
SARS-CoV-2 Infection of Airway Epithelium Triggers Pulmonary Endothelial Cell Activation and Senescence Associated with Type I IFN Production

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Airway Epithelium (HAE) Culture
2.2. Human Pulmonary Microvascular Endothelial Cells (ECs) Culture
2.3. SARS-CoV-2 Infection and HAE-EC Co-Culture
2.4. H-151 Treatment
2.5. Real-Time RT-PCR
2.6. Immunofluorescence
2.7. Cytokines Quantification
2.8. Viral Quantification
2.9. Statistical Analysis
3. Results
3.1. Co-Culture of SARS-COV-2 Infected Airway Epithelium with Microvascular Pulmonary Endothelial Cells: The Experimental Model
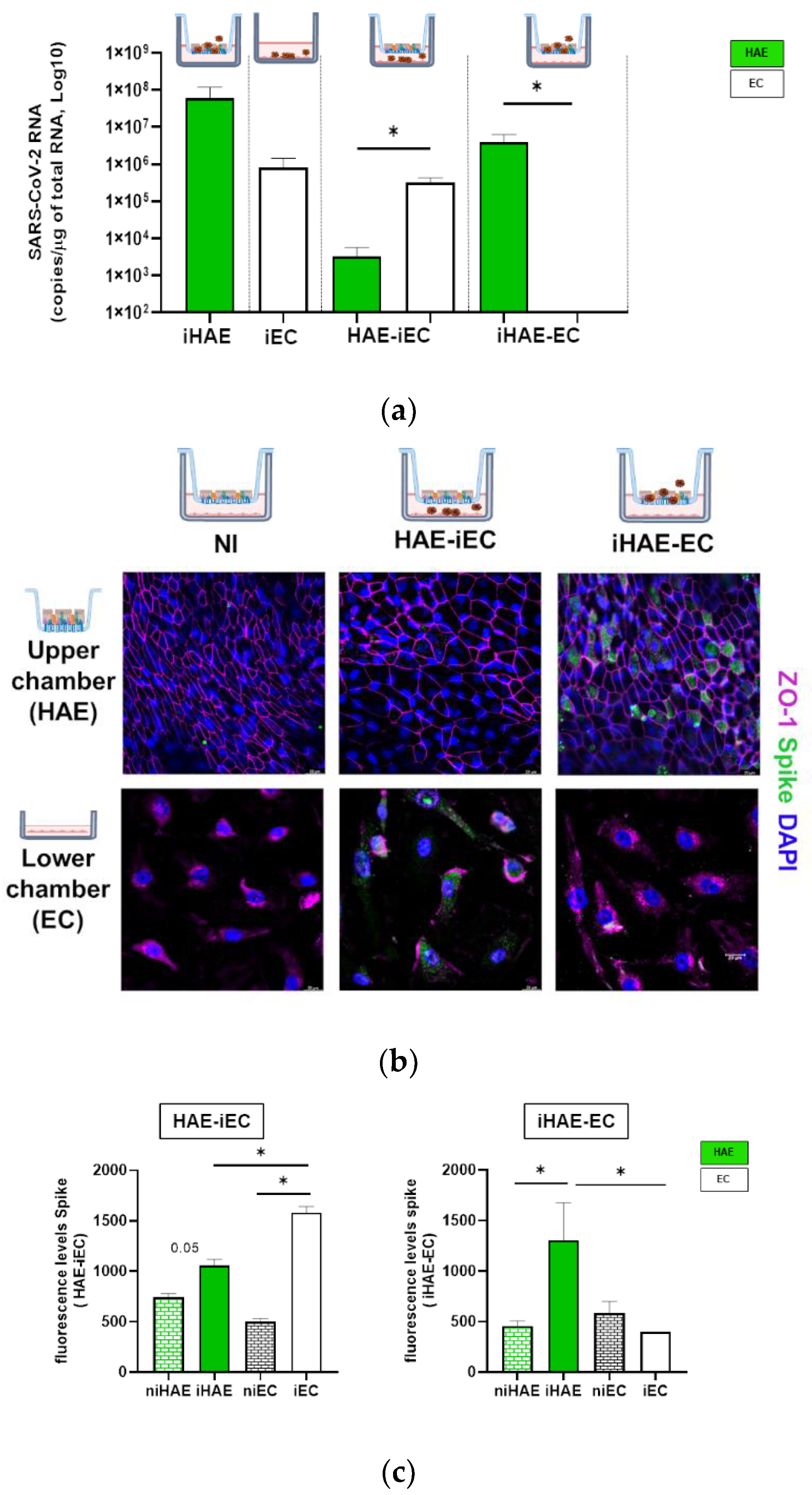
3.2. SARS-CoV-2 Infection of Human Airway Epithelial and Endothelial Cells
3.3. Viral Replication in HAE Induces IFN-β Release and Modulation of p21 Expression in EC
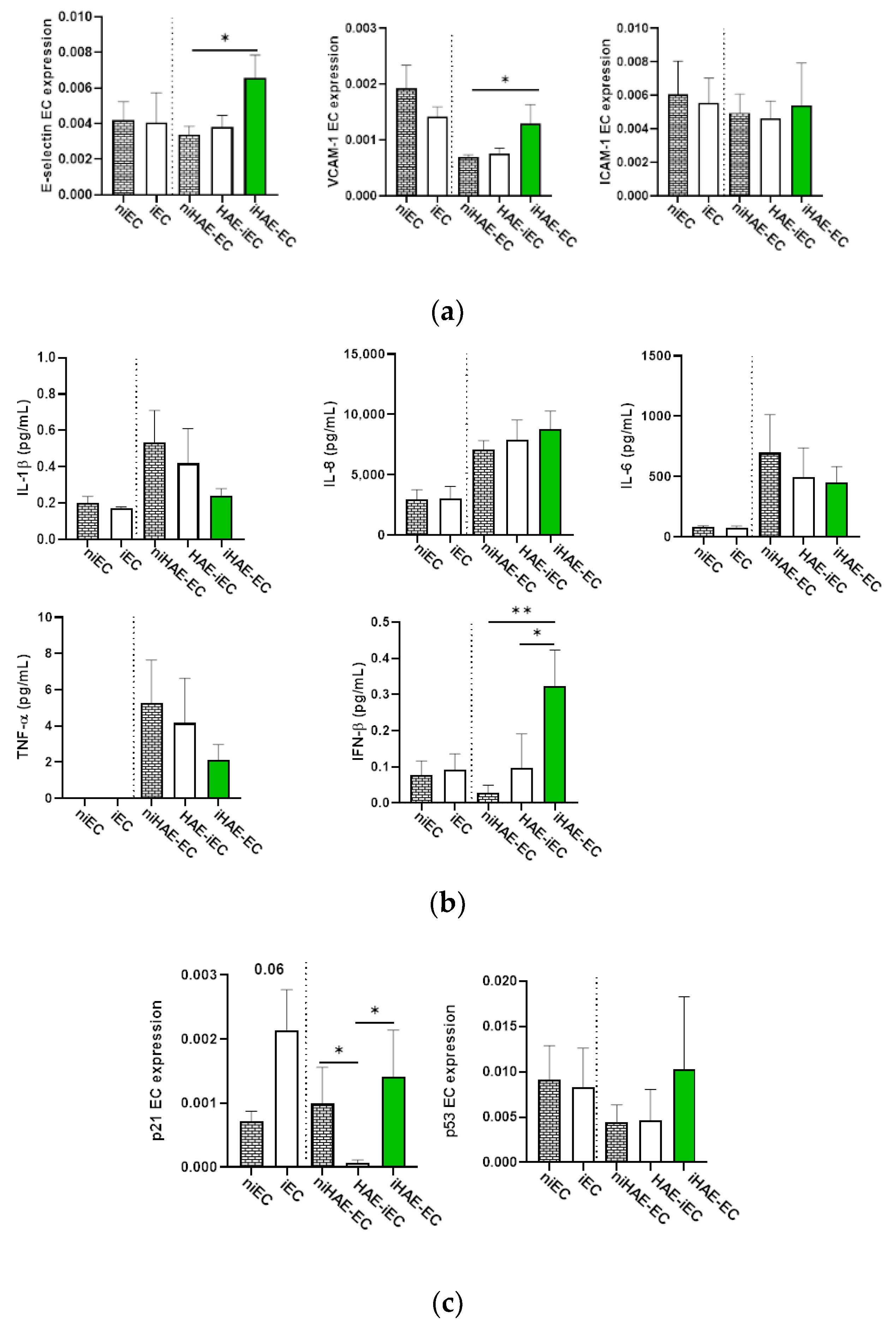
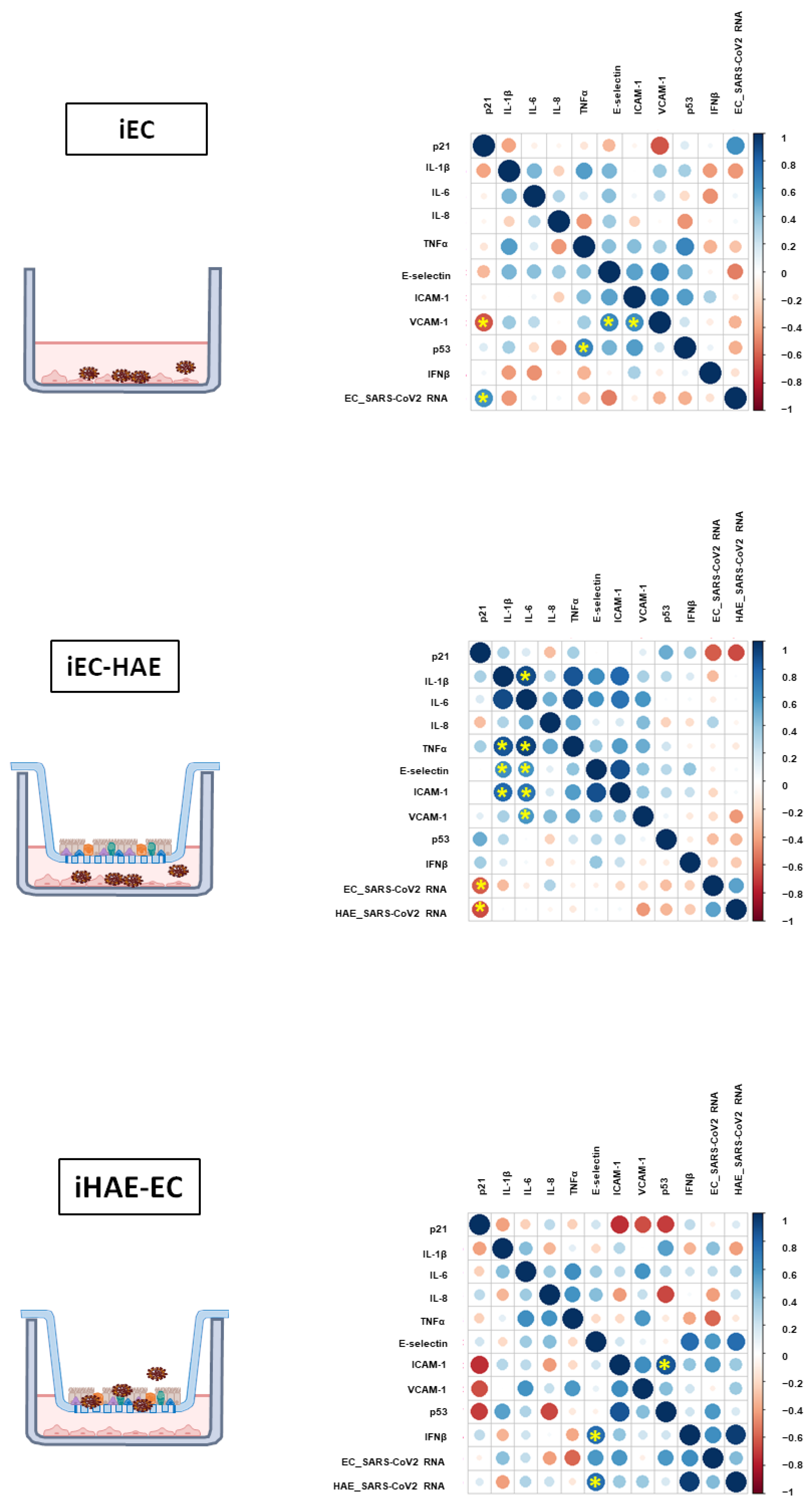
3.4. Viral Replication in HAE Correlates with EC Activation, IFN-β Release, and p21 Expression
3.5. Impact of IFN-β on EC Senescence
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Teuwen, L.A.; Geldhof, V.; Pasut, A.; Carmeliet, P. COVID-19: The vasculature unleashed. Nat. Rev. Immunol. 2020, 20, 389–391. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.-J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.-S.; Mehra, M.-R.; Schuepbach, R.-A.; Ruschitzka, F.; Mochl, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Libby, P.; Luscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef] [PubMed]
- Colmenero, I.; Santonja, C.; Alonso-Riaño, M.; Noguera-Morel, L.; Hernández-Martín, A.; Andina, D.; Wiesner, T.; Rodríguez-Peralto, J.L.; Requena, L.; Torrelo, A. SARS-CoV-2 endothelial infection causes COVID-19 chilblains: Histopathological, immunohistochemical and ultrastructural study of seven paediatric cases. Br. J. Derm. 2020, 183, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Saleh, J.; Peyssonnaux, C.; Singh, K.K.; Edeas, M. Mitochondria and microbiota dysfunction in COVID-19 pathogenesis. Mitochondrion 2020, 54, 1–7. [Google Scholar] [CrossRef]
- Lei, Y.; Zhang, J.; Schiavon, C.R.; He, M.; Chen, L.; Shen, H.; Zhang, Y.; Yin, Q.; Cho, Y.; Andrade, L. SARS-CoV-2 spike protein impairs endothelial function via downregulation of ACE 2. Circ. Res. 2021 2021, 128, 1323–1326. [Google Scholar] [CrossRef]
- Queisser, K.A.; Mellema, R.A.; Middleton, E.A.; Portier, I.; Manne, B.K.; Denorme, F.; Beswick, E.J.; Rondina, M.T.; Campbell, R.A.; Petrey, A.C. COVID-19 generates hyaluronan fragments that directly induce endothelial barrier dysfunction. Pub. Med. 2021, 6, e147472. [Google Scholar] [CrossRef]
- Xu, S.; Liu, Y.; Ding, Y.; Luo, S.; Zheng, X.; Wu, X.; Liu, Z.; Ilyas, I.; Chen, S.; Han, S. The zinc finger transcription factor, KLF2, protects against COVID-19 associated endothelial dysfunction. Signal Transduct. Target. Ther. 2021, 6, 1–9. [Google Scholar] [CrossRef]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef] [Green Version]
- Aghali, A.; Christina, M.L.K.N.; Pabelick, Y.S.P. Cellular Senescence in Aging Lungs and Diseases. Cells 2022, 11, 1781. [Google Scholar] [CrossRef]
- Barnes, P.; Baker, J.; Donnelly, L. Cellular Senescence as a Mechanism and Target in Chronic Lung Diseases. Am. J. Respir Crit. Care Med. 2019, 200, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Moreira, M.B.; Garcia-Cardeña, G.; Saffi, M.A.L.; Libby, P. Endothelium: A Coordinator of Acute and Chronic Inflammation. In Endothelium and Cardiovascular Diseases; Endothelium and Cardiovascular Diseases Academic Press: Cambridge, MA, USA, 2018; pp. 485–491. [Google Scholar]
- Dupont, A.; Rauch, A.; Staessens, S.; Moussa, M.; Rosa, M.; Corseaux, D.; Jeanpierre, E.; Goutay, J.; Caplan, M.; Varlet, P.E.A. Vascular endothelial damage in the pathogenesis of organ injury in severe COVID-19. Arter. Thromb. Vasc. Biol. 2021, 41, 1760–1773. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Ji, W.; Yang, H.; Chen, S.; Zhang, W.; Duan, G. Endothelial activation and dysfunction in COVID-19: From basic mechanisms to potential therapeutic approaches. Signal Transduct Target 2020, 5, 293. [Google Scholar] [CrossRef] [PubMed]
- Watany, M.M.; Abdou, S.; Elkolaly, R.; Elgharbawy, N.; Hodeib, H. Evaluation of admission levels of P, E and L selectins as predictors for thrombosis in hospitalized COVID-19 patients. Clin. Exp. Med. 2022, 21, 1–9. [Google Scholar] [CrossRef]
- Conde, J.N.; Schutt, W.R.; Gorbunova, E.E.; Mackow, E.R. Recombinant ACE2 Expression Is Required for SARS-CoV-2 To Infect Primary Human Endothelial Cells and Induce Inflammatory and Procoagulative Responses. MBio 2020, 11, e03185-20. [Google Scholar]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef]
- Harrington, E.O.; Stefanec, T.; Newton, J.; Rounds, S. Release of soluble E-selectin from activated endothelial cells upon apoptosis. Lung 2006, 184, 259–266. [Google Scholar] [CrossRef]
- Kelley, W.J.; Zemans, R.L.; Goldstein, D.R. Cellular senescence: Friend or foe to respiratory viral infections? Eur. Respir J. 2020, 56, 2002708. [Google Scholar] [CrossRef]
- D’Agnillo, F.; Walters, K.A.; Xiao, Y.; Sheng, Z.M.; Scherler, K.; Park, J.; Gygli, S.; Rosas, L.A.; Sadtler, K.; Kalish, H.; et al. Lung epithelial and endothelial damage, loss of tissue repair, inhibition of fibrinolysis, and cellular senescence in fatal COVID-19. Sci. Transl. Med. 2021, 13, eabj7790. [Google Scholar] [CrossRef]
- Thacker, V.V.; Sharma, K.; Dhar, N.; Mancini, G.F.; Sordet-Dessimoz, J.; McKinney, J.D. Rapid endotheliitis and vascular damage characterize SARS-CoV-2 infection in a human lung-chip model. EMBO Rep. 2021, 22, e52744. [Google Scholar] [CrossRef] [PubMed]
- Bordoni, V.; Matusali, G.; Mariotti, D.; Antonioli, M.; Cimini, E.; Sacchi, A.; Tartaglia, E.; Casetti, R.; Grassi, G.; Notari, S. The interplay between SARS-CoV-2 infected airway epithelium and immune cells modulates regulatory/inflammatory signals. iScience 2022, 25, 103854. [Google Scholar] [CrossRef] [PubMed]
- Howarth, A.G.; Hughes, M.R.; Stevenson, B.R. Detection of the tight junction-associated protein ZO-1 in astrocytes and other nonepithelial cell types. Am. J. Physiol. Cell Physiol. 1992, 262, C461–C469. [Google Scholar] [CrossRef]
- Khan, S.Y.; Awad, E.M.; Oszwald, A.; Mayr, M.; Yin, X.; Waltenberger, B.; Stuppner, H.; Lipovac, M.; Uhrin, P.; Breuss, J.M. Premature senescence of endothelial cells upon chronic exposure to TNFα can be prevented by N-acetyl cysteine and plumericin. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Gorgoulis, V.G.; Pratsinis, H.; Zacharatos, P.; Demoliou, C.; Sigala, F.; Asimacopoulos, P.J.; Papavassiliou, A.G.; Kletsas, D. Dependent ICAM-1 overexpression in senescent human cells identified in atherosclerotic lesions. Lab. Investig. 2005, 85, 502–511. [Google Scholar] [CrossRef]
- Yu, Q.; Katlinskaya, Y.V.; Carbone, C.J.; Zhao, B.; Katlinski, K.V.; Zheng, H.; Guha, M.; Li, N.; Chen, Q.; Yang, T. DNA-Damage-Induced Type I Interferon Promotes Senescence and Inhibits Stem Cell Function. Cell Rep. 2015, 11, 785–797. [Google Scholar] [CrossRef]
- Lee, P.Y.; Li, Y.; Richards, H.B.; Chan, F.S.; Zhuang, H.; Narain, S.; Butfiloski, E.J.; Sobel, E.S.; Reeves, W.H.; Segal, M.S. Type I Interferon as a Novel Risk Factor for Endothelial Progenitor Cell Depletion and Endothelial Dysfunction in Systemic Lupus Erythematosus. Arthritis Rheum. 2007, 56, 3759–3769. [Google Scholar] [CrossRef]
- Ma, Z.; Yang, K.Y.; Huang, Y.; Lui, K.O. Endothelial contribution to COVID-19: An update on mechanisms and therapeutic implications. J. Mol. Cell. Cardiol. 2022, 164, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Luo, R.; Zhang, M.; Wang, Y.; Song, T.; Tao, T.; Li, Z.; Jin, L.; Zheng, H.; Chen, W. A cross-talk between epithelium and endothelium mediates human alveolar–capillary injury during SARS-CoV-2 infection. Cell Death Dis. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Liu, F.; Han, K.; Blair, R.; Kenst, K.; Qin, Z.; Upcin, B.; Wörsdörfer, P.; Midkiff, C.C.; Mudd, J.; Belyaeva, E. SARS-CoV-2 Infects Endothelial Cells In Vivo and In Vitro. Front. Cell. Infect. Microbiol 2021, 11, 701278. [Google Scholar] [CrossRef]
- Noris, E.; Zannetti, C.; Demurtas, A.; Sinclair, J.; De Andrea, M.; Gariglio, M.; Landolfo, S. Cell cycle arrest by human cytomegalovirus 86-kDa IE2 protein resembles premature senescence. J. Virol. 2002, 76, 12135–12148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuprin, A.; Gal, H.; Biron-Shental, T.; Biran, A.; Amiel, A.; Rozenblatt, S.; Krizhanovsky, V. Cell fusion induced by ERVWE1 or measles virus causes cellular senescence. Genes Dev. 2013, 27, 2356–2366. [Google Scholar] [CrossRef] [PubMed]
- Martínez, I.; García-Carpizo, V.; Guijarro, T.; García-Gomez, A.; Navarro, D.; Aranda, A.; Zambrano, A. Induction of DNA double-strand breaks and cellular senescence by human respiratory syncytial virus. Virulence 2016, 18, 427–442. [Google Scholar] [CrossRef]
- Yan, Y.; Du, Y.Z.H.; Wang, G.; Li, R.; Chen, J.; Li, K. NS1 of H7N9 Influenza A Virus Induces NO-Mediated Cellular Senescence in Neuro2a Cells. Cell Physiol Biochem. 2017, 43, 1369–1380. [Google Scholar] [CrossRef]
- Baz-Martínez, M.; Da Silva-Álvarez, S.; Rodríguez, E.; Guerra, J.; Motiam, A.E.; Vidal, A.; García-Caballero, T.; González-Barcia, M.; Sánchez, L.; Muñoz-Fontela, C. Cell senescence is an antiviral defense mechanism. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef]
- Lee, S.; Yu, Y.; Trimpert, J.; Benthani, F.; Mairhofer, M.; Richter-Pechanska, P.; Wyler, E.; Belenki, D.; Kaltenbrunner, S.; Pammer, M. Induced senescence is a driver and therapeutic target in COVID-19. Nature 2021, 599, 283–289. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef]
- Di Domizio, J.; Gulen, M.F.; Saidoune, F.; Thacker, V.V.; Yatim, A.; Sharma, K.; Nass, T.; Guenova, E.; Schaller, M.; Conrad, C. The cGAS–STING pathway drives type I IFN immunopathology in COVID-19. Nature 2022, 603, 145–151. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bordoni, V.; Mariotti, D.; Matusali, G.; Colavita, F.; Cimini, E.; Ippolito, G.; Agrati, C. SARS-CoV-2 Infection of Airway Epithelium Triggers Pulmonary Endothelial Cell Activation and Senescence Associated with Type I IFN Production. Cells 2022, 11, 2912. https://doi.org/10.3390/cells11182912
Bordoni V, Mariotti D, Matusali G, Colavita F, Cimini E, Ippolito G, Agrati C. SARS-CoV-2 Infection of Airway Epithelium Triggers Pulmonary Endothelial Cell Activation and Senescence Associated with Type I IFN Production. Cells. 2022; 11(18):2912. https://doi.org/10.3390/cells11182912
Chicago/Turabian StyleBordoni, Veronica, Davide Mariotti, Giulia Matusali, Francesca Colavita, Eleonora Cimini, Giuseppe Ippolito, and Chiara Agrati. 2022. "SARS-CoV-2 Infection of Airway Epithelium Triggers Pulmonary Endothelial Cell Activation and Senescence Associated with Type I IFN Production" Cells 11, no. 18: 2912. https://doi.org/10.3390/cells11182912
APA StyleBordoni, V., Mariotti, D., Matusali, G., Colavita, F., Cimini, E., Ippolito, G., & Agrati, C. (2022). SARS-CoV-2 Infection of Airway Epithelium Triggers Pulmonary Endothelial Cell Activation and Senescence Associated with Type I IFN Production. Cells, 11(18), 2912. https://doi.org/10.3390/cells11182912