
Functional Intercellular Transmission of miHTT via Extracellular Vesicles: An In Vitro Proof-of-Mechanism Study


 , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
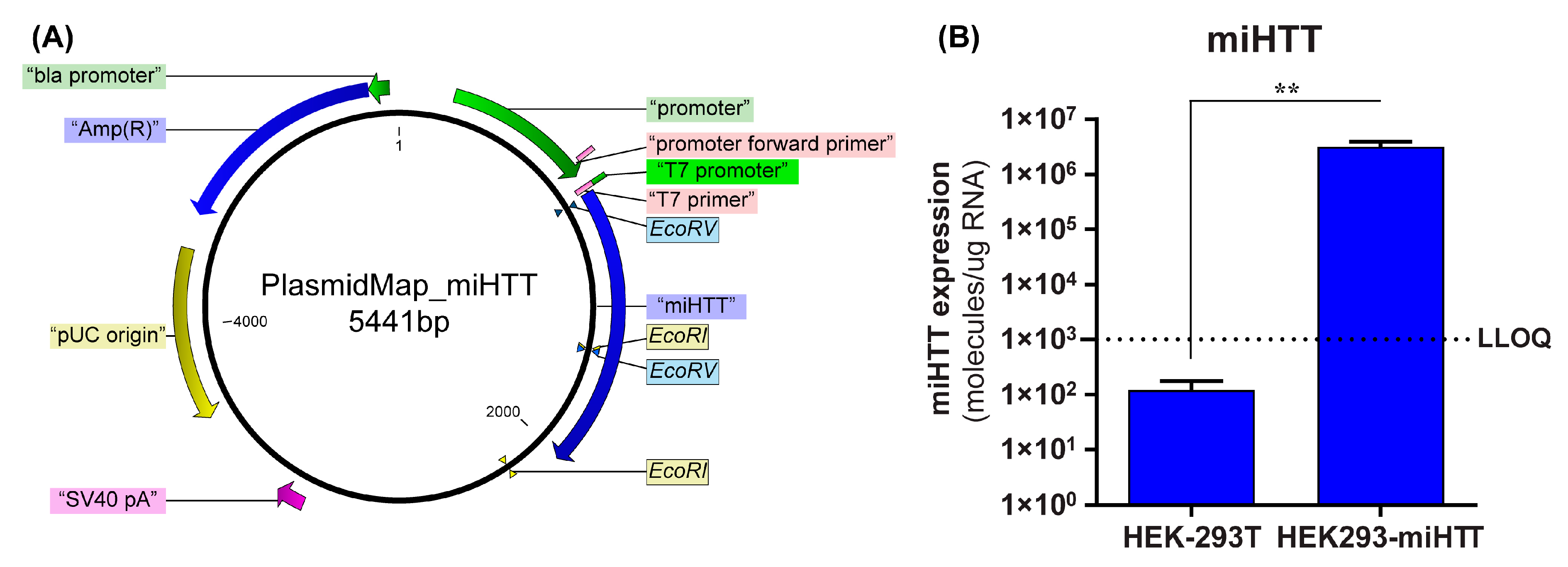
2.1. miHTT-Overexpressing Stable Cell Line Generation
2.2. Differentiation of Forebrain Neuronal Cultures from Human HD-Induced Pluripotent Stem Cells (iPSCs)
2.3. AAV5-miHTT Vector Production
2.4. Transduction of HD Patient iPSC-Derived Neurons with AA5-miHTT
2.5. EV Isolation from Culture Medium
2.5.1. EV Isolation by Precipitation
2.5.2. EV Isolation Using Size-Exclusion Chromatography (SEC)
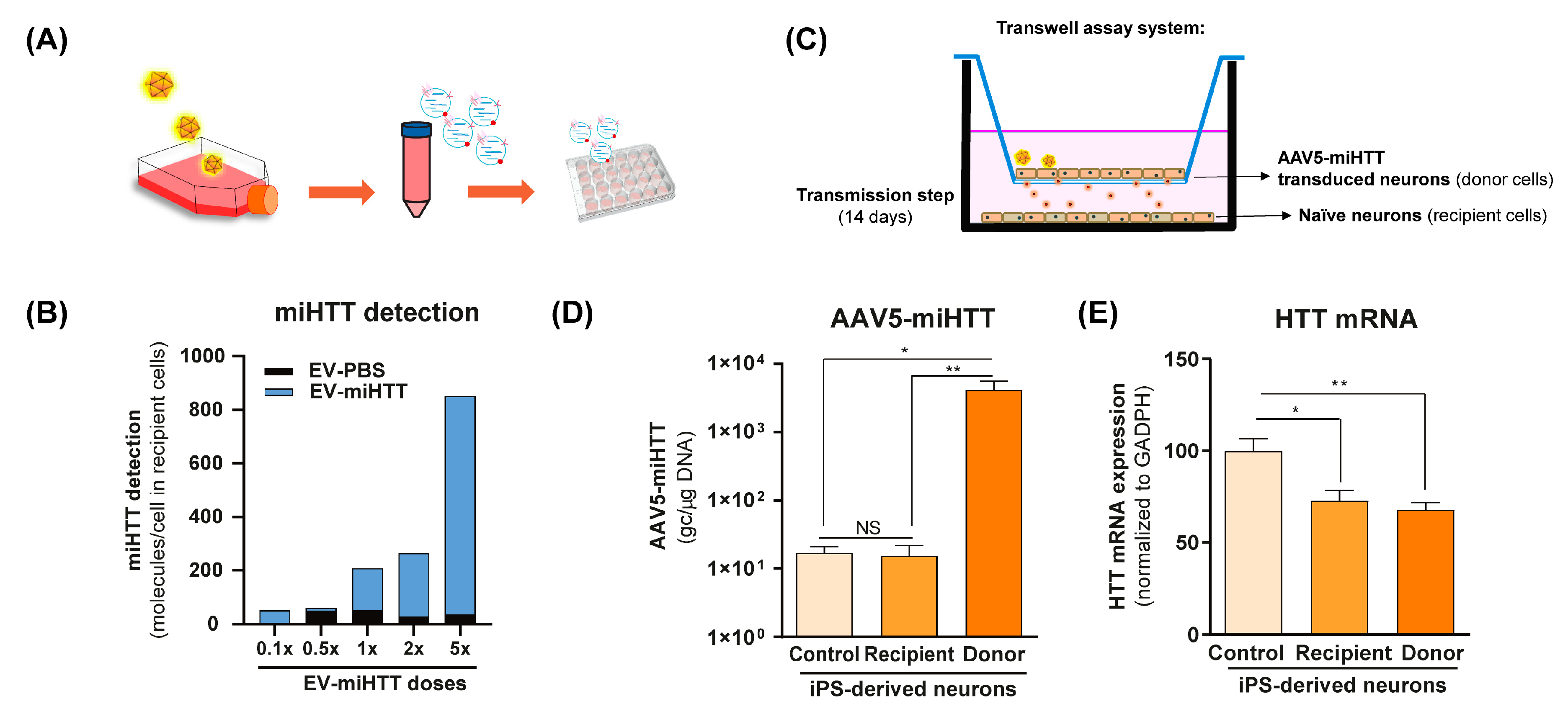
2.6. Functional miRNA Transfe—via EVs—to Naïve HD Patient iPSC-Derived Neurons
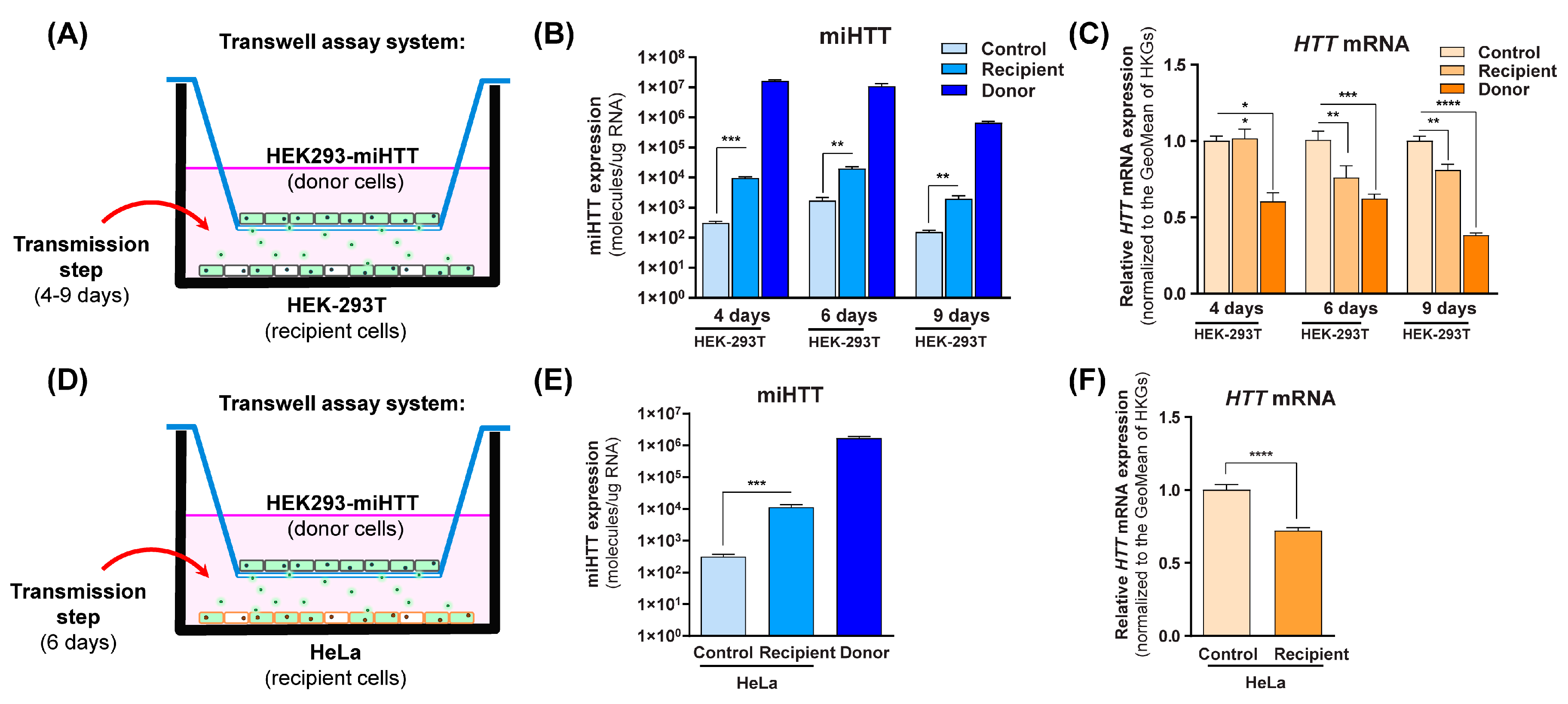
2.7. In Vitro Contactless Co-Culture Transwell Assay
2.7.1. HD Patient iPSC-Derived Neurons
2.7.2. HEK-293T and HeLa Cells
2.8. Vector DNA, miHTT, and HTT mRNA Measurement by RT-qPCR
2.9. Small RNA Sequencing
2.10. Fluorescent In Situ Hybridization (FISH) and Immunocytochemistry (ICC)
2.11. Imaging Acquisition and Quantification Analysis
2.12. Statistical Analysis
3. Results
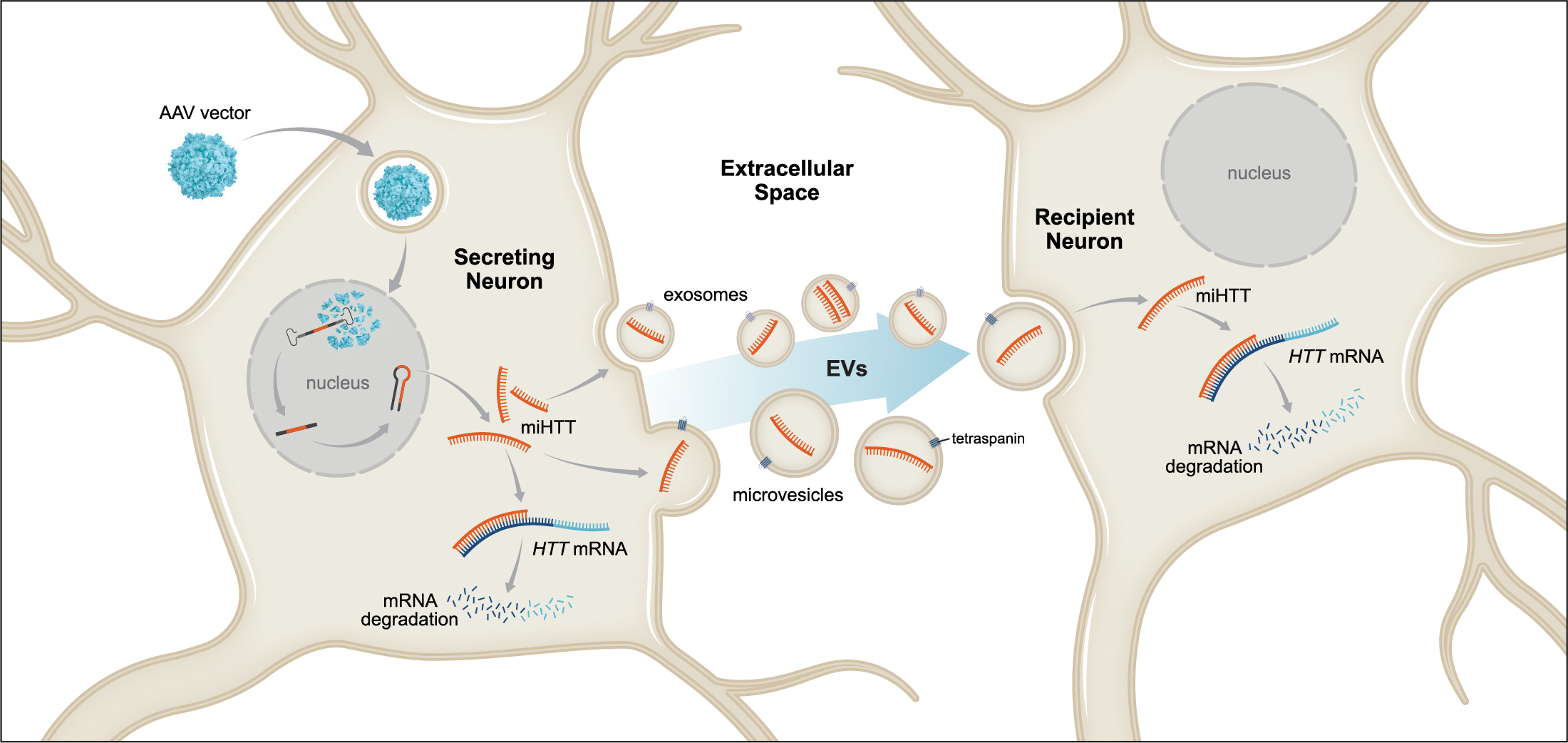
3.1. AAV-Produced miHTT Is Secreted within EVs and Transferred between Neuronal Cells
3.2. Stable Cell Line Successfully Overexpresses miHTT
3.3. miHTT Secreted by Donor Cells Effectively Mediates Endogenous HTT-mRNA Lowering in Recipient Cells
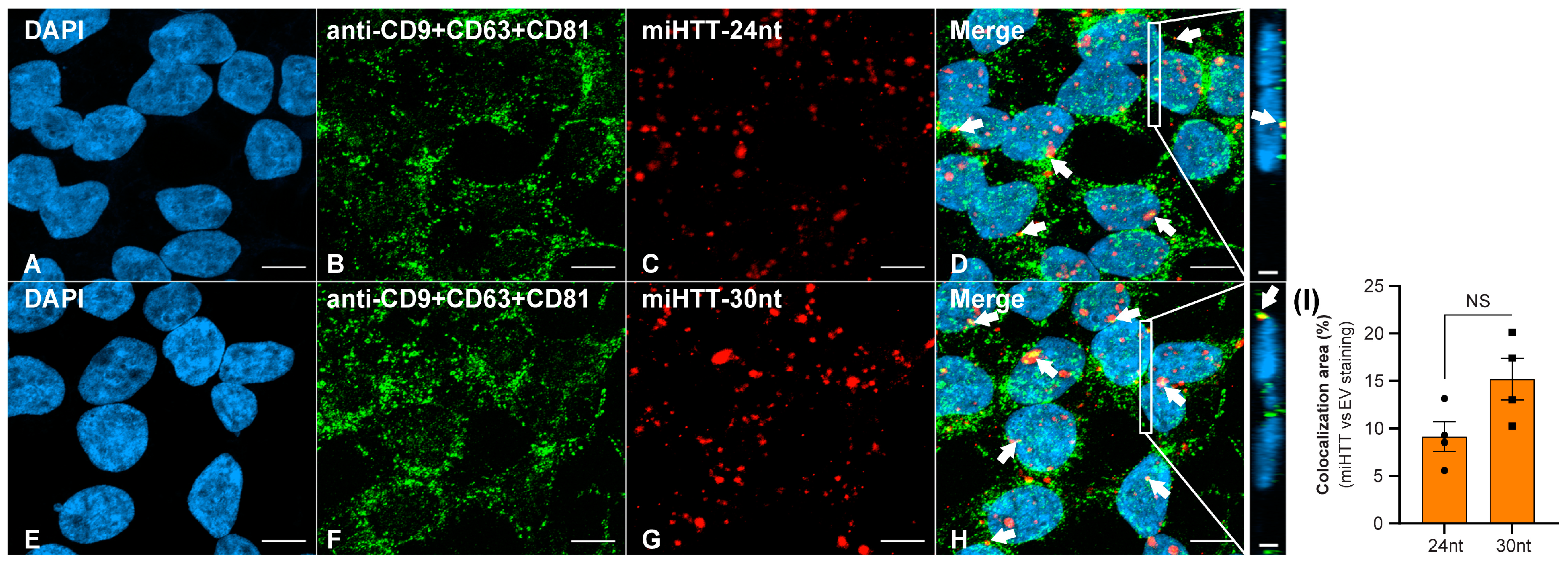
3.4. FISH and ICC Analyses Provide Evidence for miHTT Transport by Extracellular Vesicles
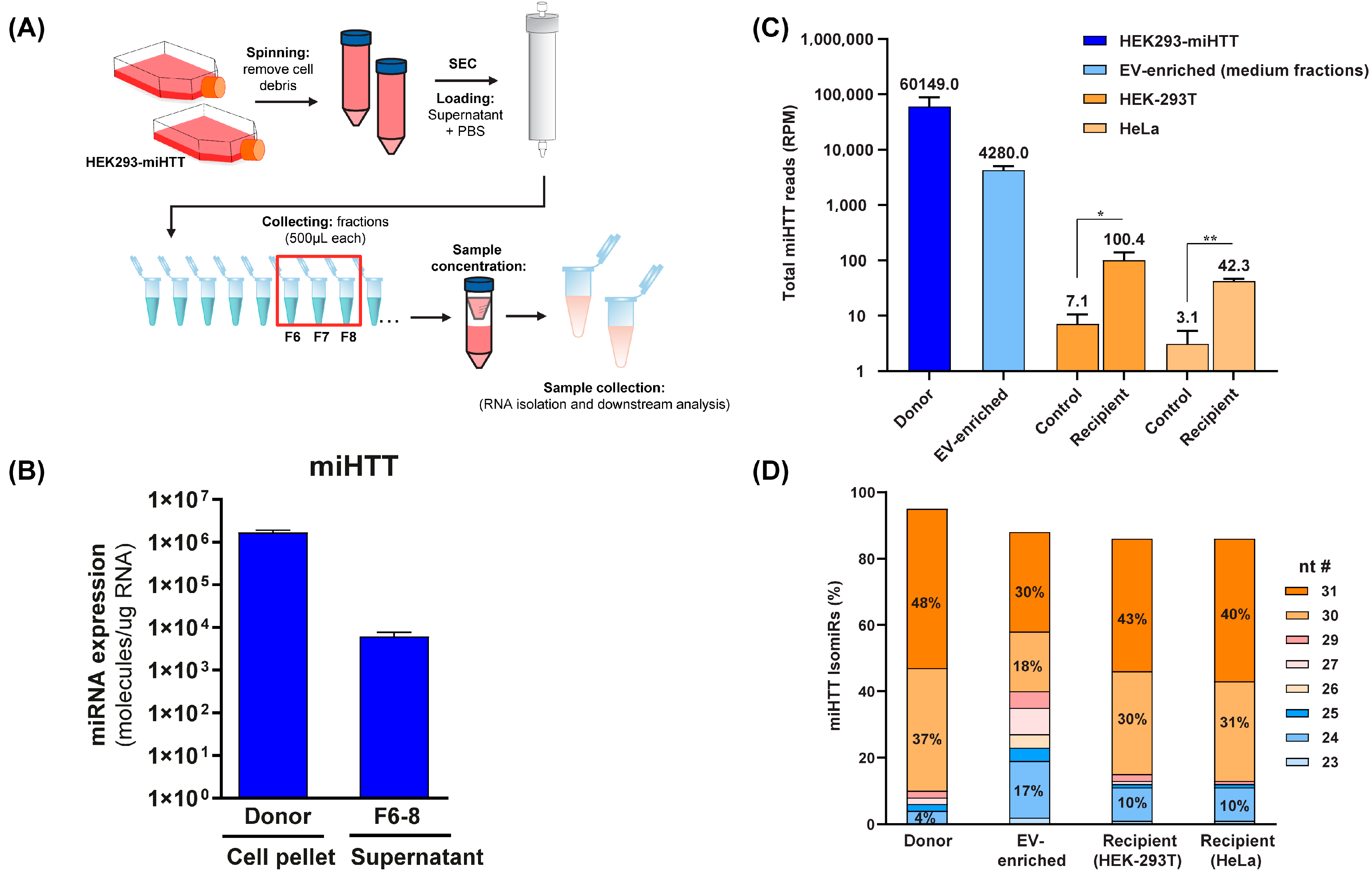
3.5. Small RNA Sequence Analysis Reveals Different miHTT isomiR Profiles in Donor and Recipient Cells and in EV-Enriched Medium Fractions
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Waldvogel, H.J.; Kim, E.H.; Tippett, L.J.; Vonsattel, J.-P.G.; Faull, R.L.M. The Neuropathology of Huntington’s Disease. Curr. Top. Behav. Neurosci. 2015, 22, 33–80. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.; Dragatsis, I.; Dietrich, P. Genetics and neuropathology of Huntington’s disease. Int. Rev. Neurobiol. 2011, 98, 325–372. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Flower, M.D.; Ross, C.A.; Wild, E.J. Huntington disease: New insights into molecular pathogenesis and therapeutic opportunities. Nat. Rev. Neurol. 2020, 16, 529–546. [Google Scholar] [CrossRef]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Caron, N.S.; Wright, G.E.; Hayden, M.R. Huntington Disease. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Mirzaa, G., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Coppen, E.M.; Roos, R.A.C. Current Pharmacological Approaches to Reduce Chorea in Huntington’s Disease. Drugs 2017, 77, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-J.; Shin, C.-W.; Jeon, B.; Park, H. Survival of Korean Huntington’s Disease Patients. J. Mov. Disord. 2016, 9, 166–170. [Google Scholar] [CrossRef]
- Foroud, T.; Gray, J.; Ivashina, J.; Conneally, P.M. Differences in duration of Huntington’s disease based on age at onset. J. Neurol. Neurosurg. Psychiatry 1999, 66, 52–56. [Google Scholar] [CrossRef]
- Rodrigues, F.B.; Abreu, D.; Damásio, J.; Goncalves, N.; Correia-Guedes, L.; Coelho, M.; Ferreira, J.J.; REGISTRY Investigators of the European Huntington’s Disease Network. Survival, Mortality, Causes and Places of Death in a European Huntington’s Disease Prospective Cohort. Mov. Disord. Clin. Pract. 2017, 4, 737–742. [Google Scholar] [CrossRef]
- Paulsen, J.S.; Langbehn, D.R.; Stout, J.C.; Aylward, E.; Ross, C.A.; Nance, M.; Guttman, M.; Johnson, S.; MacDonald, M.; Beglinger, L.J.; et al. Detection of Huntington’s disease decades before diagnosis: The Predict-HD study. J. Neurol. Neurosurg. Psychiatry 2008, 79, 874–880. [Google Scholar] [CrossRef]
- Niccolini, F.; Politis, M. Neuroimaging in Huntington’s disease. World J. Radiol. 2014, 6, 301–312. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Ghosh, R.; Leavitt, B.R. Huntingtin Lowering Strategies for Disease Modification in Huntington’s Disease. Neuron 2019, 101, 801–819. [Google Scholar] [CrossRef] [PubMed]
- Fields, E.; Vaughan, E.; Tripu, D.; Lim, I.; Shrout, K.; Conway, J.; Salib, N.; Lee, Y.; Dhamsania, A.; Jacobsen, M.; et al. Gene targeting techniques for Huntington’s disease. Ageing Res. Rev. 2021, 70, 101385. [Google Scholar] [CrossRef] [PubMed]
- Kordasiewicz, H.B.; Stanek, L.M.; Wancewicz, E.V.; Mazur, C.; McAlonis, M.M.; Pytel, K.A.; Artates, J.W.; Weiss, A.; Cheng, S.H.; Shihabuddin, L.S.; et al. Sustained Therapeutic Reversal of Huntington’s Disease by Transient Repression of Huntingtin Synthesis. Neuron 2012, 74, 1031–1044. [Google Scholar] [CrossRef]
- Boudreau, R.L.; McBride, J.L.; Martins, I.; Shen, S.; Xing, Y.; Carter, B.J.; Davidson, B.L. Nonallele-specific Silencing of Mutant and Wild-type Huntingtin Demonstrates Therapeutic Efficacy in Huntington’s Disease Mice. Mol. Ther. 2009, 17, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Stanek, L.M.; Sardi, S.P.; Mastis, B.; Richards, A.R.; Treleaven, C.M.; Taksir, T.; Misra, K.; Cheng, S.H.; Shihabuddin, L.S. Silencing Mutant Huntingtin by Adeno-Associated Virus-Mediated RNA Interference Ameliorates Disease Manifestations in the YAC128 Mouse Model of Huntington’s Disease. Hum. Gene Ther. 2014, 25, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Spronck, E.A.; Brouwers, C.C.; Vallès, A.; de Haan, M.; Petry, H.; van Deventer, S.J.; Konstantinova, P.; Evers, M.M. AAV5-miHTT Gene Therapy Demonstrates Sustained Huntingtin Lowering and Functional Improvement in Huntington Disease Mouse Models. Mol. Ther. Methods Clin. Dev. 2019, 13, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Jafar-Nejad, P.; Powers, B.; Soriano, A.; Zhao, H.; Norris, D.A.; Matson, J.; DeBrosse-Serra, B.; Watson, J.; Narayanan, P.; Chun, S.J.; et al. The atlas of RNase H antisense oligonucleotide distribution and activity in the CNS of rodents and non-human primates following central administration. Nucleic Acids Res. 2021, 49, 657–673. [Google Scholar] [CrossRef] [PubMed]
- Miniarikova, J.; Zimmer, V.; Martier, R.; Brouwers, C.C.; Pythoud, C.; Richetin, K.; Rey, M.; Lubelski, J.; Evers, M.M.; van Deventer, S.J.; et al. AAV5-miHTT gene therapy demonstrates suppression of mutant huntingtin aggregation and neuronal dysfunction in a rat model of Huntington’s disease. Gene Ther. 2017, 24, 630–639. [Google Scholar] [CrossRef]
- Vallès, A.; Evers, M.M.; Stam, A.; Sogorb-Gonzalez, M.; Brouwers, C.; Vendrell-Tornero, C.; Acar-Broekmans, S.; Paerels, L.; Klima, J.; Bohuslavova, B.; et al. Widespread and sustained target engagement in Huntington’s disease minipigs upon intrastriatal microRNA-based gene therapy. Sci. Transl. Med. 2021, 13, eabb8920. [Google Scholar] [CrossRef]
- Miniarikova, J.; Evers, M.M.; Konstantinova, P. Translation of MicroRNA-Based Huntingtin-Lowering Therapies from Preclinical Studies to the Clinic. Mol. Ther. 2018, 26, 947–962. [Google Scholar] [CrossRef] [Green Version]
- Nakai, H.; Yant, S.R.; Storm, T.A.; Fuess, S.; Meuse, L.; Kay, M.A. Extrachromosomal recombinant adeno-associated virus vector genomes are primarily responsible for stable liver transduction in vivo. J. Virol. 2001, 75, 6969–6976. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.H. Adeno-associated virus integration: Virus versus vector. Gene Ther. 2008, 15, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Miniarikova, J.; Zanella, I.; Huseinovic, A.; van der Zon, T.; Hanemaaijer, E.; Martier, R.; Koornneef, A.; Southwell, A.L.; Hayden, M.R.; van Deventer, S.J.; et al. Design, Characterization, and Lead Selection of Therapeutic miRNAs Targeting Huntingtin for Development of Gene Therapy for Huntington’s Disease. Mol. Ther. Nucleic Acids 2016, 5, e297. [Google Scholar] [CrossRef] [PubMed]
- Treiber, T.; Treiber, N.; Meister, G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat. Rev. Mol. Cell Biol. 2019, 20, 5–20. [Google Scholar] [CrossRef]
- Yang, J.-S.; Maurin, T.; Robine, N.; Rasmussen, K.D.; Jeffrey, K.L.; Chandwani, R.; Papapetrou, E.P.; Sadelain, M.; O’Carroll, D.; Lai, E.C. Conserved vertebrate mir-451 provides a platform for Dicer-independent, Ago2-mediated microRNA biogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 15163–15168. [Google Scholar] [CrossRef]
- Cifuentes, D.; Xue, H.; Taylor, D.W.; Patnode, H.; Mishima, Y.; Cheloufi, S.; Ma, E.; Mane, S.; Hannon, G.J.; Lawson, N.D.; et al. A novel miRNA processing pathway independent of Dicer requires Argonaute2 catalytic activity. Science 2010, 328, 1694–1698. [Google Scholar] [CrossRef]
- Cheloufi, S.; Dos Santos, C.O.; Chong, M.M.W.; Hannon, G.J. A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature 2010, 465, 584–589. [Google Scholar] [CrossRef]
- Keskin, S.; Brouwers, C.C.; Sogorb-Gonzalez, M.; Martier, R.; Depla, J.A.; Vallès, A.; van Deventer, S.J.; Konstantinova, P.; Evers, M.M. AAV5-miHTT Lowers Huntingtin mRNA and Protein without Off-Target Effects in Patient-Derived Neuronal Cultures and Astrocytes. Mol. Ther. Methods Clin. Dev. 2019, 15, 275–284. [Google Scholar] [CrossRef]
- Guduric-Fuchs, J.; O’Connor, A.; Camp, B.; O’Neill, C.L.; Medina, R.J.; Simpson, D.A. Selective extracellular vesicle-mediated export of an overlapping set of microRNAs from multiple cell types. BMC Genom. 2012, 13, 357. [Google Scholar] [CrossRef]
- Maas, S.L.N.; Breakefield, X.O.; Weaver, A.M. Extracellular vesicles: Unique intercellular delivery vehicles. Trends Cell Biol. 2017, 27, 172–188. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, A.; Kim, H.S.; Bojmar, L.; Gyan, K.E.; Cioffi, M.; Hernandez, J.; Zambirinis, C.P.; Rodrigues, G.; Molina, H.; Heissel, S.; et al. Extracellular Vesicle and Particle Biomarkers Define Multiple Human Cancers. Cell 2020, 182, 1044–1061.e18. [Google Scholar] [CrossRef] [PubMed]
- Andreu, Z.; Yáñez-Mó, M. Tetraspanins in Extracellular Vesicle Formation and Function. Front. Immunol. 2014, 5, 442. [Google Scholar] [CrossRef] [PubMed]
- György, B.; Hung, M.E.; Breakefield, X.O.; Leonard, J.N. Therapeutic applications of extracellular vesicles: Clinical promise and open questions. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 439–464. [Google Scholar] [CrossRef]
- Wiklander, O.P.B.; Brennan, M.Á.; Lötvall, J.; Breakefield, X.O.; El Andaloussi, S. Advances in therapeutic applications of extracellular vesicles. Sci. Transl. Med. 2019, 11, eaav8521. [Google Scholar] [CrossRef]
- Huang, X.; Yuan, T.; Tschannen, M.; Sun, Z.; Jacob, H.; Du, M.; Liang, M.; Dittmar, R.L.; Liu, Y.; Liang, M.; et al. Characterization of human plasma-derived exosomal RNAs by deep sequencing. BMC Genom. 2013, 14, 319. [Google Scholar] [CrossRef] [PubMed]
- Villarroya-Beltri, C.; Gutiérrez-Vázquez, C.; Sánchez-Cabo, F.; Pérez-Hernández, D.; Vázquez, J.; Martin-Cofreces, N.; Martinez-Herrera, D.J.; Pascual-Montano, A.; Mittelbrunn, M.; Sánchez-Madrid, F. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat. Commun. 2013, 4, 2980. [Google Scholar] [CrossRef]
- Koppers-Lalic, D.; Hackenberg, M.; Bijnsdorp, I.V.; van Eijndhoven, M.A.J.; Sadek, P.; Sie, D.; Zini, N.; Middeldorp, J.M.; Ylstra, B.; de Menezes, R.X.; et al. Nontemplated nucleotide additions distinguish the small RNA composition in cells from exosomes. Cell Rep. 2014, 8, 1649–1658. [Google Scholar] [CrossRef]
- Caron, N.S.; Southwell, A.L.; Brouwers, C.C.; Cengio, L.D.; Xie, Y.; Black, H.F.; Anderson, L.M.; Ko, S.; Zhu, X.; van Deventer, S.J.; et al. Potent and sustained huntingtin lowering via AAV5 encoding miRNA preserves striatal volume and cognitive function in a humanized mouse model of Huntington disease. Nucleic Acids Res. 2020, 48, 36–54. [Google Scholar] [CrossRef]
- Evers, M.M.; Miniarikova, J.; Juhas, S.; Vallès, A.; Bohuslavova, B.; Juhasova, J.; Skalnikova, H.K.; Vodicka, P.; Valekova, I.; Brouwers, C.; et al. AAV5-miHTT Gene Therapy Demonstrates Broad Distribution and Strong Human Mutant Huntingtin Lowering in a Huntington’s Disease Minipig Model. Mol. Ther. 2018, 26, 2163–2177. [Google Scholar] [CrossRef]
- Spronck, E.A.; Vallès, A.; Lampen, M.H.; Montenegro-Miranda, P.S.; Keskin, S.; Heijink, L.; Evers, M.M.; Petry, H.; van Deventer, S.J.; Konstantinova, P.; et al. Intrastriatal Administration of AAV5-miHTT in Non-Human Primates and Rats Is Well Tolerated and Results in miHTT Transgene Expression in Key Areas of Huntington Disease Pathology. Brain Sci. 2021, 11, 129. [Google Scholar] [CrossRef]
- Emborg, M.E.; Hurley, S.A.; Joers, V.; Tromp, D.P.M.; Swanson, C.R.; Ohshima-Hosoyama, S.; Bondarenko, V.; Cummisford, K.; Sonnemans, M.; Hermening, S.; et al. Titer and product affect the distribution of gene expression after intraputaminal convection-enhanced delivery. Stereotact. Funct. Neurosurg. 2014, 92, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Markakis, E.A.; Vives, K.P.; Bober, J.; Leichtle, S.; Leranth, C.; Beecham, J.; Elsworth, J.D.; Roth, R.H.; Samulski, R.J.; Redmond, D.E. Comparative transduction efficiency of AAV vector serotypes 1-6 in the substantia nigra and striatum of the primate brain. Mol. Ther. 2010, 18, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Samaranch, L.; Blits, B.; San Sebastian, W.; Hadaczek, P.; Bringas, J.; Sudhakar, V.; Macayan, M.; Pivirotto, P.J.; Petry, H.; Bankiewicz, K.S. MR-guided parenchymal delivery of adeno-associated viral vector serotype 5 in non-human primate brain. Gene Ther. 2017, 24, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Aschauer, D.F.; Kreuz, S.; Rumpel, S. Analysis of transduction efficiency, tropism and axonal transport of AAV serotypes 1, 2, 5, 6, 8 and 9 in the mouse brain. PLoS ONE 2013, 8, e76310. [Google Scholar] [CrossRef] [PubMed]
- Haery, L.; Deverman, B.E.; Matho, K.S.; Cetin, A.; Woodard, K.; Cepko, C.; Guerin, K.I.; Rego, M.A.; Ersing, I.; Bachle, S.M.; et al. Adeno-Associated Virus Technologies and Methods for Targeted Neuronal Manipulation. Front. Neuroanat. 2019, 13, 93. [Google Scholar] [CrossRef] [PubMed]
- Sogorb-Gonzalez, M.; Vendrell-Tornero, C.; Snapper, J.; Stam, A.; Keskin, S.; Miniarikova, J.; Spronck, E.A.; de Haan, M.; Nieuwland, R.; Konstantinova, P.; et al. Secreted therapeutics: Monitoring durability of microRNA-based gene therapies in the central nervous system. Brain Commun. 2021, 3, fcab054. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Pfister, E.L.; DiNardo, N.; Mondo, E.; Borel, F.; Conroy, F.; Fraser, C.; Gernoux, G.; Han, X.; Hu, D.; Johnson, E.; et al. Artificial miRNAs Reduce Human Mutant Huntingtin Throughout the Striatum in a Transgenic Sheep Model of Huntington’s Disease. Hum. Gene Ther. 2018, 29, 663–673. [Google Scholar] [CrossRef]
- McBride, J.L.; Pitzer, M.R.; Boudreau, R.L.; Dufour, B.; Hobbs, T.; Ojeda, S.R.; Davidson, B.L. Preclinical safety of RNAi-mediated HTT suppression in the rhesus macaque as a potential therapy for Huntington’s disease. Mol. Ther. 2011, 19, 2152–2162. [Google Scholar] [CrossRef]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [Green Version]
- Weber, J.A.; Baxter, D.H.; Zhang, S.; Huang, D.Y.; How Huang, K.; Jen Lee, M.; Galas, D.J.; Wang, K. The MicroRNA Spectrum in 12 Body Fluids. Clin. Chem. 2010, 56, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.A.; Ludwig, R.G.; Garcia-Martin, R.; Brandão, B.B.; Kahn, C.R. Extracellular miRNAs: From Biomarkers to Mediators of Physiology and Disease. Cell Metab. 2019, 30, 656–673. [Google Scholar] [CrossRef] [PubMed]
- Didiot, M.-C.; Hall, L.M.; Coles, A.H.; Haraszti, R.A.; Godinho, B.M.; Chase, K.; Sapp, E.; Ly, S.; Alterman, J.F.; Hassler, M.R.; et al. Exosome-mediated Delivery of Hydrophobically Modified siRNA for Huntingtin mRNA Silencing. Mol. Ther. 2016, 24, 1836–1847. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Dorval, T.; Chaput, N.; André, F.; Caby, M.-P.; Novault, S.; Flament, C.; Leboulaire, C.; Borg, C.; Amigorena, S.; et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: Results of thefirst phase I clinical trial. J. Transl. Med. 2005, 3, 10. [Google Scholar] [CrossRef]
- Morse, M.A.; Garst, J.; Osada, T.; Khan, S.; Hobeika, A.; Clay, T.M.; Valente, N.; Shreeniwas, R.; Sutton, M.A.; Delcayre, A.; et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J. Transl. Med. 2005, 3, 9. [Google Scholar] [CrossRef]
- Besse, B.; Charrier, M.; Lapierre, V.; Dansin, E.; Lantz, O.; Planchard, D.; Le Chevalier, T.; Livartoski, A.; Barlesi, F.; Laplanche, A.; et al. Dendritic cell-derived exosomes as maintenance immunotherapy after first line chemotherapy in NSCLC. Oncoimmunology 2016, 5, e1071008. [Google Scholar] [CrossRef]
- Dai, S.; Wei, D.; Wu, Z.; Zhou, X.; Wei, X.; Huang, H.; Li, G. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol. Ther. 2008, 16, 782–790. [Google Scholar] [CrossRef]
- Nassar, W.; El-Ansary, M.; Sabry, D.; Mostafa, M.A.; Fayad, T.; Kotb, E.; Temraz, M.; Saad, A.-N.; Essa, W.; Adel, H. Umbilical cord mesenchymal stem cells derived extracellular vesicles can safely ameliorate the progression of chronic kidney diseases. Biomater. Res. 2016, 20, 21. [Google Scholar] [CrossRef]
- Okamoto, M.; Fukushima, Y.; Kouwaki, T.; Daito, T.; Kohara, M.; Kida, H.; Oshiumi, H. MicroRNA-451a in extracellular, blood-resident vesicles attenuates macrophage and dendritic cell responses to influenza whole-virus vaccine. J. Biol. Chem. 2018, 293, 18585–18600. [Google Scholar] [CrossRef]
- Reshke, R.; Taylor, J.A.; Savard, A.; Guo, H.; Rhym, L.H.; Kowalski, P.S.; Trung, M.T.; Campbell, C.; Little, W.; Anderson, D.G.; et al. Reduction of the therapeutic dose of silencing RNA by packaging it in extracellular vesicles via a pre-microRNA backbone. Nat. Biomed. Eng. 2020, 4, 52–68. [Google Scholar] [CrossRef]
- Søkilde, R.; Newie, I.; Persson, H.; Borg, Å.; Rovira, C. Passenger strand loading in overexpression experiments using microRNA mimics. RNA Biol. 2015, 12, 787–791. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.A.; Betel, D.; Miller, M.L.; Sander, C.; Leslie, C.S.; Marks, D.S. Transfection of small RNAs globally perturbs gene regulation by endogenous microRNAs. Nat. Biotechnol. 2009, 27, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Carrillo, E.; Berkhout, B. Dicer-independent processing of small RNA duplexes: Mechanistic insights and applications. Nucleic Acids Res. 2017, 45, 10369–10379. [Google Scholar] [CrossRef] [PubMed]
- Krützfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with “antagomirs”. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Fuchs Wightman, F.; Giono, L.E.; Fededa, J.P.; de la Mata, M. Target RNAs Strike Back on MicroRNAs. Front. Genet. 2018, 9, 435. [Google Scholar] [CrossRef]
- Yang, J.-S.; Lai, E.C. Alternative miRNA Biogenesis Pathways and the Interpretation of Core miRNA Pathway Mutants. Mol. Cell 2011, 43, 892–903. [Google Scholar] [CrossRef]
- Ameres, S.L.; Horwich, M.D.; Hung, J.-H.; Xu, J.; Ghildiyal, M.; Weng, Z.; Zamore, P.D. Target RNA-directed trimming and tailing of small silencing RNAs. Science 2010, 328, 1534–1539. [Google Scholar] [CrossRef]
- Glogovitis, I.; Yahubyan, G.; Würdinger, T.; Koppers-Lalic, D.; Baev, V. isomiRs-Hidden Soldiers in the miRNA Regulatory Army, and How to Find Them? Biomolecules 2020, 11, 41. [Google Scholar] [CrossRef]
- Karlsen, T.A.; Aae, T.F.; Brinchmann, J.E. Robust Profiling of MicroRNAs and IsomiRs in Human Plasma Exosomes across 46 Individuals. Sci. Rep. 2019, 9, 19999. [Google Scholar] [CrossRef]
- Cloonan, N.; Wani, S.; Xu, Q.; Gu, J.; Lea, K.; Heater, S.; Barbacioru, C.; Steptoe, A.L.; Martin, H.C.; Nourbakhsh, E.; et al. MicroRNAs and Their IsomiRs Function Cooperatively to Target Common Biological Pathways. Genome Biol. 2011, 12, R126. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibodies | Description | Reference Number | Dilution | Secondary Antibodies | Reference Number | Dilution |
|---|---|---|---|---|---|---|
| Anti-CD9 | Rabbit monoclonal | ab92726 | 1:500 | Alexa Fluor® 647 | A-31573 | 1:750 |
| Anti-CD63 | Rabbit monoclonal | ab252919 | 1:500 | Alexa Fluor® 647 | A-31573 | 1:750 |
| Anti-CD81 | Mouse monoclonal | ab70559 | 1:500 | Alexa Fluor® 488 | R37114 | 1:750 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morais, R.D.V.S.; Sogorb-González, M.; Bar, C.; Timmer, N.C.; Van der Bent, M.L.; Wartel, M.; Vallès, A. Functional Intercellular Transmission of miHTT via Extracellular Vesicles: An In Vitro Proof-of-Mechanism Study. Cells 2022, 11, 2748. https://doi.org/10.3390/cells11172748
Morais RDVS, Sogorb-González M, Bar C, Timmer NC, Van der Bent ML, Wartel M, Vallès A. Functional Intercellular Transmission of miHTT via Extracellular Vesicles: An In Vitro Proof-of-Mechanism Study. Cells. 2022; 11(17):2748. https://doi.org/10.3390/cells11172748
Chicago/Turabian StyleMorais, Roberto D. V. S., Marina Sogorb-González, Citlali Bar, Nikki C. Timmer, M. Leontien Van der Bent, Morgane Wartel, and Astrid Vallès. 2022. "Functional Intercellular Transmission of miHTT via Extracellular Vesicles: An In Vitro Proof-of-Mechanism Study" Cells 11, no. 17: 2748. https://doi.org/10.3390/cells11172748
APA StyleMorais, R. D. V. S., Sogorb-González, M., Bar, C., Timmer, N. C., Van der Bent, M. L., Wartel, M., & Vallès, A. (2022). Functional Intercellular Transmission of miHTT via Extracellular Vesicles: An In Vitro Proof-of-Mechanism Study. Cells, 11(17), 2748. https://doi.org/10.3390/cells11172748