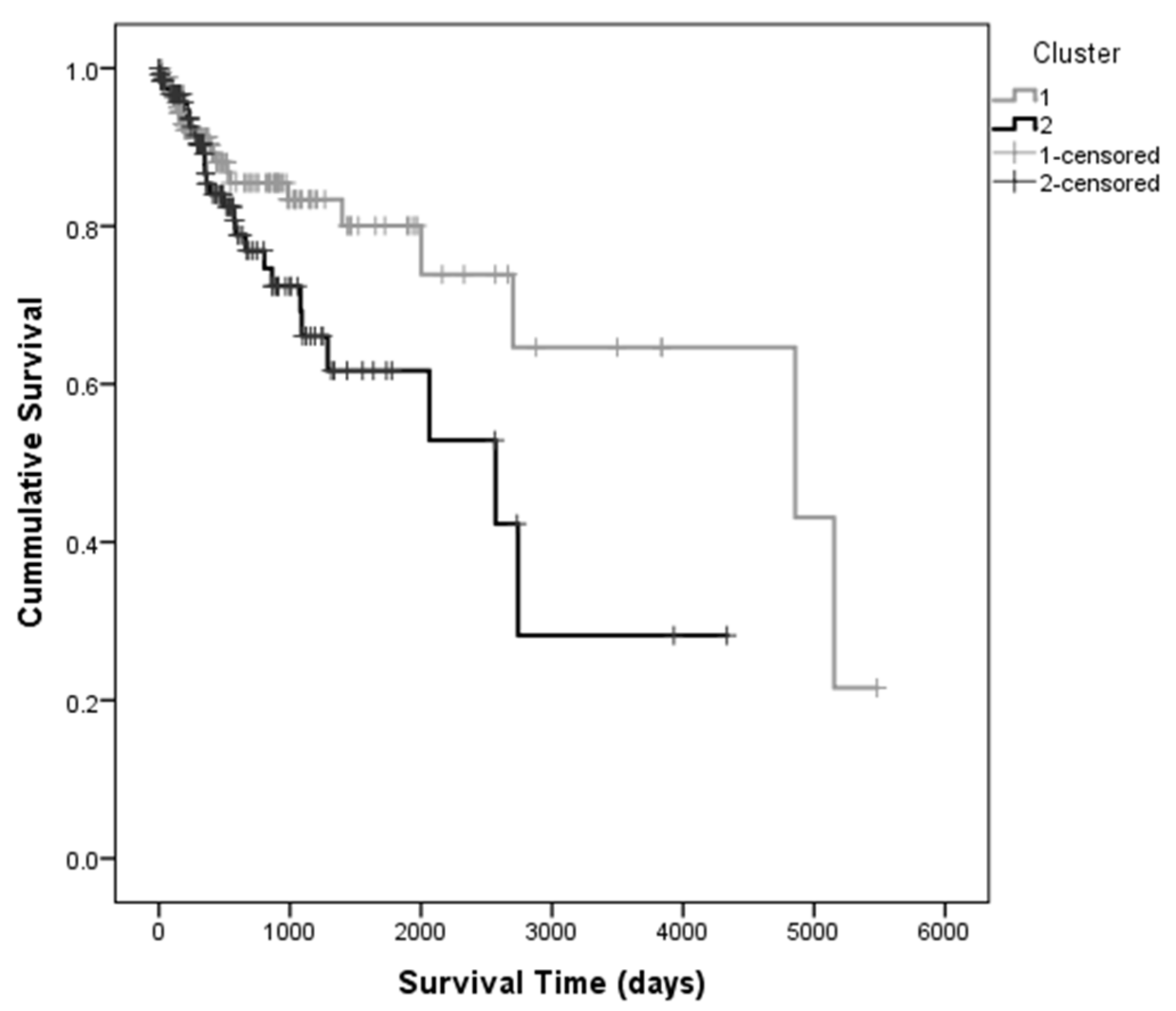
3.3. Classification Using the First Ten PCs as Predictors for Survival
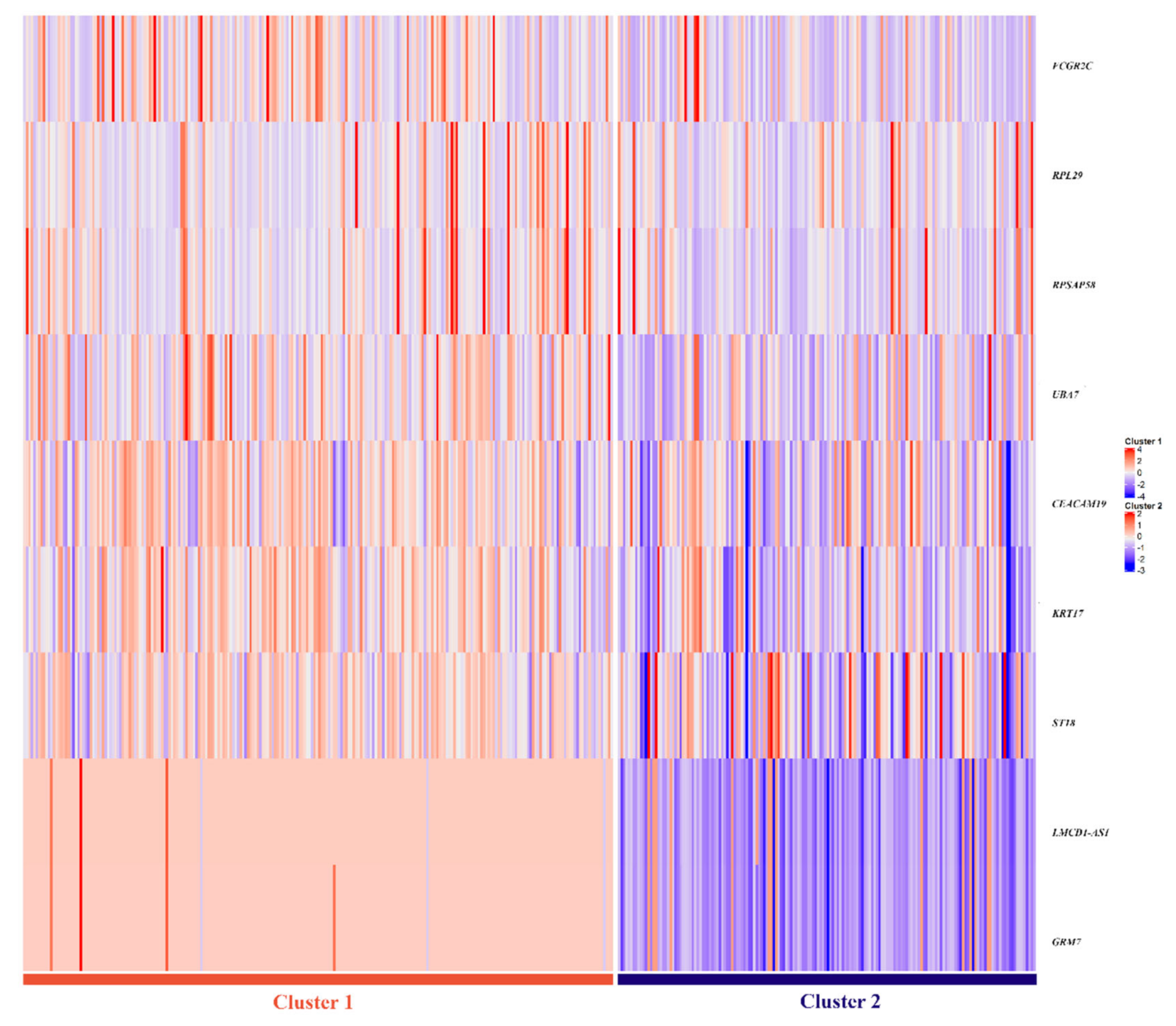
3.4. Selection of Genes for the Multi-Omics Signature
3.5. Assessment of Prediction Value of Proposed Multi-Omics Signature
3.6. Association of the Obtained Clusters with Metastatic Status of the Patients
4. Discussion
- (i)
- copy number alterations in LMCD1-A1S (3p26.1) and GRM7 (3p26.1) genes;
- (ii)
- gene expression of FCGR2C (1q23.2), RPL29 (3p21.1), UBA7(3p21.31), and RPSAP58 (19p12);
- (iii)
- methylation of ST18 (8q11.23), KRT17 (17q21.2), and CEACAM19 (19q13.31) genes,
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Plath, M.; Gass, J.; Hlevnjak, M.; Li, Q.; Feng, B.; Hostench, X.P.; Bieg, M.; Schroeder, L.; Holzinger, D.; Zapatka, M.; et al. Unraveling most abundant mutational signatures in head and neck cancer. Int. J. Cancer 2021, 148, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Global Burden of Disease Cancer Collaboration; Fitzmaurice, C.; Allen, C.; Barber, R.M.; Barregard, L.; Bhutta, Z.A.; Brenner, H.; Dicker, D.J.; Chimed-Orchir, O.; Dandona, R.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-years for 32 Cancer Groups, 1990 to 2015: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2017, 3, 524–548. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.Q.M. Head and Neck Cancer. N. Engl. J. Med. 2020, 382, 60–72. [Google Scholar] [CrossRef]
- Braakhuis, B.J.; Brakenhoff, R.H.; Leemans, C.R. Second field tumors: A new opportunity for cancer prevention? Oncologist 2005, 10, 493–500. [Google Scholar] [CrossRef]
- Ribeiro, I.P.; Esteves, L.; Santos, A.; Barroso, L.; Marques, F.; Caramelo, F.; Melo, J.B.; Carreira, I.M. A seven-gene signature to predict the prognosis of oral squamous cell carcinoma. Oncogene 2021, 40, 3859–3869. [Google Scholar] [CrossRef]
- Ribeiro, I.P.; Barroso, L.; Marques, F.; Melo, J.B.; Carreira, I.M. Early detection and personalized treatment in oral cancer: The impact of omics approaches. Mol. Cytogenet. 2016, 9, 85. [Google Scholar] [CrossRef]
- Costa, R.L.; Boroni, M.; Soares, M.A. Distinct co-expression networks using multi-omic data reveal novel interventional targets in HPV-positive and negative head-and-neck squamous cell cancer. Sci. Rep. 2018, 8, 15254. [Google Scholar] [CrossRef]
- Ribeiro, I.P.; Caramelo, F.; Esteves, L.; Menoita, J.; Marques, F.; Barroso, L.; Migueis, J.; Melo, J.B.; Carreira, I.M. Genomic predictive model for recurrence and metastasis development in head and neck squamous cell carcinoma patients. Sci. Rep. 2017, 7, 13897. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef]
- Yang, J.; Chen, Y.; Luo, H.; Cai, H. The Landscape of Somatic Copy Number Alterations in Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2020, 10, 321. [Google Scholar] [CrossRef]
- You, G.R.; Cheng, A.J.; Lee, L.Y.; Huang, Y.C.; Liu, H.; Chen, Y.J.; Chang, J.T. Prognostic signature associated with radioresistance in head and neck cancer via transcriptomic and bioinformatic analyses. BMC Cancer 2019, 19, 64. [Google Scholar] [CrossRef]
- Guo, H.; Li, C.; Su, X.; Huang, X. A Five-mRNA Expression Signature to Predict Survival in Oral Squamous Cell Carcinoma by Integrated Bioinformatic Analyses. Genet. Test. Mol. Biomark. 2021, 25, 517–527. [Google Scholar] [CrossRef]
- Ribeiro, I.P.; Esteves, L.; Anjo, S.I.; Marques, F.; Barroso, L.; Manadas, B.; Carreira, I.M.; Melo, J.B. Proteomics-based Predictive Model for the Early Detection of Metastasis and Recurrence in Head and Neck Cancer. Cancer Genom. Proteom. 2020, 17, 259–269. [Google Scholar] [CrossRef]
- Bohnenberger, H.; Kaderali, L.; Strobel, P.; Yepes, D.; Plessmann, U.; Dharia, N.V.; Yao, S.; Heydt, C.; Merkelbach-Bruse, S.; Emmert, A.; et al. Comparative proteomics reveals a diagnostic signature for pulmonary head-and-neck cancer metastasis. EMBO Mol. Med. 2018, 10, e8428. [Google Scholar] [CrossRef]
- Ribeiro, I.P.; Caramelo, F.; Marques, F.; Domingues, A.; Mesquita, M.; Barroso, L.; Prazeres, H.; Juliao, M.J.; Baptista, I.P.; Ferreira, A.; et al. WT1, MSH6, GATA5 and PAX5 as epigenetic oral squamous cell carcinoma biomarkers—A short report. Cell. Oncol. 2016, 39, 573–582. [Google Scholar] [CrossRef]
- Worsham, M.J.; Chen, K.M.; Datta, I.; Stephen, J.K.; Chitale, D.; Gothard, A.; Divine, G. The biological significance of methylome differences in human papilloma virus associated head and neck cancer. Oncol. Lett. 2016, 12, 4949–4956. [Google Scholar] [CrossRef]
- Bai, G.; Song, J.; Yuan, Y.; Chen, Z.; Tian, Y.; Yin, X.; Niu, Y.; Liu, J. Systematic analysis of differentially methylated expressed genes and site-speci fi c methylation as potential prognostic markers in head and neck cancer. J. Cell. Physiol. 2019, 234, 22687–22702. [Google Scholar] [CrossRef]
- Bioconductor Core Team. Homo. Sapiens: Annotation Package for the Homo.Sapiens Object; R package version 1.3.1. 2015. Available online: https://bioconductor.org/packages/release/data/annotation/html/Homo.sapiens.html (accessed on 18 July 2022).
- Demsar, J.C.T.; Erjavec, A.; Gorup, C.; Hocevar, T.; Milutinovic, M.; Mozina, M.; Polajnar, M.; Toplak, M.; Staric, A.; Stajdohar, M.; et al. Orange: Data Mining Toolbox in Python. J. Mach. Learn. Res. 2013, 14, 2349–2353. [Google Scholar]
- Bersanelli, M.; Mosca, E.; Remondini, D.; Giampieri, E.; Sala, C.; Castellani, G.; Milanesi, L. Methods for the integration of multi-omics data: Mathematical aspects. BMC Bioinform. 2016, 17 (Suppl. S2), 15. [Google Scholar] [CrossRef]
- Vincent-Chong, V.K.; Salahshourifar, I.; Woo, K.M.; Anwar, A.; Razali, R.; Gudimella, R.; Rahman, Z.A.; Ismail, S.M.; Kallarakkal, T.G.; Ramanathan, A.; et al. Genome wide profiling in oral squamous cell carcinoma identifies a four genetic marker signature of prognostic significance. PLoS ONE 2017, 12, e0174865. [Google Scholar] [CrossRef]
- Ribeiro, I.P.; Marques, F.; Caramelo, F.; Pereira, J.; Patricio, M.; Prazeres, H.; Ferrao, J.; Juliao, M.J.; Castelo-Branco, M.; de Melo, J.B.; et al. Genetic gains and losses in oral squamous cell carcinoma: Impact on clinical management. Cell. Oncol. 2014, 37, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Califano, J.; van der Riet, P.; Westra, W.; Nawroz, H.; Clayman, G.; Piantadosi, S.; Corio, R.; Lee, D.; Greenberg, B.; Koch, W.; et al. Genetic progression model for head and neck cancer: Implications for field cancerization. Cancer Res. 1996, 56, 2488–2492. [Google Scholar] [CrossRef]
- Estiar, M.A.; Esmaeili, R.; Zare, A.A.; Farahmand, L.; Fazilaty, H.; Zekri, A.; Jafarbeik-Iravani, N.; Majidzadeh, A.K. High expression of CEACAM19, a new member of carcinoembryonic antigen gene family, in patients with breast cancer. Clin. Exp. Med. 2017, 17, 547–553. [Google Scholar] [CrossRef]
- Zhao, H.; Xu, J.; Wang, Y.; Jiang, R.; Li, X.; Zhang, L.; Che, Y. Knockdown of CEACAM19 suppresses human gastric cancer through inhibition of PI3K/Akt and NF-kappaB. Surg. Oncol. 2018, 27, 495–502. [Google Scholar] [CrossRef]
- Zhong, Q.; Zhou, L.; Zhu, D. Identification of prognostic marker genes in head and neck squamous cell carcinoma: A study based on TCGA database and experimental validation. J. Oral Pathol. Med. 2021, 50, 891–901. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, M.; Lei, L.; Fei, L.; Zheng, Y.; Huang, W.; Li, Z.; Liu, C.; Xu, H. Overexpression of KRT17 promotes proliferation and invasion of non-small cell lung cancer and indicates poor prognosis. Cancer Manag. Res. 2019, 11, 7485–7497. [Google Scholar] [CrossRef]
- Liu, Z.; Yu, S.; Ye, S.; Shen, Z.; Gao, L.; Han, Z.; Zhang, P.; Luo, F.; Chen, S.; Kang, M. Keratin 17 activates AKT signalling and induces epithelial-mesenchymal transition in oesophageal squamous cell carcinoma. J. Proteom. 2020, 211, 103557. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, S.; Yones, S.A.; Garbulowski, M.; Sun, J.; Skaftason, A.; Mayrhofer, M.; Norgren, N.; Herlin, M.K.; Sundström, C.; Eriksson, A.; et al. Transcriptomic analysis reveals proinflammatory signatures associated with acute myeloid leukemia progression. Blood Adv. 2022, 11, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Li, Y.; Qin, S.; Jiao, Y.; Hua, F. Ubiquitin-like modifier-activating enzyme 7 as a marker for the diagnosis and prognosis of breast cancer. Oncol. Lett. 2020, 19, 2773–2784. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients (n = 410) | |||
|---|---|---|---|
| n (%) | n (%) | ||
| Gender | HPV | ||
| Male | 304 (74) | Positive | 79 (19.5) |
| Female | 106 (26) | Negative NA | 329 (80) 2 (0.5) |
| Age at diagnosis (Years) | Anatomic Subsite | ||
| <60 | 185 (45) | Oral Tongue Larynx Oral Cavity Floor of mouth Tonsil Base of tongue Buccal Mucosa Alveolar Ridge Hypopharynx Oropharynx Hard Palate Lip | 104 (25) |
| ≥60 | 225 (55) | 88 (21) | |
| Tobacco | 54 (13) | ||
| Yes | 306 (75) | 45 (11) | |
| No NA | 96 (23) 8 (2) | 37 (9) 23 (6) | |
| Alcohol | 19 (5) | ||
| Yes | 280(68) | 15 (4) | |
| No | 120 (29) | 8 (2) | |
| NA | 10 (2) | 8 (2) | |
| TNM stage | 7 (2) | ||
| I | 19 (5) | 2 (0.5) | |
| II | 72 (18) | ||
| III | 82 (20) | ||
| IV | 226 (55) | ||
| NA | 11 (3) | ||
| Metastasis | |||
| Yes | 102 (25) | ||
| No | 308 (75) | ||
| Mean | Median | |||||||
|---|---|---|---|---|---|---|---|---|
| 95% Confidence Interval | 95% Confidence Interval | |||||||
| Cluster | Estimate | Std. Error | Lower Bound | Upper Bound | Estimate | Std. Error | Lower Bound | Upper Bound |
| 1 | 2879.300 | 276.379 | 2337.598 | 3421.003 | 2900.000 | 702.434 | 1523.229 | 4276.771 |
| 2 | 2085.443 | 252.901 | 1589.757 | 2581.128 | 2064.000 | 652.289 | 785.513 | 3342.487 |
| Overall | 2633.589 | 214.999 | 2212.191 | 3054.987 | 2319.000 | 378.696 | 1576.757 | 3061.243 |
| Minimum | 1st Quantile | Median | Mean | 3rd Quantile | Maximum | |
|---|---|---|---|---|---|---|
| Accuracy | 0.8654 | 0.9519 | 0.9615 | 0.9624 | 0.9808 | 1.0000 |
| Sensitivity | 0.7500 | 0.9038 | 0.9423 | 0.9338 | 0.9615 | 1.0000 |
| Specificity | 0.8846 | 0.9808 | 1.0000 | 0.9910 | 1.0000 | 1.0000 |
| Minimum | 1st Quantile | Median | Mean | 3rd Quantile | Maximum | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Model | SVM | RF | SVM | RF | SVM | RF | SVM | RF | SVM | RF | SVM | RF |
| Accuracy | 0.8654 | 0.8365 | 0.9519 | 0.9423 | 0.9615 | 0.9519 | 0.9624 | 0.9548 | 0.9808 | 0.9712 | 1.0000 | 1.0000 |
| Sensitivity | 0.7500 | 0.6923 | 0.9038 | 0.9231 | 0.9423 | 0.9423 | 0.9338 | 0.9373 | 0.9615 | 0.9615 | 1.0000 | 1.0000 |
| Specificity | 0.8846 | 0.7500 | 0.9808 | 0.9615 | 1.0000 | 0.9808 | 0.9910 | 0.9723 | 1.0000 | 0.9808 | 1.0000 | 1.0000 |
| 95% CI | |||
|---|---|---|---|
| Gene | AUC | Lower | Higher |
| GRM7 (CNA) | 0.959 | 0.935 | 0.983 |
| LMCD1-AS1 (CNA) | 0.956 | 0.931 | 0.981 |
| RPL29 (RNASeq) | 0.658 | 0.605 | 0.711 |
| UBA7 (RNAseq) | 0.726 | 0.677 | 0.774 |
| FCGR2C (RNASeq) | 0.721 | 0.672 | 0.770 |
| RPSAP58 (RNASeq) | 0.717 | 0.667 | 0.767 |
| CEACAM19 (Methylation) | 0.723 | 0.674 | 0.772 |
| KRT17 (Methylation) | 0.767 | 0.721 | 0.813 |
| ST18 (Methylation) | 0.703 | 0.651 | 0.755 |
| Gene | GRM7 | LMCD1-AS1 | RPL29 | RPSAP58 | FCGR2C | UBA7 | CEACAM19 | KR17 | ST18 |
| Cut-off point | −0.150 | −0.150 | 6885.475 | 7082.378 | 33.152 | 618.547 | 0.625 | 0.344 | 0.573 |
| B | S.E. | p-Value | ORadj | 95% CI ORadj | ||
|---|---|---|---|---|---|---|
| Lower | Upper | |||||
| RPL29 | 2.209 | 0.668 | 0.001 | 9.105 | 2.456 | 33.749 |
| FCGR2C | 1.729 | 0.596 | 0.004 | 5.635 | 1.752 | 18.120 |
| LMCD1-AS1 | 7.528 | 0.896 | <0.001 | 1859.334 | 321.161 | 10,764.458 |
| CEACAM19 | 2.668 | 0.812 | 0.001 | 14.405 | 2.935 | 70.707 |
| Constant | −7.116 | 1.138 | <0.001 | 0.001 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro, I.P.; Esteves, L.; Caramelo, F.; Carreira, I.M.; Melo, J.B. Integrated Multi-Omics Signature Predicts Survival in Head and Neck Cancer. Cells 2022, 11, 2536. https://doi.org/10.3390/cells11162536
Ribeiro IP, Esteves L, Caramelo F, Carreira IM, Melo JB. Integrated Multi-Omics Signature Predicts Survival in Head and Neck Cancer. Cells. 2022; 11(16):2536. https://doi.org/10.3390/cells11162536
Chicago/Turabian StyleRibeiro, Ilda Patrícia, Luísa Esteves, Francisco Caramelo, Isabel Marques Carreira, and Joana Barbosa Melo. 2022. "Integrated Multi-Omics Signature Predicts Survival in Head and Neck Cancer" Cells 11, no. 16: 2536. https://doi.org/10.3390/cells11162536
APA StyleRibeiro, I. P., Esteves, L., Caramelo, F., Carreira, I. M., & Melo, J. B. (2022). Integrated Multi-Omics Signature Predicts Survival in Head and Neck Cancer. Cells, 11(16), 2536. https://doi.org/10.3390/cells11162536