
A Novel Homozygous Founder Variant of RTN4IP1 in Two Consanguineous Saudi Families

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Recruitment and Sample Collection
2.2. DNA Isolation, PCR and Sanger Sequencing
2.3. Genome-Wide SNP Genotyping and Autozygosity Mapping
2.4. Next Generation Sequencing Using Illumina Platform
2.5. Next Generation Sequencing Using Ion Torrent Platform
2.6. Variant Analysis and Bioinformatics Analysis
2.7. Cell Passaging and Harvesting
2.8. Immunoblotting
3. Results
3.1. Clinical Evaluations
3.1.1. Patient 1 (Family 1)
3.1.2. Patient 2 (Family 1)
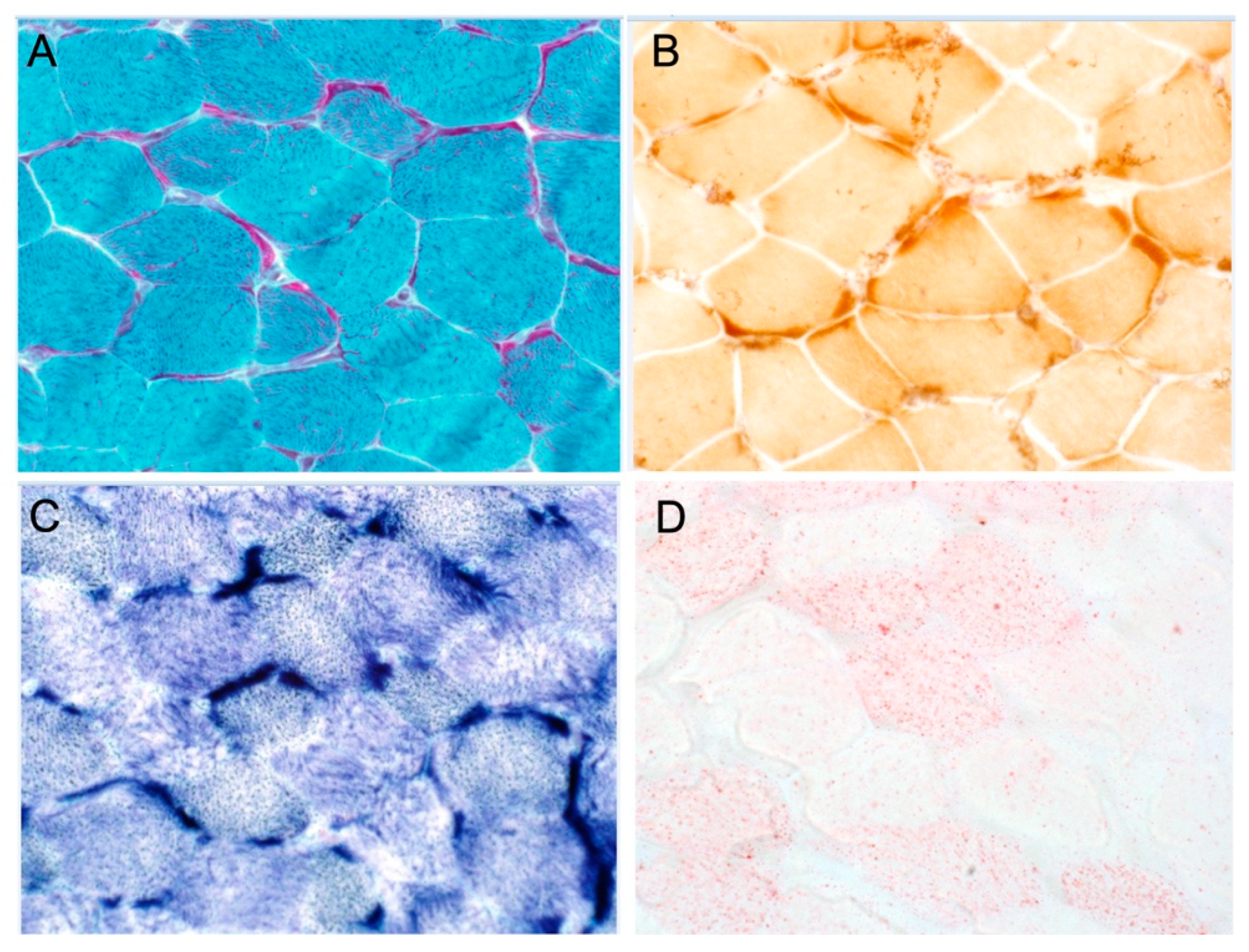
3.1.3. Patient 3 (Family 1)
3.1.4. Patient 4 (Family 2)
3.1.5. Patient 5 (Family 2)
3.1.6. Patient 6 (Family 2)
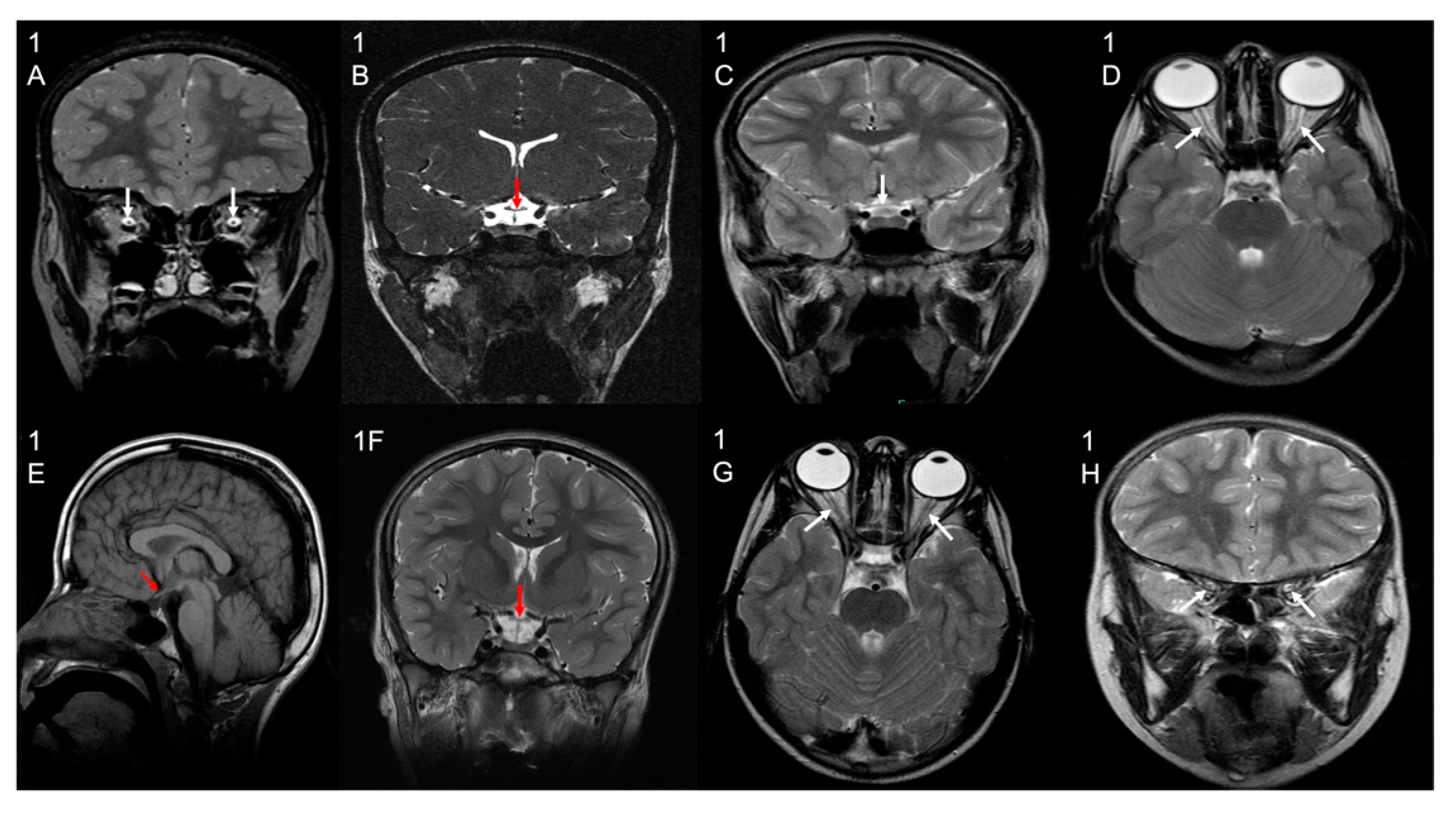
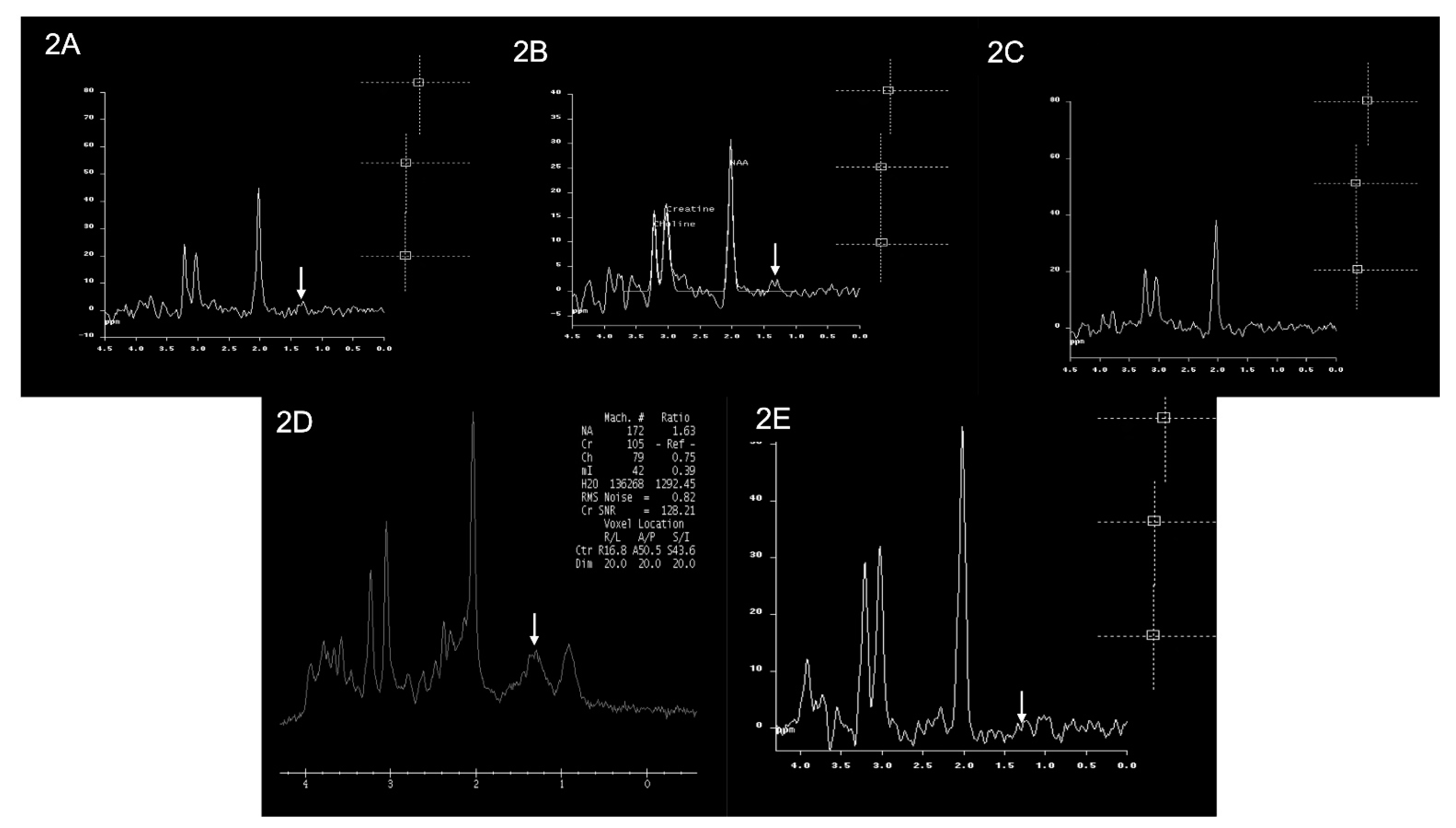
3.2. Brain Imaging Findings
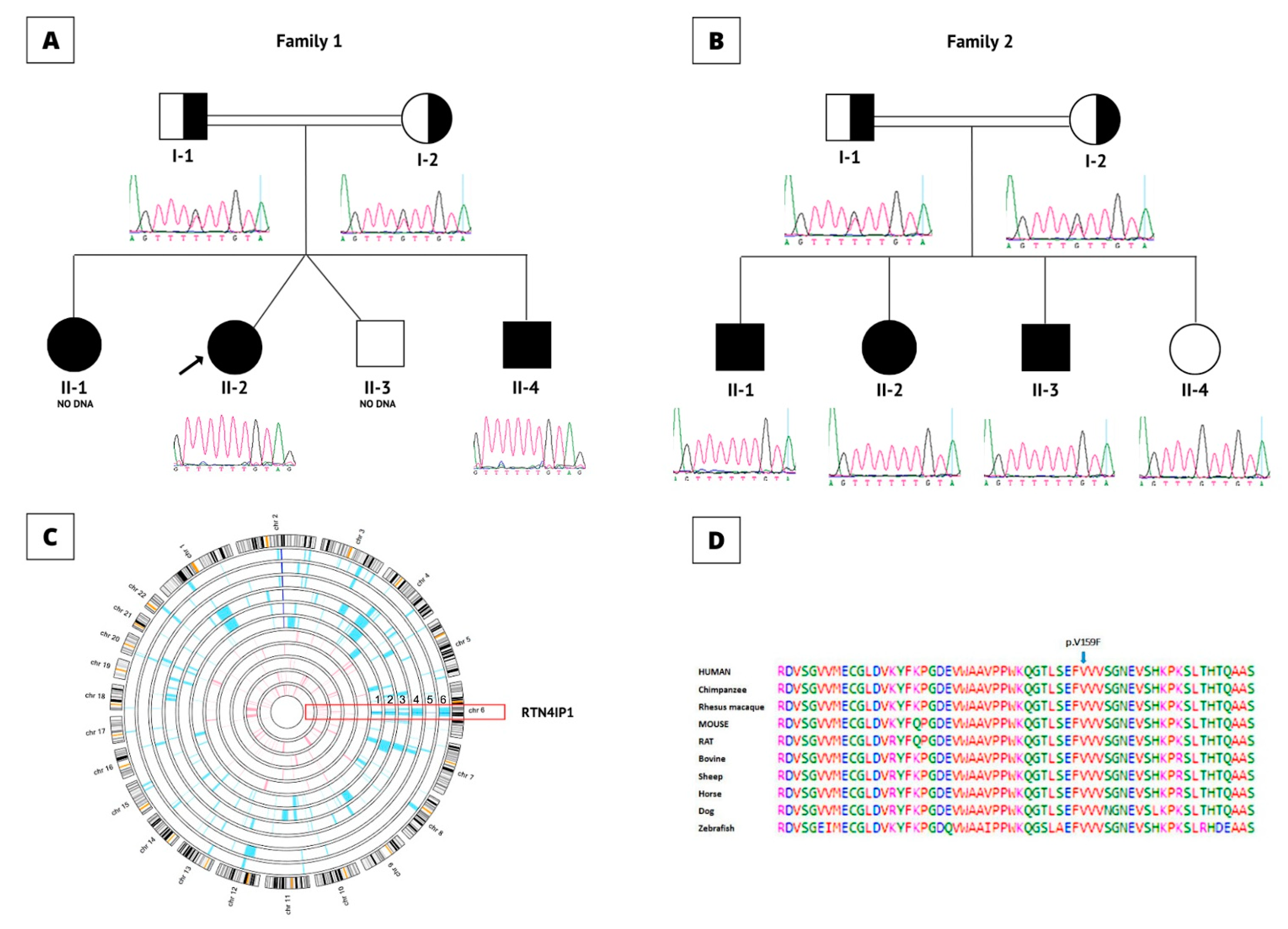
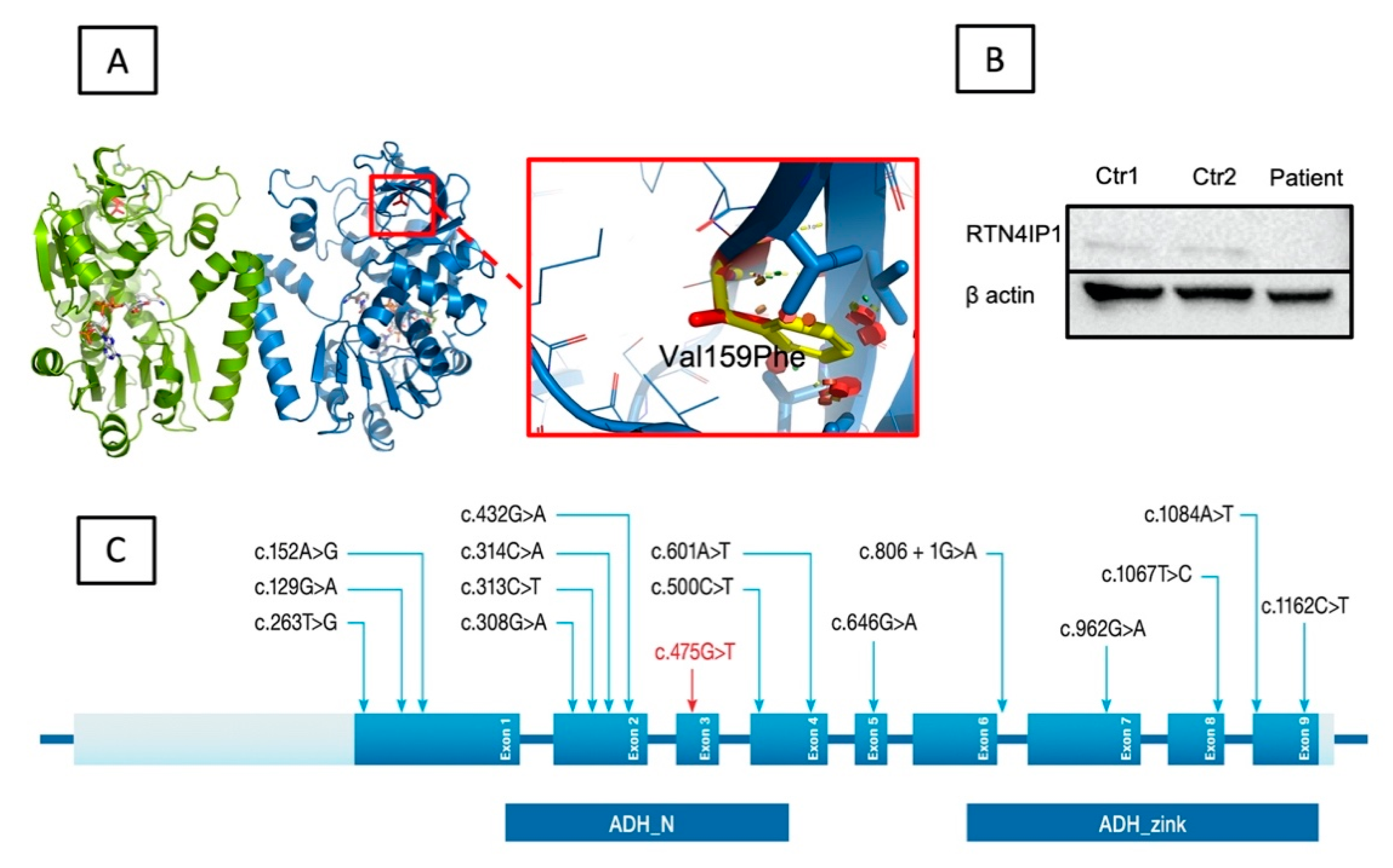
3.3. Genetic and Molecular Results Evaluations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Al-Hassnan, Z.N.; Al-Dosary, M.; Alfadhel, M.; Faqeih, E.A.; Alsagob, M.; Kenana, R.; Almass, R.; Al-Harazi, O.S.; Al-Hindi, H.; Malibari, O.I.; et al. ISCA2 mutation causes infantile neurodegenerative mitochondrial disorder. J. Med. Genet. 2015, 52, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Kerrison, J.B. Hereditary optic neuropathies. Ophthalmol. Clin. N. Am. 2001, 14, 99–107. [Google Scholar]
- Angebault, C.; Guichet, P.O.; Talmat-Amar, Y.; Charif, M.; Gerber, S.; Fares-Taie, L.; Gueguen, N.; Halloy, F.; Moore, D.; Amati-Bonneau, P.; et al. Recessive Mutations in RTN4IP1 Cause Isolated and Syndromic Optic Neuropathies. Am. J. Hum. Genet. 2015, 97, 754–760. [Google Scholar] [CrossRef] [Green Version]
- D’Gama, A.M.; England, E.; Madden, J.A.; Shi, J.; Chao, K.R.; Wojcik, M.H.; Torres, A.R.; Tan, W.H.; Berry, G.T.; Prabhu, S.P.; et al. Exome sequencing identifies novel missense and deletion variants in RTN4IP1 associated with optic atrophy, global developmental delay, epilepsy, ataxia, and choreoathetosis. Am. J. Med. Genet. A 2021, 185, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.H.; Hausmann, O.N.; Yan, M.S.; Walters, W.M.; Wong, P.K.; Bethea, J.R. Identification and characterization of a novel Nogo-interacting mitochondrial protein (NIMP). J. Neurochem. 2002, 81, 36–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.S.; Huber, A.B.; van der Haar, M.E.; Frank, M.; Schnell, L.; Spillmann, A.A.; Christ, F.; Schwab, M.E. Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature 2000, 403, 434–439. [Google Scholar] [CrossRef]
- GrandPre, T.; Nakamura, F.; Vartanian, T.; Strittmatter, S.M. Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature 2000, 403, 439–444. [Google Scholar] [CrossRef]
- Prinjha, R.; Moore, S.E.; Vinson, M.; Blake, S.; Morrow, R.; Christie, G.; Michalovich, D.; Simmons, D.L.; Walsh, F.S. Inhibitor of neurite outgrowth in humans. Nature 2000, 403, 383–384. [Google Scholar] [CrossRef]
- Park, I.; Kim, K.-e.; Kim, J.; Bae, S.; Jung, M.; Choi, J.; Kwak, C.; Kang, M.-G.; Yoo, C.-M.; Mun, J.Y. In vivo mitochondrial matrix proteome profiling reveals RTN4IP1/OPA10 as an antioxidant NADPH oxidoreductase. bioRxiv 2021. [Google Scholar] [CrossRef]
- Charif, M.; Nasca, A.; Thompson, K.; Gerber, S.; Makowski, C.; Mazaheri, N.; Bris, C.; Goudenege, D.; Legati, A.; Maroofian, R.; et al. Neurologic Phenotypes Associated with Mutations in RTN4IP1 (OPA10) in Children and Young Adults. JAMA Neurol. 2018, 75, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Meunier, I.; Bocquet, B.; Charif, M.; Dhaenens, C.M.; Manes, G.; Amati-Bonneau, P.; Roubertie, A.; Zanlonghi, X.; Lenaers, G. A Rod-Cone Dystrophy Is Systematically Associated to the Rtn4ip1 Recessive Optic Atrophy. Retina 2021, 41, 1771–1779. [Google Scholar] [CrossRef]
- Okamoto, N.; Miya, F.; Hatsukawa, Y.; Suzuki, Y.; Kawato, K.; Yamamoto, Y.; Tsunoda, T.; Kato, M.; Saitoh, S.; Yamasaki, M.; et al. Siblings with optic neuropathy and RTN4IP1 mutation. J. Hum. Genet. 2017, 62, 927–929. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, T.; Gamucci, A.; Pisciotta, L.; Nesti, C.; Fiorillo, C.; Doccini, S.; Morana, G.; Nobili, L.; Santorelli, F.M.; Mancardi, M.M.; et al. Optic Atrophy and Generalized Chorea in a Patient Harboring an OPA10/RTN4IP1 Pathogenic Variant. Neuropediatrics 2020, 51, 425–429. [Google Scholar] [CrossRef]
- Zou, X.H.; Guo, X.X.; Su, H.Z.; Wang, C.; Dong, E.L.; Wang, N.; Chen, W.J.; Zhang, Q.J. Whole Exome Sequencing Identifies Two Novel Mutations in the Reticulon 4-Interacting Protein 1 Gene in a Chinese Family with Autosomal Recessive Optic Neuropathies. J. Mol. Neurosci. 2019, 68, 640–646. [Google Scholar] [CrossRef]
- Carr, I.M.; Flintoff, K.J.; Taylor, G.R.; Markham, A.F.; Bonthron, D.T. Interactive visual analysis of SNP data for rapid autozygosity mapping in consanguineous families. Hum. Mutat. 2006, 27, 1041–1046. [Google Scholar] [CrossRef] [PubMed]
- Perenthaler, E.; Nikoncuk, A.; Yousefi, S.; Berdowski, W.M.; Alsagob, M.; Capo, I.; van der Linde, H.C.; van den Berg, P.; Jacobs, E.H.; Putar, D.; et al. Loss of UGP2 in brain leads to a severe epileptic encephalopathy, emphasizing that bi-allelic isoform-specific start-loss mutations of essential genes can cause genetic diseases. Acta Neuropathol. 2020, 139, 415–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldhalaan, H.; AlBakheet, A.; AlRuways, S.; AlMutairi, N.; AlNakiyah, M.; AlGhofaili, R.; Cardona-Londono, K.J.; Alahmadi, K.O.; AlQudairy, H.; AlRasheed, M.M.; et al. A Novel GEMIN4 Variant in a Consanguineous Family Leads to Neurodevelopmental Impairment with Severe Microcephaly, Spastic Quadriplegia, Epilepsy, and Cataracts. Genes 2021, 13, 92. [Google Scholar] [CrossRef]
- AlMuhaizea, M.; AlMass, R.; AlHargan, A.; AlBader, A.; Medico Salsench, E.; Howaidi, J.; Ihinger, J.; Karachunski, P.; Begtrup, A.; Segura Castell, M.; et al. Truncating mutations in YIF1B cause a progressive encephalopathy with various degrees of mixed movement disorder, microcephaly, and epilepsy. Acta Neuropathol. 2020, 139, 791–794. [Google Scholar] [CrossRef] [Green Version]
- Salih, M.A.; Hamad, M.H.; Savarese, M.; Alorainy, I.A.; Al-Jarallah, A.S.; Alkhalidi, H.; AlQudairy, H.; Albader, A.; Alotaibi, A.J.; Alsagob, M.; et al. Exome Sequencing Reveals Novel TTN Variants in Saudi Patients with Congenital Titinopathies. Genet. Test. Mol. Biomark. 2021, 25, 757–764. [Google Scholar] [CrossRef]
- Sanderson, L.E.; Lanko, K.; Alsagob, M.; Almass, R.; Al-Ahmadi, N.; Najafi, M.; Al-Muhaizea, M.A.; Alzaidan, H.; AlDhalaan, H.; Perenthaler, E.; et al. Bi-allelic variants in HOPS complex subunit VPS41 cause cerebellar ataxia and abnormal membrane trafficking. Brain 2021, 144, 769–780. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Rodelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef]
- Gonzalez-Perez, A.; Lopez-Bigas, N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am. J. Hum. Genet. 2011, 88, 440–449. [Google Scholar] [CrossRef] [Green Version]
- Wong, K.-C.; Zhang, Z. SNPdryad: Predicting deleterious non-synonymous human SNPs using only orthologous protein sequences. Bioinformatics 2014, 30, 1112–1119. [Google Scholar] [CrossRef] [Green Version]
- Chelban, V.; Alsagob, M.; Kloth, K.; Chirita-Emandi, A.; Vandrovcova, J.; Maroofian, R.; Davagnanam, I.; Bakhtiari, S.; AlSayed, M.D.; Rahbeeni, Z.; et al. Genetic and phenotypic characterization of NKX6-2-related spastic ataxia and hypomyelination. Eur. J. Neurol. 2020, 27, 334–342. [Google Scholar] [CrossRef] [Green Version]
- Seidahmed, M.Z.; Hamad, M.H.; AlBakheet, A.; Elmalik, S.A.; AlDrees, A.; Al-Sufayan, J.; Alorainy, I.; Ghozzi, I.M.; Colak, D.; Salih, M.A.; et al. Ancient founder mutation in RUBCN: A second unrelated family confirms Salih ataxia (SCAR15). BMC Neurol 2020, 20, 207. [Google Scholar] [CrossRef]
- Medico Salsench, E.; Maroofian, R.; Deng, R.; Lanko, K.; Nikoncuk, A.; Perez, B.; Sanchez-Lijarcio, O.; Ibanez-Mico, S.; Wojcik, A.; Vargas, M.; et al. Expanding the mutational landscape and clinical phenotype of the YIF1B related brain disorder. Brain 2021, 144, e85. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Shaw, K.; Phillips, A.; Cooper, D.N. The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 2014, 133, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Al-Muhaizea, M.A.; Aldeeb, H.; Almass, R.; Jaber, H.; Binhumaid, F.; Alquait, L.; Abukhalid, M.; Aldhalaan, H.; Alsagob, M.; Al-Bakheet, A.; et al. Genetics of ataxia telangiectasia in a highly consanguineous population. Ann. Hum. Genet. 2022, 86, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef] [PubMed]
- Jurkute, N.; Arno, G.; Webster, A.R.; Yu-Wai-Man, P.; Genomics England Research, C. Whole Genome Sequencing Identifies a Partial Deletion of RTN4IP1 in a Patient with Isolated Optic Atrophy. J. Neuroophthalmol. 2022; online ahead of print. [Google Scholar] [CrossRef]
- Rajabian, F.; Manitto, M.P.; Palombo, F.; Caporali, L.; Grazioli, A.; Starace, V.; Arrigo, A.; Cascavilla, M.L.; La Morgia, C.; Barboni, P.; et al. Combined Optic Atrophy and Rod-Cone Dystrophy Expands the RTN4IP1 (Optic Atrophy 10) Phenotype. J. Neuroophthalmol. 2021, 41, e290–e292. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Family 1 | Family 2 | ||||
|---|---|---|---|---|---|---|
| Patient Number | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 |
| Patient Codes | F1-II-1 | F1-II-2 | F1-II-3 | F2-II-1 | F2-II-2 | F2-II-3 |
| Gender | Male | Female | Male | Male | Female | Male |
| Age (years) | 29 | 27 | 25 | 26 | 17 | 19 |
| Parental Consanguinity | Yes | Yes | Yes | Yes | Yes | Yes |
| Age of onset (years) | Not available | Not available | Not available | 2 | 1.5 | 1.5 |
| Microcephaly | No | No | No | No | No | No |
| Developmental delay | Yes | Yes | Yes | Yes | Yes | Yes |
| Axial Hypertonia | No | No | No | No | No | No |
| Axial Hypotonia | No | No | No | No | No | No |
| Other movement disorder | Unknown | Unknown | Unknown | Unbalanced and delayed walking | Lost ability to walk | Unbalanced and delayed walking |
| Optalmological findings | Nystagmus, optic atrophy | Nystagmus, optic atrophy | Nystagmus, optic atrophy | Optic atrophy | Optic atrophy | Optic atrophy |
| Seizure | Generalized tonic clonic | Generalized tonic clonic | Generalized tonic clonic | Generalized tonic clonic | Generalized tonic clonic | Generalized tonic clonic |
| White matter abnormalities | No | No | No | No | No | No |
| Left hemispheres abnormalities | No | No | No | No | No | No |
| Cerebral encephalopathy | Yes | Yes | Yes | Yes | Yes | Yes |
| Temporal areas abnormalities | No | No | No | No | No | No |
| Subdural area abnormalities | No | No | No | No | No | No |
| Levetiracetam | No | No | No | No | No | No |
| Phenytoin | No | No | No | No | No | No |
| Carbamazepine | No | Yes | No | No | Yes | No |
| Phenobarbital | No | No | No | No | No | No |
| Clonazepam | No | No | No | No | No | No |
| Topiramate | No | No | No | No | No | No |
| NCBI Reference ID | NM:032730 | NM:032730; | NM:032730 | NM:032730 | NM:032730 | NM:032730 |
| cDNA Change | c.475G>T | c.475G>T | c.475G>T | c.475G>T | c.475G>T | c.475G>T |
| Amino Acid Changes | p.Val159Phe | p.Val159Phe | p.Val159Phe | p.Val159Phe | p.Val159Phe | p.Val159Phe |
| Others | - | - | - | - | Bulbous nose, curved relatively small ears, hypertelorism, synopsis large upper lip, short philtrum and skin eczema in limbs | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aldosary, M.; Alsagob, M.; AlQudairy, H.; González-Álvarez, A.C.; Arold, S.T.; Dababo, M.A.; Alharbi, O.A.; Almass, R.; AlBakheet, A.; AlSarar, D.; et al. A Novel Homozygous Founder Variant of RTN4IP1 in Two Consanguineous Saudi Families. Cells 2022, 11, 3154. https://doi.org/10.3390/cells11193154
Aldosary M, Alsagob M, AlQudairy H, González-Álvarez AC, Arold ST, Dababo MA, Alharbi OA, Almass R, AlBakheet A, AlSarar D, et al. A Novel Homozygous Founder Variant of RTN4IP1 in Two Consanguineous Saudi Families. Cells. 2022; 11(19):3154. https://doi.org/10.3390/cells11193154
Chicago/Turabian StyleAldosary, Mazhor, Maysoon Alsagob, Hanan AlQudairy, Ana C. González-Álvarez, Stefan T. Arold, Mohammad Anas Dababo, Omar A. Alharbi, Rawan Almass, AlBandary AlBakheet, Dalia AlSarar, and et al. 2022. "A Novel Homozygous Founder Variant of RTN4IP1 in Two Consanguineous Saudi Families" Cells 11, no. 19: 3154. https://doi.org/10.3390/cells11193154
APA StyleAldosary, M., Alsagob, M., AlQudairy, H., González-Álvarez, A. C., Arold, S. T., Dababo, M. A., Alharbi, O. A., Almass, R., AlBakheet, A., AlSarar, D., Qari, A., Al-Ansari, M. M., Oláhová, M., Al-Shahrani, S. A., AlSayed, M., Colak, D., Taylor, R. W., AlOwain, M., & Kaya, N. (2022). A Novel Homozygous Founder Variant of RTN4IP1 in Two Consanguineous Saudi Families. Cells, 11(19), 3154. https://doi.org/10.3390/cells11193154