
Human iPSC-Cardiomyocytes as an Experimental Model to Study Epigenetic Modifiers of Electrophysiology



Abstract
:1. Introduction
1.1. hiPSC-CMs as a Scalable Model of Cardiac Electrophysiology
1.2. Human Experimental Models Are Needed for Functional Cardiac Studies
1.3. hiPSC-CMs for Drug Cardiotoxicity Screening
2. Epigenetic Modulators of the Cardiovascular System
2.1. Epigenetic Regulators in the Heart and Control of Cardiac Electrophysiology
2.2. Differential Gene Expression in hiPSC-CMs and the Adult Heart Relevant to Epigenetic Modifiers and Electrophysiological Function
3. Pharmacological HDAC Inhibition
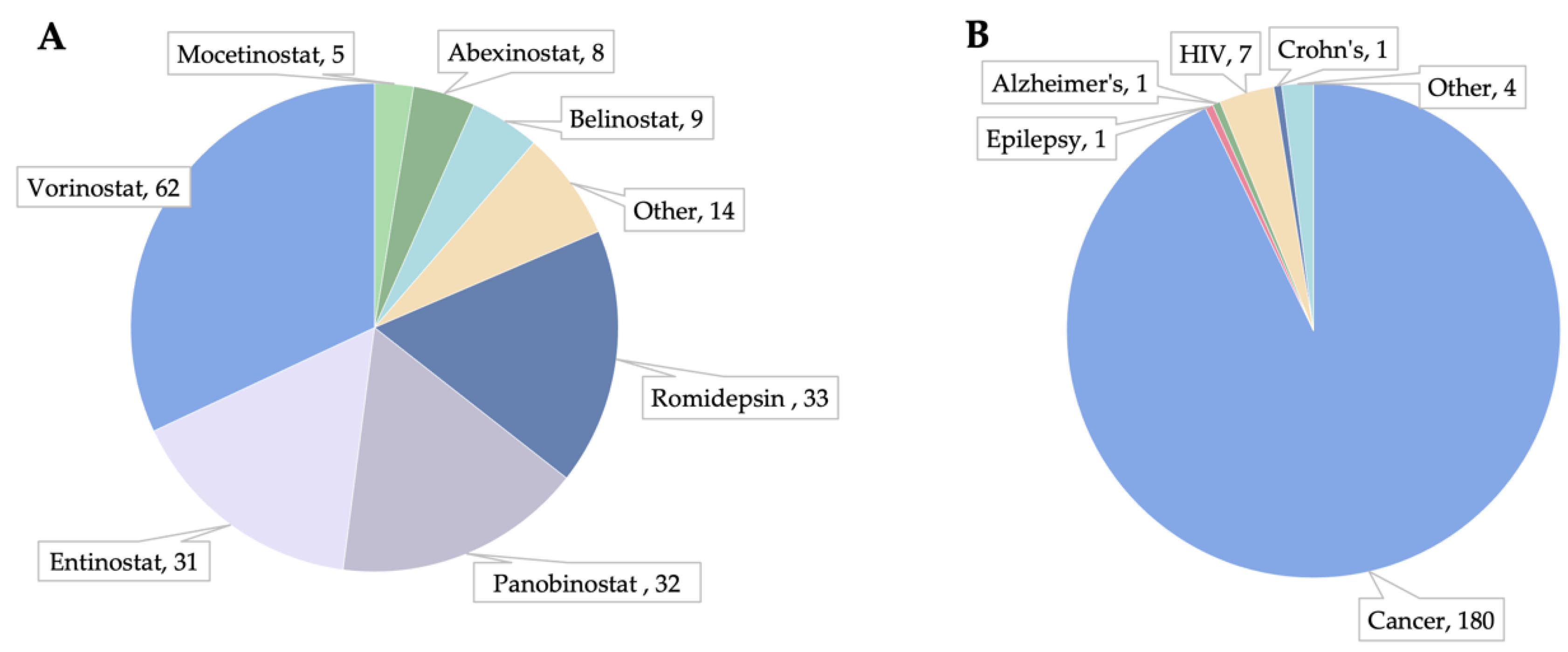
3.1. HDACi in Clinical Trials and Post-Market Observations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class I | Class IIa | Class IIb | Class IV | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Inhibitor Name | HDAC1 | HDAC2 | HDAC3 | HDAC8 | HDAC4 | HDAC5 | HDAC7 | HDAC9 | HDAC6 | HDAC10 | HDAC11 | Clinicaltrials.gov (02/22/2019) | |
| Vorinostat (SAHA) | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | Merck (FDA) | 251 |
| Panobinostat | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | Novartis (FDA) | 133 |
| Trichostatin A | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | 15 | |
| Belinostat | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | TopoTarget (FDA) | 44 |
| Dacinostat | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | Novartis | - |
| M344 | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | - | |
| AR-42 | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | Arno Therapeutics | 5 |
| Quisinostat | ++++ | ++++ | ++++ | ++++ | ++++ | ++++ | ++ | +++ | ++ | ++++ | ++++ | 6 | |
| CUDC-907 | ++++ | ++++ | ++++ | ++ | ++ | + | ++ | ++ | +++ | ++++ | ++++ | 6 | |
| Pracinostat | +++ | ++ | +++ | ++ | +++ | +++ | ++ | ++ | + | +++ | ++ | MEI Pharma | 12 |
| CUDC-101 | ++++ | +++ | ++++ | ++ | +++ | +++ | ++ | ++ | ++++ | +++ | Curis | 4 | |
| Ricolinostat | ++ | +++ | +++ | ++ | + | + | + | ++++ | Celgene/Acetylon | 9 | |||
| Abexinostat | ++++ | +++ | ++++ | ++ | +++ | +++ | Pharmacyclics | 9 | |||||
| HPOB | + | + | + | + | +++ | + | 1 | ||||||
| MC1568 | ++ | ++ | ++ | ++ | - | ||||||||
| Mocetinostat | ++ | ++ | + | + | Mirati | 22 | |||||||
| TMP269 | ++ | ++ | +++ | +++ | - | ||||||||
| PCI-34051 | + | ++++ | + | + | - | ||||||||
| Droxinostat | + | + | + | - | |||||||||
| Resminostat | +++ | +++ | ++ | 4SC | 5 | ||||||||
| BRD72954 | + | ++ | +++ | - | |||||||||
| BG45 | + | + | ++ | - | |||||||||
| 4SC-202 | + | + | + | 4SC | 3 | ||||||||
| Tacedinaline | + | + | + | 3 | |||||||||
| LMK-235 | +++ | ++++ | - | ||||||||||
| Romidepsin | +++ | +++ | Celgene (FDA) | 88 | |||||||||
| RG2833 | +++ | Replign | - | ||||||||||
| Entinostat | ++ | + | Syndax | 60 | |||||||||
| CAY10603 | ++ | ++++ | - | ||||||||||
| Tubacin | ++++ | - | |||||||||||
| RGFP966 | ++ | - | |||||||||||
| Tubastatin A | +++ | - | |||||||||||
| Nexturastat A | ++++ | - | |||||||||||
| SS-2-08 | ++++ | - | |||||||||||
3.2. HDACi Have Cardiac Therapeutic Potential
4. Methodologies for Quantifying Effects of HDACi in hiPSC-CMs
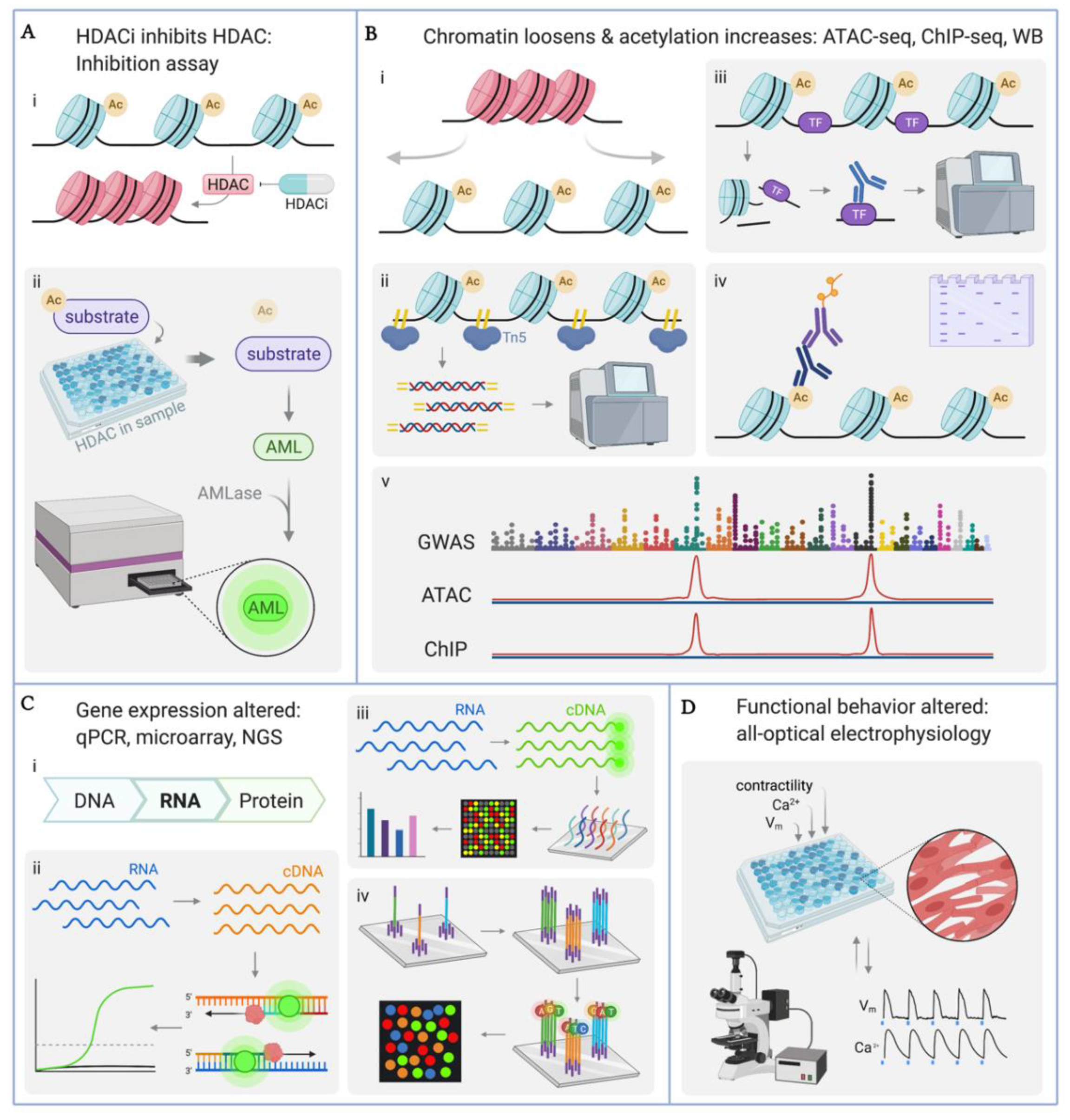
4.1. Quantification of HDAC Enzyme Inhibition
4.2. Quantification of Histone Acetylation: ATAC-seq, ChIP-seq, Western Blot
4.3. Quantification of Gene Transcription: qPCR, Microarray, RNA-seq
4.4. Quantitative Functional Studies
5. Epigenetic Studies in hiPSC-CMs
5.1. Epigenetic Characterization of hiPSC-CMs
| Cell Line(s) Used | HDACi Applied | Chm | Ac-H | Gene Exp | Fxnl | Major Findings |
|---|---|---|---|---|---|---|
| In-house-derived hiPSC-CM | ✓ | – | WB | qPCR | MEA, optical Ca2+ | TSA improved differentiation towards the cardiac lineage [175]. |
| In-house-derived hiPSC-CM | ✓ | – | WB | qRT-PCR, microarray | MEA | TSA treatment and suspension culture improve maturity (expression of cardiac genes, homogenous response to hERG blocker) [176]. |
| hiPSC-CM (Axiogenesis) | ✓ | – | – | microarray | impedance recordings, MEA | HDACi had delayed cardiotoxicity (reduced beat rate, arrhythmic events), HDACi modified pathways related to cell contraction, microtubule/cytoskeleton-based transport, and Z-disc binding [133]. |
| hiPSC-CM (Axiogenesis) | ✓ | – | – | microarray | – | Panobinostat diminished contractile properties (beat area, beat rate, contraction velocity), increased levels of cardiotoxicity biomarkers (cTnI, FABP3, and NT-proBNP), downregulated cardiac structural and functional genes) [177]. |
| hiPSC-CM (iCell, CDI) | ✓ | – | – | – | whole-cell patch clamp | Vorinostat reduced INa current density [178]. |
| 26 in-house-derived hiPSC-CM lines | ATAC-seq | ChIP-seq (H3K27ac, NKX2-5) | RNA-seq, WGS | – | NKX2-5 (TF), H3K27ac, and ATAC peaks are associated with enrichment for EKG characteristics such as heart rate, QT interval, QRS duration, and atrial fibrillation. Histone acetylation and TF info from ChIP-seq can be cross-referenced with ATAC-seq peaks and GWAS to illuminate mechanisms of phenotypic effects. dbGaP: phs000924; NCBI: PRJNA285375; GEO: GSE125540, GSE133833 [155]. | |
| 27 in-house-derived hiPSC-CM lines | Hi-C, ATAC-seq | ChIP-seq (H3K27ac, NKX2-5) | RNA-seq, WGS | – | Contact propensity is a mechanism of regulating gene expression and is positively associated with H3K27 acetylation and gene expression. dbGaP: phs000924 [169]. | |
| In-house derived hiPSC-CM | ATAC-seq, DNA methylation | – | RNA-seq | – | Hypoxia and subsequent reoxygenation alter chromatin accessibility (both positively and negatively in various regions), particularly at transcription start sites, indicating the role of hypoxia-induced chromatin reorganization in regulating gene expression. GEO: GSE144426 [174]. |
5.2. HDACi in hiPSC-CM Differentiation and Maturation
5.3. HDACi Cardiotoxicity Testing in hiPSC-CMs
5.3.1. Transcriptional Effects of HDACi in hiPSC-CMs
5.3.2. Functional Effects of HDACi in hiPSC-CMs
5.3.3. Transcriptional Changes Corroborate Functional Outputs
5.3.4. In Vitro Results Are Consistent with Clinical Observations
6. Conclusions and Future Outlook
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Rakyan, V.K.; Hildmann, T.; Novik, K.L.; Lewin, J.; Tost, J.; Cox, A.V.; Andrews, T.D.; Howe, K.L.; Otto, T.; Olek, A.; et al. DNA Methylation Profiling of the Human Major Histocompatibility Complex: A Pilot Study for the Human Epigenome Project. PLoS Biol. 2004, 2, e405. [Google Scholar] [CrossRef]
- Murrell, A.; Rakyan, V.K.; Beck, S. From Genome to Epigenome. Hum. Mol. Genet. 2005, 14, R3–R10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, L.W.; Ferguson, B.S. Food Bioactive HDAC Inhibitors in the Epigenetic Regulation of Heart Failure. Nutrients 2018, 10, 1120. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced Pluripotent Stem Cell Technology: A Decade of Progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef]
- Staerk, J.; Dawlaty, M.M.; Gao, Q.; Maetzel, D.; Hanna, J.; Sommer, C.A.; Mostoslavsky, G.; Jaenisch, R. Reprogramming of Human Peripheral Blood Cells to Induced Pluripotent Stem Cells. Cell Stem Cell 2010, 7, 20–24. [Google Scholar] [CrossRef] [Green Version]
- Zwi, L.; Caspi, O.; Arbel, G.; Huber, I.; Gepstein, A.; Park, I.H.; Gepstein, L. Cardiomyocyte Differentiation of Human Induced Pluripotent Stem Cells. Circulation 2009, 120, 1513–1523. [Google Scholar] [CrossRef] [Green Version]
- Sayed, N.; Ameen, M.; Wu, J.C. Personalized Medicine in Cardio-Oncology: The Role of Induced Pluripotent Stem Cell. Cardiovasc. Res. 2019, 115, 949–959. [Google Scholar] [CrossRef]
- Stack, J.P.; Moslehi, J.; Sayed, N.; Wu, J.C. Cancer Therapy-Induced Cardiomyopathy: Can Human Induced Pluripotent Stem Cell Modelling Help Prevent It? Eur. Heart J. 2019, 40, 1764–1770. [Google Scholar] [CrossRef] [PubMed]
- Burridge, P.W.; Diecke, S.; Matsa, E.; Sharma, A.; Wu, H.; Wu, J.C. Modeling Cardiovascular Diseases with Patient-Specific Human Pluripotent Stem Cell-Derived Cardiomyocytes. Methods Mol. Biol. 2016, 1353, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Shinozawa, T.; Nakamura, K.; Shoji, M.; Morita, M.; Kimura, M.; Furukawa, H.; Ueda, H.; Shiramoto, M.; Matsuguma, K.; Kaji, Y.; et al. Recapitulation of Clinical Individual Susceptibility to Drug-Induced QT Prolongation in Healthy Subjects Using IPSC-Derived Cardiomyocytes. Stem Cell Rep. 2017, 8, 226–234. [Google Scholar] [CrossRef] [Green Version]
- Da Rocha, A.M.; Creech, J.; Thonn, E.; Mironov, S.; Herron, T.J. Detection of Drug-Induced Torsades de Pointes Arrhythmia Mechanisms Using HiPSC-CM Syncytial Monolayers in a High-Throughput Screening Voltage Sensitive Dye Assay. Toxicol. Sci. 2020, 173, 402–415. [Google Scholar] [CrossRef]
- Blinova, K.; Schocken, D.; Patel, D.; Daluwatte, C.; Vicente, J.; Wu, J.C.; Strauss, D.G. Clinical Trial in a Dish: Personalized Stem Cell–Derived Cardiomyocyte Assay Compared With Clinical Trial Results for Two QT-Prolonging Drugs. Clin. Transl. Sci. 2019, 12, 687–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; Tien, N.T.; de Haan, L.; Louisse, J.; Rietjens, I.M.C.M.; Bouwmeester, H. Evaluation of in Vitro Models of Stem Cell-Derived Cardiomyocytes to Screen for Potential Cardiotoxicity of Chemicals. Toxicol. In Vitro 2020, 67, 104891. [Google Scholar] [CrossRef] [PubMed]
- Klimas, A.; Ambrosi, C.M.; Yu, J.; Williams, J.C.; Bien, H.; Entcheva, E. OptoDyCE as an Automated System for High-Throughput All-Optical Dynamic Cardiac Electrophysiology. Nat. Commun. 2016, 7, 11542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klimas, A.; Ortiz, G.; Boggess, S.C.; Miller, E.W.; Entcheva, E. Multimodal On-Axis Platform for All-Optical Electrophysiology with near-Infrared Probes in Human Stem-Cell-Derived Cardiomyocytes. Prog. Biophys. Mol. Biol. 2020, 154, 62–70. [Google Scholar] [CrossRef]
- Biendarra-Tiegs, S.M.; Li, X.; Ye, D.; Brandt, E.B.; Ackerman, M.J.; Nelson, T.J. Single-Cell RNA-Sequencing and Optical Electrophysiology of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes Reveal Discordance Between Cardiac Subtype-Associated Gene Expression Patterns and Electrophysiological Phenotypes. Stem Cells Dev. 2019, 28, 659–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Entcheva, E.; Kay, M.W. Cardiac Optogenetics: A Decade of Enlightenment. Nat. Rev. Cardiol. 2021, 18, 349–367. [Google Scholar] [CrossRef]
- Shaheen, N.; Shiti, A.; Huber, I.; Shinnawi, R.; Arbel, G.; Gepstein, A.; Setter, N.; Goldfracht, I.; Gruber, A.; Chorna, S.V. Human Induced Pluripotent Stem Cell-Derived Cardiac Cell Sheets Expressing Genetically Encoded Voltage Indicator for Pharmacological and Arrhythmia Studies. Stem Cell Rep. 2018, 10, 1879–1894. [Google Scholar] [CrossRef] [PubMed]
- Shinnawi, R.; Huber, I.; Maizels, L.; Shaheen, N.; Gepstein, A.; Arbel, G.; Tijsen, A.J.; Gepstein, L. Monitoring Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes with Genetically Encoded Calcium and Voltage Fluorescent Reporters. Stem Cell Rep. 2015, 5, 582–596. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Awari, D.W.; Han, E.Y.; Uche-Anya, E.; Park, S.-H.E.; Yabe, Y.A.; Chung, W.K.; Yazawa, M. Dual Optical Recordings for Action Potentials and Calcium Handling in Induced Pluripotent Stem Cell Models of Cardiac Arrhythmias Using Genetically Encoded Fluorescent Indicators. Stem Cells Transl. Med. 2015, 4, 468–475. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Li, W.; Entcheva, E.; Li, Z. Microfluidics-Enabled 96-Well Perfusion System for High-Throughput Tissue Engineering and Long-Term All-Optical Electrophysiology. Lab Chip 2020, 20, 4031–4042. [Google Scholar] [CrossRef] [PubMed]
- Potthoff, M.J.; Wu, H.; Arnold, M.A.; Shelton, J.M.; Backs, J.; McAnally, J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Histone Deacetylase Degradation and MEF2 Activation Promote the Formation of Slow-Twitch Myofibers. J. Clin. Investig. 2007, 117, 2459–2467. [Google Scholar] [CrossRef] [Green Version]
- Jansen, K.; Pou Casellas, C.; Groenink, L.; Wever, K.E.; Masereeuw, R. Humans Are Animals, but Are Animals Human Enough? A Systematic Review and Meta-Analysis on Interspecies Differences in Renal Drug Clearance. Drug Discov. Today 2020, 25, 706–717. [Google Scholar] [CrossRef]
- Tanner, M.R.; Beeton, C. Differences in Ion Channel Phenotype and Function between Humans and Animal Models. Front. Biosci. 2018, 23, 43. [Google Scholar] [CrossRef] [Green Version]
- Zicha, S.; Moss, I.; Allen, B.; Varro, A.; Papp, J.; Dumaine, R.; Antzelevich, C.; Nattel, S. Molecular Basis of Species-Specific Expression of Repolarizing K+ Currents in the Heart. Am. J. Physiol.-Heart Circ. Physiol. 2003, 285, 1641–1649. [Google Scholar] [CrossRef] [Green Version]
- Boukens, B.J.; Rivaud, M.R.; Rentschler, S.; Coronel, R. Misinterpretation of the Mouse ECG: “Musing the Waves of Mus Musculus”. J. Physiol. 2014, 592, 4613–4626. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Xie, D.; Cao, X.; Yu, P.; Xing, X.; Chen, C.C.; Musselman, M.; Xie, M.; West, F.D.; Lewin, H.A.; et al. Comparative Epigenomic Annotation of Regulatory DNA. Cell 2012, 149, 1381–1392. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Lin, Y.; Nery, J.R.; Urich, M.A.; Breschi, A.; Davis, C.A.; Dobin, A.; Zaleski, C.; Beer, M.A.; Chapman, W.C.; et al. Comparison of the Transcriptional Landscapes between Human and Mouse Tissues. Proc. Natl. Acad. Sci. USA 2014, 111, 17224–17229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legato, M.J.; Leghe, J.K. Chapter 14—Gender and the Heart: Sex-Specific Differences in the Normal Myocardial Anatomy and Physiology. In Principles of Gender-Specific Medicine; Elsevier Inc.: Amsterdam, The Netherlands, 2010; pp. 151–161. [Google Scholar] [CrossRef]
- Hrdina, R.; Gersl, V.; Klimtová, I.; Simůnek, T.; Machácková, J.; Adamcová, M. Anthracycline-Induced Cardiotoxicity. Acta Medica (Hradec Kralove) 2000, 43, 75–82. [Google Scholar] [CrossRef] [Green Version]
- Robert, J. Preclinical Assessment of Anthracycline Cardiotoxicity in Laboratory Animals: Predictiveness and Pitfalls. Cell Biol. Toxicol. 2007, 23, 27–37. [Google Scholar] [CrossRef]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to Improve R&D Productivity: The Pharmaceutical Industry’s Grand Challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [CrossRef]
- Tiburcy, M.; Hudson, J.E.; Balfanz, P.; Schlick, S.; Meyer, T.; Chang Liao, M.-L.; Levent, E.; Raad, F.; Zeidler, S.; Wingender, E.; et al. Defined Engineered Human Myocardium With Advanced Maturation for Applications in Heart Failure Modeling and Repair. Circulation 2017, 135, 1832–1847. [Google Scholar] [CrossRef]
- Wang, G.; McCain, M.L.; Yang, L.; He, A.; Pasqualini, F.S.; Agarwal, A.; Yuan, H.; Jiang, D.; Zhang, D.; Zangi, L.; et al. Modeling the Mitochondrial Cardiomyopathy of Barth Syndrome with Induced Pluripotent Stem Cell and Heart-on-Chip Technologies. Nat. Med. 2014, 20, 616–623. [Google Scholar] [CrossRef]
- Mosqueira, D.; Mannhardt, I.; Bhagwan, J.R.; Lis-Slimak, K.; Katili, P.; Scott, E.; Hassan, M.; Prondzynski, M.; Harmer, S.C.; Tinker, A.; et al. CRISPR/Cas9 Editing in Human Pluripotent Stem Cell-Cardiomyocytes Highlights Arrhythmias, Hypocontractility, and Energy Depletion as Potential Therapeutic Targets for Hypertrophic Cardiomyopathy. Eur. Heart J. 2018, 39, 3879–3892. [Google Scholar] [CrossRef]
- Malan, D.; Zhang, M.; Stallmeyer, B.; Müller, J.; Fleischmann, B.K.; Schulze-Bahr, E.; Sasse, P.; Greber, B. Human IPS Cell Model of Type 3 Long QT Syndrome Recapitulates Drug-Based Phenotype Correction. Basic Res. Cardiol. 2016, 111, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burridge, P.W.; Li, Y.F.; Matsa, E.; Wu, H.; Ong, S.G.; Sharma, A.; Holmström, A.; Chang, A.C.; Coronado, M.J.; Ebert, A.D.; et al. Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes Recapitulate the Predilection of Breast Cancer Patients to Doxorubicin-Induced Cardiotoxicity. Nat. Med. 2016, 22, 547–556. [Google Scholar] [CrossRef] [Green Version]
- Tohyama, S.; Fujita, J.; Fujita, C.; Yamaguchi, M.; Kanaami, S.; Ohno, R.; Sakamoto, K.; Kodama, M.; Kurokawa, J.; Kanazawa, H.; et al. Efficient Large-Scale 2D Culture System for Human Induced Pluripotent Stem Cells and Differentiated Cardiomyocytes. Stem Cell Rep. 2017, 9, 1406–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldfracht, I.; Efraim, Y.; Shinnawi, R.; Kovalev, E.; Huber, I.; Gepstein, A.; Arbel, G.; Shaheen, N.; Tiburcy, M.; Zimmermann, W.H.; et al. Engineered Heart Tissue Models from HiPSC-Derived Cardiomyocytes and Cardiac ECM for Disease Modeling and Drug Testing Applications. Acta Biomater. 2019, 92, 145–159. [Google Scholar] [CrossRef]
- Herron, T.J. Calcium and Voltage Mapping in HiPSC-CM Monolayers. Cell Calcium 2016, 59, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, P.; Jackson, C.B.; Ozhathil, L.C.; Agarkova, I.; Galindo, C.L.; Sawyer, D.B.; Suter, T.M.; Zuppinger, C. 3D Co-Culture of HiPSC-Derived Cardiomyocytes with Cardiac Fibroblasts Improves Tissue-Like Features of Cardiac Spheroids. Front. Mol. Biosci. 2020, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Mannhardt, I.; Saleem, U.; Mosqueira, D.; Loos, M.F.; Ulmer, B.M.; Lemoine, M.D.; Larsson, C.; Améen, C.; de Korte, T.; Vlaming, M.L.H.; et al. Comparison of 10 Control HPSC Lines for Drug Screening in an Engineered Heart Tissue Format. Stem Cell Rep. 2020, 15, 983–998. [Google Scholar] [CrossRef]
- Arai, K.; Murata, D.; Takao, S.; Nakamura, A.; Itoh, M.; Kitsuka, T.; Nakayama, K. Drug Response Analysis for Scaffold-Free Cardiac Constructs Fabricated Using Bio-3D Printer. Sci. Rep. 2020, 10, 8972. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.L.; Xiang, Y.; Yu, C.; Pustelnik, J.; Wu, J.; Ma, X.; Matsui, T.; Imahashi, K.; Chen, S. Rapid 3D BioPrinting of a Human IPSC-Derived Cardiac Micro-Tissue for High-Throughput Drug Testing. Organs-on-a-Chip 2021, 3, 100007. [Google Scholar] [CrossRef]
- Wei, F.; Pourrier, M.; Strauss, D.G.; Stockbridge, N.; Pang, L. Effects of Electrical Stimulation on HiPSC-CM Responses to Classic Ion Channel Blockers. Toxicol. Sci. 2020, 174, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Gintant, G.; Burridge, P.; Gepstein, L.; Harding, S.; Herron, T.; Hong, C.; Jalife, J.; Wu, J.C. Use of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes in Preclinical Cancer Drug Cardiotoxicity Testing: A Scientific Statement from the American Heart Association. Circ. Res. 2019, 125, e75–e92. [Google Scholar] [CrossRef]
- Magdy, T.; Schuldt, A.J.T.; Wu, J.C.; Bernstein, D.; Burridge, P.W. Human Induced Pluripotent Stem Cell (HiPSC)-Derived Cells to Assess Drug Cardiotoxicity: Opportunities and Problems. Annu. Rev. Pharmacol. Toxicol. Rev. Pharmacol. Toxicol 2018, 58, 83–103. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Wu, J.C.; Wu, S.M. Induced Pluripotent Stem Cell-Derived Cardiomyocytes for Cardiovascular Disease Modeling and Drug Screening. Stem Cell Res. Ther. 2013, 4, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillette, T.; Hill, J. Readers, Writers and Erasers: Chromatin as the Whiteboard of Heart Disease. Circ. Res. 2015, 176, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Trotter, K.W.; Archer, T.K. The BRG1 Transcriptional Coregulator. Nucl. Recept. Signal. 2008, 6, e004. [Google Scholar] [CrossRef] [Green Version]
- Marmorstein, R.; Zhou, M.M. Writers and Readers of Histone Acetylation: Structure, Mechanism, and Inhibition. Cold Spring Harb. Perspect. Biol. 2014, 6, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Ohtani, K.; Zhao, C.; Dobreva, G.; Manavski, Y.; Kluge, B.; Braun, T.; Rieger, M.A.; Zeiher, A.M.; Dimmeler, S. Jmjd3 Controls Mesodermal and Cardiovascular Differentiation of Embryonic Stem Cells. Circ. Res. 2013, 113, 856–862. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, H.; Wang, L.; Liu, D.; Hill, J.A.; Liu, Z. The Histone Trimethyllysine Demethylase JMJD2A Promotes Cardiac Hypertrophy in Response to Hypertrophic Stimuli in Mice. J. Clin. 2011, 121, 2447–2456. [Google Scholar] [CrossRef] [Green Version]
- Hohl, M.; Wagner, M.; Reil, J.C.; Müller, S.A.; Tauchnitz, M.; Zimmer, A.M.; Lehmann, L.H.; Thiel, G.; Böhm, M.; Backs, J.; et al. HDAC4 Controls Histone Methylation in Response to Elevated Cardiac Load. J. Clin. Investig. 2013, 123, 1359–1370. [Google Scholar] [CrossRef] [Green Version]
- Lyons, D.B.; Lomvardas, S. Repressive Histone Methylation: A Case Study in Deterministic versus Stochastic Gene Regulation. Biochim. Biophys. Acta-Gene Regul. Mech. 2014, 1839, 1373–1384. [Google Scholar] [CrossRef]
- Greco, C.M.; Condorelli, G. Epigenetic Modifications and Noncoding RNAs in Cardiac Hypertrophy and Failure. Nat. Rev. Cardiol. 2015, 12, 488–497. [Google Scholar] [CrossRef]
- Eom, G.H.; Nam, Y.S.; Oh, J.G.; Choe, N.; Min, H.K.; Yoo, E.K.; Kang, G.; Nguyen, V.H.; Min, J.J.; Kim, J.K.; et al. Regulation of Acetylation of Histone Deacetylase 2 by P300/CBP-Associated Factor/Histone Deacetylase 5 in the Development of Cardiac Hypertrophy. Circ. Res. 2014, 114, 1133–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gusterson, R.J.; Jazrawi, E.; Adcock, I.M.; Latchman, D.S. The Transcriptional Co-Activators CREB-Binding Protein (CBP) and P300 Play a Critical Role in Cardiac Hypertrophy That Is Dependent on Their Histone Acetyltransferase Activity. J. Biol. Chem. 2003, 278, 6838–6847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shikama, N.; Lutz, W.; Kretzschmar, R.; Sauter, N.; Roth, J.F.; Marino, S.; Wittwer, J.; Scheidweiler, A.; Eckner, R. Essential Function of P300 Acetyltransferase Activity in Heart, Lung and Small Intestine Formation. EMBO J. 2003, 22, 5175–5185. [Google Scholar] [CrossRef] [Green Version]
- Yao, T.P.; Oh, S.P.; Fuchs, M.; Zhou, N.D.; Ch’ng, L.E.; Newsome, D.; Bronson, R.T.; Li, E.; Livingston, D.M.; Eckner, R. Gene Dosage-Dependent Embryonic Development and Proliferation Defects in Mice Lacking the Transcriptional Integrator P300. Cell 1998, 93, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine Acetylation Targets Protein Complexes and Co-Regulates Major Cellular Functions. Science 2009, 325, 834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milstone, Z.J.; Sherin, S.; Bourke, L.M.; Tomer, S.; Haynes, C.M.; Trivedi, C.M. Histone Deacetylases 1 and 2 Silence Cryptic Transcription to Promote Mitochondrial Function during Cardiogenesis. Sci. Adv. 2020, 6, eaax5150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, D.; Nomura, Y.; Nakano, M.; Han, M.; Bhatta, A.; Chen, K.; Akiyoshi, K.; Pandey, D. Endothelial-Specific Overexpression of Histone Deacetylase 2 Protects Mice against Endothelial Dysfunction and Atherosclerosis. Cell. Physiol. Biochem. 2020, 54, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, C.M.; Luo, Y.; Yin, Z.; Zhang, M.; Zhu, W.; Wang, T.; Floss, T.; Goettlicher, M.; Noppinger, P.R.; Wurst, W.; et al. Hdac2 Regulates the Cardiac Hypertrophic Response by Modulating Gsk3β Activity. Nat. Med. 2007, 13, 324–331. [Google Scholar] [CrossRef]
- Trivedi, C.M.; Min, M.L.; Wang, Q.; Epstein, J.A. Transgenic Overexpression of Hdac3 in the Heart Produces Increased Postnatal Cardiac Myocyte Proliferation but Does Not Induce Hypertrophy. J. Biol. Chem. 2008, 283, 26484–26489. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, R.L.; Potthoff, M.J.; Haberland, M.; Qi, X.; Matsuzaki, S.; Humphries, K.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Maintenance of Cardiac Energy Metabolism by Histone Deacetylase 3 in Mice. J. Clin. Investig. 2008, 118, 3588–3597. [Google Scholar] [CrossRef] [Green Version]
- Saito, S.; Zhuang, Y.; Suzuki, T.; Ota, Y.; Bateman, M.E.; Alkhatib, A.L.; Morris, G.F.; Lasky, J.A. HDAC8 Inhibition Ameliorates Pulmonary Fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L175–L186. [Google Scholar] [CrossRef]
- Zhang, L.X.; DeNicola, M.; Qin, X.; Du, J.; Ma, J.; Zhao, Y.T.; Zhuang, S.; Liu, P.Y.; Wei, L.; Qin, G.; et al. Specific Inhibition of HDAC4 in Cardiac Progenitor Cells Enhances Myocardial Repairs. Am. J. Physiol.-Cell Physiol. 2014, 307, 358–372. [Google Scholar] [CrossRef] [Green Version]
- Granger, A.; Abdullah, I.; Huebner, F.; Stout, A.; Wang, T.; Huebner, T.; Epstein, J.A.; Gruber, P.J. Histone Deacetylase Inhibition Reduces Myocardial Ischemia-Reperfusion Injury in Mice. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2008, 22, 3549–3560. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.; McKinsey, T.A.; Zhang, C.L.; Richardson, J.A.; Hill, J.A.; Olson, E.N. Histone Deacetylases 5 and 9 Govern Responsiveness of the Heart to a Subset of Stress Signals and Play Redundant Roles in Heart Development. Mol. Cell. Biol. 2004, 24, 8467–8476. [Google Scholar] [CrossRef] [Green Version]
- Hsu, A.; Duan, Q.; McMahon, S.; Huang, Y.; Wood, S.A.B.; Gray, N.S.; Wang, B.; Bruneau, B.G.; Haldar, S.M. Salt-Inducible Kinase 1 Maintains HDAC7 Stability to Promote Pathologic Cardiac Remodeling. J. Clin. Investig. 2020, 130, 2966–2977. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.L.; McKinsey, T.A.; Chang, S.; Antos, C.L.; Hill, J.A.; Olson, E.N. Class II Histone Deacetylases Act as Signal-Responsive Repressors of Cardiac Hypertrophy. Cell 2002, 110, 479–488. [Google Scholar] [CrossRef] [Green Version]
- Azghandi, S.; Prell, C.; Van Der Laan, S.W.; Schneider, M.; Malik, R.; Berer, K.; Gerdes, N.; Pasterkamp, G.; Weber, C.; Haffner, C.; et al. Deficiency of the Stroke Relevant HDAC9 Gene Attenuates Atherosclerosis in Accord with Allele-Specific Effects at 7p21.1. Stroke 2015, 46, 197–202. [Google Scholar] [CrossRef] [Green Version]
- Tao, H.; Yang, J.-J.; Hu, W.; Shi, K.-H.; Li, J. HDAC6 Promotes Cardiac Fibrosis Progression through Suppressing RASSF1A Expression. Cardiology 2016, 133, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Planavila, A.; Iglesias, R.; Giralt, M.; Villarroya, F. Sirt1 Acts in Association with PPARα to Protect the Heart from Hypertrophy, Metabolic Dysregulation, and Inflammation. Cardiovasc. Res. 2011, 90, 276–284. [Google Scholar] [CrossRef] [Green Version]
- Alcendor, R.R.; Gao, S.; Zhai, P.; Zablocki, D.; Holle, E.; Yu, X.; Tian, B.; Wagner, T.; Vatner, S.F.; Sadoshima, J. Sirt1 Regulates Aging and Resistance to Oxidative Stress in the Heart. Circ. Res. 2007, 100, 1512–1521. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Chen, X.F.; Wang, N.Y.; Wang, X.M.; Liang, S.T.; Zheng, W.; Lu, Y.B.; Zhao, X.; Hao, D.L.; Zhang, Z.Q.; et al. SIRT2 Acts as a Cardioprotective Deacetylase in Pathological Cardiac Hypertrophy. Circulation 2017, 136, 2051–2067. [Google Scholar] [CrossRef]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 Blocks the Cardiac Hypertrophic Response by Augmenting Foxo3a-Dependent Antioxidant Defense Mechanisms in Mice. J. Clin. Investig. 2009, 119, 2758–2771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koentges, C.; Pfeil, K.; Schnick, T.; Wiese, S.; Dahlbock, R.; Cimolai, M.C.; Meyer-Steenbuck, M.; Cenkerova, K.; Hoffmann, M.M.; Jaeger, C.; et al. SIRT3 Deficiency Impairs Mitochondrial and Contractile Function in the Heart. Basic Res. Cardiol. 2015, 110, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.X.; Tang, X.; An, X.Z.; Xie, X.M.; Chen, X.F.; Zhao, X.; Hao, D.L.; Chen, H.Z.; Liu, D.P. SIRT4 Accelerates Ang II-Induced Pathological Cardiac Hypertrophy by Inhibiting Manganese Superoxide Dismutase Activity. Eur. Heart J. 2017, 38, 1389–1398. [Google Scholar] [CrossRef] [Green Version]
- Sadhukhan, S.; Liu, X.; Ryu, D.; Nelson, O.D.; Stupinski, J.A.; Li, Z.; Chen, W.; Zhang, S.; Weiss, R.S.; Locasale, J.W.; et al. Metabolomics-Assisted Proteomics Identifies Succinylation and SIRT5 as Important Regulators of Cardiac Function. Proc. Natl. Acad. Sci. USA 2016, 113, 4320–4325. [Google Scholar] [CrossRef] [Green Version]
- Sundaresan, N.R.; Vasudevan, P.; Zhong, L.; Kim, G.; Samant, S.; Parekh, V.; Pillai, V.B.; Ravindra, P.V.; Gupta, M.; Jeevanandam, V.; et al. The Sirtuin SIRT6 Blocks IGF-Akt Signaling and Development of Cardiac Hypertrophy by Targeting c-Jun. Nat. Med. 2012, 18, 1643–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.X.; Wang, X.L.; Tong, M.M.; Gan, L.; Chen, H.; Wu, S.S.; Chen, J.X.; Li, R.L.; Wu, Y.; Zhang, H.Y.; et al. SIRT6 Protects Cardiomyocytes against Ischemia/Reperfusion Injury by Augmenting FoxO3α-Dependent Antioxidant Defense Mechanisms. Basic Res. Cardiol. 2016, 111, 1–19. [Google Scholar] [CrossRef]
- Vakhrusheva, O.; Smolka, C.; Gajawada, P.; Kostin, S.; Boettger, T.; Kubin, T.; Braun, T.; Bober, E. Sirt7 Increases Stress Resistance of Cardiomyocytes and Prevents Apoptosis and Inflammatory Cardiomyopathy in Mice. Circ. Res. 2008, 102, 703–710. [Google Scholar] [CrossRef] [Green Version]
- Fischle, W.; Dequiedt, F.; Hendzel, M.J.; Guenther, M.G.; Lazar, M.A.; Voelter, W.; Verdin, E. Enzymatic Activity Associated with Class II HDACs Is Dependent on a Multiprotein Complex Containing HDAC3 and SMRT/N-CoR. Mol. Cell 2002, 9, 45–57. [Google Scholar] [CrossRef] [Green Version]
- Mihaylova, M.M.; Vasquez, D.S.; Ravnskjaer, K.; Denechaud, P.-D.; Yu, R.T.; Alvarez, J.G.; Downes, M.; Evans, R.M.; Montminy, M.; Shaw, R.J. Class IIa Histone Deacetylases Are Hormone-Activated Regulators of FOXO and Mammalian Glucose Homeostasis. Cell 2011, 145, 607–621. [Google Scholar] [CrossRef] [Green Version]
- Abraham, W.T.; Gilbert, E.M.; Lowes, B.D.; Minobe, W.A.; Larrabee, P.; Roden, R.L.; Dutcher, D.; Sederberg, J.; Lindenfeld, J.A.; Wolfel, E.E.; et al. Coordinate Changes in Myosin Heavy Chain Isoform Gene Expression Are Selectively Associated with Alterations in Dilated Cardiomyopathy Phenotype. Mol. Med. 2002, 8, 750–760. [Google Scholar] [CrossRef] [Green Version]
- Krenz, M.; Robbins, J. Impact of Beta-Myosin Heavy Chain Expression on Cardiac Function during Stress. J. Am. Coll. Cardiol. 2004, 44, 2390–2397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, J.M.; Wang, D.J.; Shettigar, V.; Roof, S.R.; Canan, B.D.; Bakkar, N.; Shintaku, J.; Jin-Mo, G.; Little, S.C.; Ratnam, N.M.; et al. NF-κB Inhibition Rescues Cardiac Function by Remodeling Calcium Genes in a Duchenne Muscular Dystrophy Model. Nat. Commun. 2018, 9, 3431. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.G.; Wang, S.H.; Mani, S.K.; Kasiganesan, H.; Chou, C.J.; Menick, D.R. Evidence for a Non-Canonical Role of HDAC5 in Regulation of the Cardiac Ncx1 and Bnp Genes. Nucleic Acids Res. 2016, 44, 3610–3617. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, S.; Peterson, R.E.; Mani, S.K.; Addy, B.; Buchholz, A.L.; Xu, L.; Thiyagarajan, T.; Kasiganesan, H.; Kern, C.B.; Menick, D.R. Histone Deacetylases Facilitate Sodium/Calcium Exchanger up-Regulation in Adult Cardiomyocytes. FASEB J. 2009, 23, 3851–3864. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, L.H.; Jebessa, Z.H.; Kreusser, M.M.; Horsch, A.; He, T.; Kronlage, M.; Dewenter, M.; Sramek, V.; Oehl, U.; Krebs-Haupenthal, J.; et al. A Proteolytic Fragment of Histone Deacetylase 4 Protects the Heart from Failure by Regulating the Hexosamine Biosynthetic Pathway. Nat. Med. 2018, 24, 62–72. [Google Scholar] [CrossRef]
- Gupta, M.P.; Samant, S.A.; Smith, S.H.; Shroff, S.G. HDAC4 and PCAF Bind to Cardiac Sarcomeres and Play a Role in Regulating Myofilament Contractile Activity. J. Biol. Chem. 2008, 283, 10135–10146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, D.B.; Liu, T.; Rucker, J.; O’Meally, R.N.; Devine, L.R.; Cole, R.N.; O’Rourke, B. The Cardiac Acetyl-Lysine Proteome. PLoS ONE 2013, 8, e67513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, R.M.; Fay, A.J.; Puthenveedu, M.A.; von Zastrow, M.; Jan, Y.-N.; Jan, L.Y. Microtubule Plus-End-Tracking Proteins Target Gap Junctions Directly from the Cell Interior to Adherens Junctions. Cell 2007, 128, 547–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, D.; Kuhn, C.; Katus, H.A.; Frey, N. The Sarcomeric Z-Disc: A Nodal Point in Signalling and Disease. J. Mol. Med. 2006, 84, 446. [Google Scholar] [CrossRef]
- Zhang, X.; Yuan, Z.; Zhang, Y.; Yong, S.; Salas-Burgos, A.; Koomen, J.; Olashaw, N.; Parsons, J.T.; Yang, X.-J.; Dent, S.R.; et al. HDAC6 Modulates Cell Motility by Altering the Acetylation Level of Cortactin. Mol. Cell 2007, 27, 197–213. [Google Scholar] [CrossRef] [Green Version]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 Is a Microtubule-Associated Deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef]
- Zhang, D.; Wu, C.T.; Qi, X.Y.; Meijering, R.A.M.; Hoogstra-Berends, F.; Tadevosyan, A.; Deniz, G.C.; Durdu, S.; Akar, A.R.; Sibon, O.C.M.; et al. Activation of Histone Deacetylase-6 Induces Contractile Dysfunction through Derailment of α-Tubulin Proteostasis in Experimental and Human Atrial Fibrillation. Circulation 2014, 129, 346–358. [Google Scholar] [CrossRef] [Green Version]
- Sequeira, V.; Nijenkamp, L.L.A.M.; Regan, J.A.; van der Velden, J. The Physiological Role of Cardiac Cytoskeleton and Its Alterations in Heart Failure. Biochim. Biophys. Acta-Biomembr. 2014, 1838, 700–722. [Google Scholar] [CrossRef] [Green Version]
- Hein, S.; Kostin, S.; Heling, A.; Maeno, Y.; Schaper, J. The Role of the Cytoskeleton in Heart Failure. Cardiovasc. Res. 2000, 45, 273–278. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Yung, A.; Covarrubias, M.; Radice, G.L. Cortactin Is Required for N-Cadherin Regulation of Kv1.5 Channel Function. J. Biol. Chem. 2011, 286, 20478–20489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brundel, B.J.J.M.; Li, J.; Zhang, D. Role of HDACs in Cardiac Electropathology: Therapeutic Implications for Atrial Fibrillation. Biochim. Biophys. Acta-Mol. Cell Res. 2020, 1867, 118459. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.L.; Davis, C.A.; Potthoff, M.J.; Haberland, M.; Fielitz, J.; Qi, X.; Hill, J.A.; Richardson, J.A.; Olson, E.N. Histone Deacetylases 1 and 2 Redundantly Regulate Cardiac Morphogenesis, Growth, and Contractility. Genes Dev. 2007, 21, 1790–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antos, C.L.; McKinsey, T.A.; Dreitz, M.; Hollingsworth, L.M.; Zhang, C.L.; Schreiber, K.; Rindt, H.; Gorczynski, R.J.; Olson, E.N. Dose-Dependent Blockade to Cardiomyocyte Hypertrophy by Histone Deacetylase Inhibitors. J. Biol. Chem. 2003, 278, 28930–28937. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.; Tannous, P.; Lu, G.; Berenji, K.; Rothermel, B.A.; Olson, E.N.; Hill, J.A. Suppression of Class I and II Histone Deacetylases Blunts Pressure-Overload Cardiac Hypertrophy. Circulation 2006, 113, 2579–2588. [Google Scholar] [CrossRef]
- Kee, H.J.; Sohn, I.S.; Nam, K.I.; Park, J.E.; Qian, Y.R.; Yin, Z.; Ahn, Y.; Jeong, M.H.; Bang, Y.J.; Kim, N.; et al. Inhibition of Histone Deacetylation Blocks Cardiac Hypertrophy Induced by Angiotensin II Infusion and Aortic Banding. Circulation 2006, 113, 51–59. [Google Scholar] [CrossRef]
- Gallo, P.; Latronico, M.V.G.; Gallo, P.; Grimaldi, S.; Borgia, F.; Todaro, M.; Jones, P.; Gallinari, P.; De Francesco, R.; Ciliberto, G.; et al. Inhibition of Class I Histone Deacetylase with an Apicidin Derivative Prevents Cardiac Hypertrophy and Failure. Cardiovasc. Res. 2008, 80, 416–424. [Google Scholar] [CrossRef]
- Cao, D.J.; Wang, Z.V.; Battiprolu, P.K.; Jiang, N.; Morales, C.R.; Kong, Y.; Rothermel, B.A.; Gillette, T.G.; Hill, J.A. Histone Deacetylase (HDAC) Inhibitors Attenuate Cardiac Hypertrophy by Suppressing Autophagy. Proc. Natl. Acad. Sci. USA 2011, 108, 4123–4128. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Singh, N.; Mullican, S.E.; Everett, L.J.; Li, L.; Yuan, L.; Liu, X.; Epstein, J.A.; Lazar, M.A. Diet-Induced Lethality Due to Deletion of the Hdac3 Gene in Heart and Skeletal Muscle. J. Biol. Chem. 2011, 286, 33301–33309. [Google Scholar] [CrossRef] [Green Version]
- McKinsey, T.A.; Olson, E.N. Cardiac Histone Acetylation–Therapeutic Opportunities Abound. Trends Genet. 2004, 20, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Ago, T.; Liu, T.; Zhai, P.; Chen, W.; Li, H.; Molkentin, J.D.; Vatner, S.F.; Sadoshima, J. A Redox-Dependent Pathway for Regulating Class II HDACs and Cardiac Hypertrophy. Cell 2008, 133, 978–993. [Google Scholar] [CrossRef] [Green Version]
- McKinsey, T.A. The Biology and Therapeutic Implications of HDACs in the Heart. In Histone Deacetylases: The Biology and Clinical Implication; Yao, T.-P., Seto, E., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 57–78. ISBN 978-3-642-21631-2. [Google Scholar]
- Grant, A.O. Cardiac Ion Channels. Circ. Arrhythmia Electrophysiol. 2009, 2, 185–194. [Google Scholar] [CrossRef] [Green Version]
- Babiarz, J.E.; Ravon, M.; Sridhar, S.; Ravindran, P.; Swanson, B.; Bitter, H.; Weiser, T.; Chiao, E.; Certa, U.; Kolaja, K.L. Determination of the Human Cardiomyocyte MRNA and MiRNA Differentiation Network by Fine-Scale Profiling. Stem Cells Dev. 2012, 21, 1956–1965. [Google Scholar] [CrossRef] [Green Version]
- Gupta, M.K.; Illich, D.J.; Gaarz, A.; Matzkies, M.; Nguemo, F.; Pfannkuche, K.; Liang, H.; Classen, S.; Reppel, M.; Schultze, J.L.; et al. Global Transcriptional Profiles of Beating Clusters Derived from Human Induced Pluripotent Stem Cells and Embryonic Stem Cells Are Highly Similar. BMC Dev. Biol. 2010, 10, 98. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Guo, L.; Fiene, S.J.; Anson, B.D.; Thomson, J.A.; Kamp, T.J.; Kolaja, K.L.; Swanson, B.J.; January, C.T. High Purity Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes: Electrophysiological Properties of Action Potentials and Ionic Currents. Am. J. Physiol. Circ. Physiol. 2011, 301, H2006–H2017. [Google Scholar] [CrossRef]
- Li, M.; Kanda, Y.; Ashihara, T.; Sasano, T.; Nakai, Y.; Kodama, M.; Hayashi, E.; Sekino, Y.; Furukawa, T.; Kurokawa, J. Overexpression of KCNJ2 in Induced Pluripotent Stem Cell-Derived Cardiomyocytes for the Assessment of QT-Prolonging Drugs. J. Pharmacol. Sci. 2017, 134, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Sedighi, A.; Wang, L. Microarray Technology: Methods and Applications; Methods in Molecular Biology; Springer: Berlin/Heidelberg, Germany, 2016; Volume 1368. [Google Scholar] [CrossRef]
- Ahmed, R.E.; Anzai, T.; Chanthra, N.; Uosaki, H. A Brief Review of Current Maturation Methods for Human Induced Pluripotent Stem Cells-Derived Cardiomyocytes. Front. Cell Dev. Biol. 2020, 8, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, A.C.; Johnstone, R.W. New and Emerging HDAC Inhibitors for Cancer Treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, 1–34. [Google Scholar] [CrossRef] [Green Version]
- Ho, T.C.S.; Chan, A.H.Y.; Ganesan, A. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem. 2020, 63, 12460–12484. [Google Scholar] [CrossRef] [PubMed]
- Szyf, M. Epigenetic Therapeutics in Autoimmune Disease. Clin. Rev. Allergy Immunol. 2010, 39, 62–77. [Google Scholar] [CrossRef] [PubMed]
- Richon, V.M.; Emiliani, S.; Verdin, E.; Webb, Y.; Breslow, R.; Rifkind, R.A.; Marks, P.A. A Class of Hybrid Polar Inducers of Transformed Cell Differentiation Inhibits Histone Deacetylases. Proc. Natl. Acad. Sci. USA 1998, 95, 3003–3007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greshock, T.J.; Johns, D.M.; Noguchi, Y.; Williams, R.M. Improved Total Synthesis of the Potent HDAC Inhibitor FK228 (FR-901228). Org. Lett. 2008, 10, 613–616. [Google Scholar] [CrossRef] [Green Version]
- Plumb, J.A.; Finn, P.W.; Williams, R.J.; Bandara, M.J.; Romero, M.R.; Watkins, C.J.; La Thangue, N.B.; Brown, R. Pharmacodynamic Response and Inhibition of Growth of Human Tumor Xenografts by the Novel Histone Deacetylase Inhibitor PXD101. Mol. Cancer Ther. 2003, 2, 721–728. [Google Scholar]
- George, P.; Bali, P.; Annavarapu, S.; Scuto, A.; Fiskus, W.; Guo, F.; Sigua, C.; Sondarva, G.; Moscinski, L.; Atadja, P.; et al. Combination of the Histone Deacetylase Inhibitor LBH589 and the Hsp90 Inhibitor 17-AAG Is Highly Active against Human CML-BC Cells and AML Cells with Activating Mutation of FLT-3. Blood 2005, 105, 1768–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiattarella, G.G.; Sannino, A.; Toscano, E.; Cattaneo, F.; Trimarco, B.; Esposito, G.; Perrino, C. Cardiovascular Effects of Histone Deacetylase Inhibitors Epigenetic Therapies: Systematic Review of 62 Studies and New Hypotheses for Future Research. Int. J. Cardiol. 2016, 219, 396–403. [Google Scholar] [CrossRef]
- Albini, A.; Pennesi, G.; Donatelli, F.; Cammarota, R.; De Flora, S.; Noonan, D.M. Cardiotoxicity of Anticancer Drugs: The Need for Cardio-Oncology and Cardio-Oncological Prevention. J. Natl. Cancer Inst. 2010, 102, 14–25. [Google Scholar] [CrossRef] [Green Version]
- Ferdinandy, P.; Baczkó, I.; Bencsik, P.; Giricz, Z.; Görbe, A.; Pacher, P.; Varga, Z.V.; Varró, A.; Schulz, R. Definition of Hidden Drug Cardiotoxicity: Paradigm Change in Cardiac Safety Testing and Its Clinical Implications. Eur. Heart J. 2019, 40, 1771–1777. [Google Scholar] [CrossRef]
- Kopljar, I.; Gallacher, D.; De Bondt, A.; Cougnaud, L.; Vlaminckx, E.; Van Den Wyngaert, I.; Lu, H. Functional and Transcriptional Characterization of Histone Deacetylase Inhibitor-Mediated Cardiac Adverse Effects in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Stem Cells Transl. Med. 2016, 5, 505–509. [Google Scholar] [CrossRef] [Green Version]
- Banik, D.; Moufarrij, S.; Villagra, A. Immunoepigenetics Combination Therapies: An Overview of the Role of HDACs in Cancer Immunotherapy. Int. J. Mol. Sci. 2019, 20, 2241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, L.H.; Worst, B.C.; Stanmore, D.A.; Backs, J. Histone Deacetylase Signaling in Cardioprotection. Cell. Mol. Life Sci. 2014, 71, 1673–1690. [Google Scholar] [CrossRef] [Green Version]
- McKinsey, T.A. Therapeutic Potential for HDAC Inhibitors in the Heart. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 303–319. [Google Scholar] [CrossRef] [PubMed]
- Kook, H.; Lepore, J.J.; Gitler, A.D.; Lu, M.M.; Yung, W.W.-M.; Mackay, J.; Zhou, R.; Ferrari, V.; Gruber, P.; Epstein, J.A. Cardiac Hypertrophy and Histone Deacetylase-Dependent Transcriptional Repression Mediated by the Atypical Homeodomain Protein Hop. J. Clin. Investig. 2003, 112, 863–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.-M.; Lin, M.-S.; Chang, N.-C. Inhibition of Histone Deacetylase on Ventricular Remodeling in Infarcted Rats. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H968–H977. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.C.; Cheng, G.; Zhang, L.X.; Tseng, Y.T.; Padbury, J.F. Inhibition of Histone Deacetylases Triggers Pharmacologic Preconditioning Effects against Myocardial Ischemic Injury. Cardiovasc. Res. 2007, 76, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.; Fenning, A.; Lim, J.; Le, G.T.; Reid, R.C.; Halili, M.A.; Fairlie, D.P.; Brown, L. Antifibrotic Activity of an Inhibitor of Histone Deacetylases in DOCA-Salt Hypertensive Rats. Br. J. Pharmacol. 2010, 159, 1408–1417. [Google Scholar] [CrossRef] [Green Version]
- Cardinale, J.P.; Sriramula, S.; Pariaut, R.; Guggilam, A.; Mariappan, N.; Elks, C.M.; Francis, J. HDAC Inhibition Attenuates Inflammatory, Hypertrophic, and Hypertensive Responses in Spontaneously Hypertensive Rats. Hypertension 2010, 56, 437–444. [Google Scholar] [CrossRef] [Green Version]
- Rothermel, B.A.; Hill, J.A. Autophagy in Load-Induced Heart Disease. Circ. Res. 2008, 103, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Wallner, M.; Eaton, D.M.; Berretta, R.M.; Liesinger, L.; Schittmayer, M.; Gindlhuber, J.; Wu, J.; Jeong, M.Y.; Lin, Y.H.; Borghetti, G.; et al. HDAC Inhibition Improves Cardiopulmonary Function in a Feline Model of Diastolic Dysfunction. Sci. Transl. Med. 2020, 12, eaay7205. [Google Scholar] [CrossRef]
- Woosley, R.; Heise, C.; Gallo, T.; Tate, J.; Woosley, D.; Romero, K. QTdrugs List. Available online: www.CredibleMeds.org (accessed on 22 November 2021).
- Klein, D.C.; Hainer, S.J. Genomic Methods in Profiling DNA Accessibility and Factor Localization. Chromosome Res. 2019, 28, 69–85. [Google Scholar] [CrossRef] [Green Version]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of Native Chromatin for Fast and Sensitive Epigenomic Profiling of Open Chromatin, DNA-Binding Proteins and Nucleosome Position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-Seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 109, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Albert, I.; Mavrich, T.N.; Tomsho, L.P.; Qi, J.; Zanton, S.J.; Schuster, S.C.; Pugh, B.F. Translational and Rotational Settings of H2A.Z Nucleosomes across the Saccharomyces cerevisiae Genome. Nature 2007, 446, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Gilmour, D.S.; Lis, J.T. Detecting protein-DNA interactions in vivo: Distribution of RNA polymerase on specific bacterial genes. Proc. Natl. Acad. Sci. USA 1984, 81, 4275–4279. [Google Scholar] [CrossRef] [Green Version]
- Solomon, M.J.; Varshavsky, A. Formaldehyde-Mediated DNA-Protein Crosslinking: A Probe for in Vivo Chromatin Structures. Proc. Natl. Acad. Sci. USA 1985, 82, 6470–6474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Towbin, H.; Staehelin, T.; Gordon, J. Electrophoretic Transfer of Proteins from Polyacrylamide Gels to Nitrocellulose Sheets: Procedure and Some Applications. Proc. Natl. Acad. Sci. USA 1979, 76, 4350–4354. [Google Scholar] [CrossRef] [Green Version]
- Burnette, W.N. “Western Blotting”: Electrophoretic Transfer of Proteins from Sodium Dodecyl Sulfate-Polyacrylamide Gels to Unmodified Nitrocellulose and Radiographic Detection with Antibody and Radioiodinated Protein A. Anal. Biochem. 1981, 112, 195–203. [Google Scholar] [CrossRef]
- Li, W.; Han, J.L.; Entcheva, E. Syncytium Cell Growth Increases Kir2.1 Contribution in Human IPSC-Cardiomyocytes. Am. J. Physiol. Circ. Physiol. 2020, 319, H1112–H1122. [Google Scholar] [CrossRef] [PubMed]
- Benaglio, P.; D’Antonio-Chronowska, A.; Ma, W.; Yang, F.; Young Greenwald, W.W.; Donovan, M.K.R.; DeBoever, C.; Li, H.; Drees, F.; Singhal, S.; et al. Allele-Specific NKX2-5 Binding Underlies Multiple Genetic Associations with Human Electrocardiographic Traits. Nat. Genet. 2019, 51, 1506–1517. [Google Scholar] [CrossRef]
- Higuchi, R.; Fockler, C.; Dollinger, G.; Watson, R. Kinetic PCR Analysis: Real-Time Monitoring of DNA Amplification Reactions. Bio/Technology 1993, 11, 1026–1030. [Google Scholar] [CrossRef]
- Heid, C.A.; Stevens, J.; Livak, K.J.; Williams, P.M. Real Time Quantitative PCR. Genome Res. 1996, 6, 986–994. [Google Scholar] [CrossRef] [Green Version]
- Xia, C.; Fan, J.; Emanuel, G.; Hao, J.; Zhuang, X. Spatial Transcriptome Profiling by MERFISH Reveals Subcellular RNA Compartmentalization and Cell Cycle-Dependent Gene Expression. Proc. Natl. Acad. Sci. USA 2019, 116, 19490–19499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. MRNA-Seq Whole-Transcriptome Analysis of a Single Cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, G.T.; Chaudhary, K.W.; Atwater, N.; Nguyen, C.; Brown, B.S.; McNeish, J.D.; Cohen, A.E.; Kralj, J.M. Cardiotoxicity Screening with Simultaneous Optogenetic Pacing, Voltage Imaging and Calcium Imaging. J. Pharmacol. Toxicol. Methods 2016, 81, 240–250. [Google Scholar] [CrossRef]
- Werley, C.A.; Chien, M.-P.; Cohen, A.E. Ultrawidefield Microscope for High-Speed Fluorescence Imaging and Targeted Optogenetic Stimulation. Biomed. Opt. Express 2017, 8, 5794–5813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Araki, S.; Wu, J.; Teramoto, T.; Chang, Y.-F.; Nakano, M.; Abdelfattah, A.S.; Fujiwara, M.; Ishihara, T.; Nagai, T.; et al. An Expanded Palette of Genetically Encoded Ca2+ Indicators. Science 2011, 333, 1888–1891. [Google Scholar] [CrossRef] [Green Version]
- Dana, H.; Mohar, B.; Sun, Y.; Narayan, S.; Gordus, A.; Hasseman, J.P.; Tsegaye, G.; Holt, G.T.; Hu, A.; Walpita, D.; et al. Sensitive Red Protein Calcium Indicators for Imaging Neural Activity. Elife 2016, 5, e12727. [Google Scholar] [CrossRef]
- Nagel, G.; Szellas, T.; Huhn, W.; Kateriya, S.; Adeishvili, N.; Berthold, P.; Ollig, D.; Hegemann, P.; Bamberg, E. Channelrhodopsin-2, a Directly Light-Gated Cation-Selective Membrane Channel. Proc. Natl. Acad. Sci. USA 2003, 100, 13940–13945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagel, G.; Brauner, M.; Liewald, J.F.; Adeishvili, N.; Bamberg, E.; Gottschalk, A. Light Activation of Channelrhodopsin-2 in Excitable Cells of Caenorhabditis Elegans Triggers Rapid Behavioral Responses. Curr. Biol. 2005, 15, 2279–2284. [Google Scholar] [CrossRef] [Green Version]
- Hochbaum, D.R.; Zhao, Y.; Farhi, S.L.; Klapoetke, N.; Werley, C.A.; Kapoor, V.; Zou, P.; Kralj, J.M.; Maclaurin, D.; Smedemark-Margulies, N.; et al. All-Optical Electrophysiology in Mammalian Neurons Using Engineered Microbial Rhodopsins. Nat. Methods 2014, 11, 825–833. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.-L.; Walker, A.S.; Miller, E.W. A Photostable Silicon Rhodamine Platform for Optical Voltage Sensing. J. Am. Chem. Soc. 2015, 137, 10767–10776. [Google Scholar] [CrossRef] [Green Version]
- Matiukas, A.; Mitrea, B.G.; Qin, M.; Pertsov, A.M.; Shvedko, A.G.; Warren, M.D.; Zaitsev, A.V.; Wuskell, J.P.; Wei, M.; Watras, J.; et al. Near-Infrared Voltage-Sensitive Fluorescent Dyes Optimized for Optical Mapping in Blood-Perfused Myocardium. Heart Rhythm 2007, 4, 1441–1451. [Google Scholar] [CrossRef] [Green Version]
- Haase, A.; Kohrn, T.; Fricke, V.; Ricci Signorini, M.E.; Witte, M.; Göhring, G.; Gruh, I.; Martin, U. Establishment of MHHi001-A-5, a GCaMP6f and RedStar dual reporter human iPSC line for in vitro and in vivo characteri-zation and in situ tracing of iPSC derivatives. Stem Cell Res. 2021, 52, 102206. [Google Scholar] [CrossRef]
- Björk, S.; Ojala, E.A.; Nordström, T.; Ahola, A.; Liljeström, M.; Hyttinen, J.; Kankuri, E.; Mervaala, E. Evaluation of Optogenetic Electrophysiology Tools in Human Stem Cell-Derived Cardiomyocytes. Front. Physiol. 2017, 8, 884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.-F.; Broyles, C.N.; Brook, F.A.; Davies, M.J.; Turtle, C.W.; Nagai, T.; Daniels, M.J. Non-Invasive Phenotyping and Drug Testing in Single Cardiomyocytes or Beta-Cells by Calcium Imaging and Optogenetics. PLoS ONE 2017, 12, e0174181. [Google Scholar] [CrossRef] [PubMed]
- Paci, M.; Passini, E.; Klimas, A.; Severi, S.; Hyttinen, J.; Rodriguez, B.; Entcheva, E. All-Optical Electrophysiology Refines Populations of In Silico Human IPSC-CMs for Drug Evaluation. Biophys. J. 2020, 118, 2596–2611. [Google Scholar] [CrossRef]
- Greenwald, W.W.; Li, H.; Benaglio, P.; Jakubosky, D.; Matsui, H.; Schmitt, A.; Selvaraj, S.; D’Antonio, M.; D’Antonio-Chronowska, A.; Smith, E.N.; et al. Subtle Changes in Chromatin Loop Contact Propensity Are Associated with Differential Gene Regulation and Expression. Nat. Commun. 2019, 10, 1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, M.C.; Banovich, N.E.; Sarkar, A.; Stephens, M.; Gilad, Y. Dynamic Effects of Genetic Variation on Gene Expression Revealed Following Hypoxic Stress in Cardiomyocytes. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.Y.; Sivakumaran, P.; Crombie, D.E.; Dusting, G.J.; Pébay, A.; Dilley, R.J. Trichostatin A Enhances Differentiation of Human Induced Pluripotent Stem Cells to Cardiogenic Cells for Cardiac Tissue Engineering. Stem Cells Transl. Med. 2013, 2, 715–725. [Google Scholar] [CrossRef]
- Otsuji, T.G.; Kurose, Y.; Suemori, H.; Tada, M.; Nakatsuji, N. Dynamic Link between Histone H3 Acetylation and an Increase in the Functional Characteristics of Human ESC/IPSC-Derived Cardiomyocytes. PLoS ONE 2012, 7, e45010. [Google Scholar] [CrossRef] [Green Version]
- Kopljar, I.; De Bondt, A.; Vinken, P.; Teisman, A.; Damiano, B.; Goeminne, N.; Van den Wyngaert, I.; Gallacher, D.J.; Lu, H.R. Chronic Drug-Induced Effects on Contractile Motion Properties and Cardiac Biomarkers in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Br. J. Pharmacol. 2017, 174, 3766–3779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; Patel, D.; Zhang, X.; Veenstra, R.D. Changes in Cardiac NAV1.5 Expression, Function, and Acetylation by Pan-Histone Deacetylase Inhibitors. Am. J. Physiol.-Heart Circ. Physiol. 2016, 311, H1139–H1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, N.; Liu, Z.; Chen, Z.; Wang, J.; Chen, T.; Zhao, X.; Ma, Y.; Qin, L.; Kang, J.; Wei, B.; et al. Ascorbic Acid Enhances the Cardiac Differentiation of Induced Pluripotent Stem Cells through Promoting the Proliferation of Cardiac Progenitor Cells. Cell Res. 2012, 22, 219–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geuss, L.R.; Suggs, L.J. Making Cardiomyocytes: How Mechanical Stimulation Can Influence Differentiation of Pluripotent Stem Cells. Biotechnol. Prog. 2013, 29, 1089–1096. [Google Scholar] [CrossRef]
- Nunes, S.S.; Miklas, J.W.; Liu, J.; Aschar-Sobbi, R.; Xiao, Y.; Zhang, B.; Jiang, J.; Massé, S.; Gagliardi, M.; Hsieh, A.; et al. Biowire: A Platform for Maturation of Human Pluripotent Stem Cell–Derived Cardiomyocytes. Nat. Methods 2013, 10, 781–787. [Google Scholar] [CrossRef] [Green Version]
- Fan, D.; Takawale, A.; Basu, R.; Patel, V.; Lee, J.; Kandalam, V.; Wang, X.; Oudit, G.Y.; Kassiri, Z. Differential Role of TIMP2 and TIMP3 in Cardiac Hypertrophy, Fibrosis, and Diastolic Dysfunction. Cardiovasc. Res. 2014, 103, 268–280. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Petrashevskaya, N.; Ren, S.; Zhao, A.; Chakir, K.; Gao, E.; Chuprun, J.K.; Wang, Y.; Talan, M.; Dorn, G.W.; et al. Gi-Biased Β2AR Signaling Links GRK2 Upregulation to Heart Failure. Circ. Res. 2012, 110, 265–274. [Google Scholar] [CrossRef] [Green Version]
- Maillet, M.; Purcell, N.H.; Sargent, M.A.; York, A.J.; Bueno, O.F.; Molkentin, J.D. DUSP6 (MKP3) Null Mice Show Enhanced ERK1/2 Phosphorylation at Baseline and Increased Myocyte Proliferation in the Heart Affecting Disease Susceptibility. J. Biol. Chem. 2008, 283, 31246–31255. [Google Scholar] [CrossRef] [Green Version]
- Bostrom, P.; Mann, N.; Wu, J.; Quintero, P.; Plovie, E.; Panakova, D.; Gupta, R.K.; Xiao, C.; Macrae, C.A.; Rosenzweig, A.; et al. C/EBPB Controls Exercise-Induced Cardiac Growth and Protects against Pathological Cardiac Remodeling. Cell 2011, 143, 617–632. [Google Scholar] [CrossRef]
- Bovill, E.; Westaby, S.; Reji, S.; Sayeed, R.; Crisp, A.; Shaw, T. Induction by Left Ventricular Overload and Left Ventricular Failure of the Human Jumonji Gene (JARID2) Encoding a Protein That Regulates Transcription and Reexpression of a Protective Fetal Program. J. Thorac. Cardiovasc. Surg. 2008, 136, 709–716. [Google Scholar] [CrossRef] [Green Version]
- Sheikh-Hamad, D.; Bick, R.; Wu, G.Y.; Christensen, B.M.; Razeghi, P.; Poindexter, B.; Taegtmeyer, H.; Wamsley, A.; Padda, R.; Entman, M.; et al. Stanniocalcin-1 Is a Naturally Occurring L-Channel Inhibitor in Cardiomyocytes: Relevance to Human Heart Failure. Am. J. Physiol.-Heart Circ. Physiol. 2003, 285, 442–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wang, H.; Chen, J.; Wang, X.; Sun, K.; Wang, Y.; Wang, J.; Yang, X.; Song, X.; Xin, Y.; et al. GADD45B Inhibits MKK7-Induced Cardiac Hypertrophy and the Polymorphisms of GADD45B Is Associated with Inter-Ventricular Septum Hypertrophy. Biochem. Biophys. Res. Commun. 2008, 372, 623–628. [Google Scholar] [CrossRef] [PubMed]






| Class | Gene | Chr | Subcellular Localization | Heart Exp | Known Effects on TF | Known Action | Known Cardiac Involvement |
|---|---|---|---|---|---|---|---|
| I | HDAC1 | 1 | nucleus | low | NF-kb, KLF5, YY1, NKX2.5, NR1D2, PER1 | H2A, H2B, H3, H4 | Promotes cardiogenesis [63] |
| HDAC2 | 6 | nucleus | high | YY1, KLF4, CRY1 | H2A, H2B, H3, H4 | Promotes cardiogenesis [63], aids in atherosclerosis models [64], KO increases resistance to hypertrophy [65] | |
| HDAC3 | 5 | nucleus, cytoplasm (shuttles between) | medium | NKX2.5, TBX5, PRARa, YY1, ARNTL/BMAL1-CRY1 | H23K27, H3, H4 | Promotes cardiomyocyte proliferation [66], KO linked to hypertrophy [67] | |
| HDAC8 | X | nucleus (excluded from nucleoli) | medium | TGFb1, RUNX1 | H2A, H2B, H3, H4 | KO ameliorates pulmonary fibrosis [68] | |
| IIa | HDAC4 | 2 | nucleus, cytoplasm (shuttles between) | medium | MEF2, FOXO, TGF-b1 | H2A, H2B, H3, H4 | KO increases myocardial regeneration, overexpression inhibits cardiomyogenesis [69], inhibition ameliorates I/R injury [70] |
| HDAC5 | 17 | nucleus, cytoplasm (shuttles between) | low | MEF2, YY1, NKX2.5, PGC-1a, FOXO | H2A, H2B, H3, H4 | KO linked to hypertrophy with age [71] | |
| HDAC7 | 12 | nucleus, cytoplasm (shuttles between) | – | MEF2, FOXP3, RARA | H2A, H2B, H3, H4 | Promotes hypertrophy [72] | |
| HDAC9 | 7 | nucleus | low | MEF2 | H2A, H2B, H3, H4 | Suppresses hypertrophy [73], KO attenuates atherosclerosis [74] | |
| IIb | HDAC6 | X | nucleus, microtubules | low | TGFb1, GATA6 | H2A, H2B, H3, H4; misfolded proteins | Promotes fibrosis, KO linked to inhibited fibroblast proliferation [75] |
| HDAC10 | 22 | nucleus | high | NOTCH1, PAX3, KAP1 | – | – | |
| III | SIRT1 | 10 | nucleus, mitochondria | low | FOXO, MEF2, HIF1a, PER2, BMAL1 | H2A, H3K14, H4K16 | Protective against hypertrophy [76], severe overexpression promotes cardiomyopathy [77] |
| SIRT2 | 19 | plasma membrane, cytoskeleton, nucleus | low | NFAT, FOXO3, HIF1a | H3K56, H4K16 | KO increases hypertrophy and fibrosis, decreases ejection fraction [78] | |
| SIRT3 | 11 | mitochondria | high | FOXO, CERS | – | KO promotes hypertrophy and fibrosis [79], KO decreases ejection fraction [80] | |
| SIRT4 | 12 | mitochondria | medium | PPARa | – | Promotes hypertrophy and fibrosis [81] | |
| SIRT5 | 6 | mitochondria, cytoplasm | medium | CPS1, SOD1, SHMT2, CYCS | H3K9 | KO promotes hypertrophic cardiomyopathy [82] | |
| SIRT6 | 19 | nucleus | high | NF-kb, HIF1a | H3K9, H3K56 | KO promotes hypertrophy [83], protective against I/R injury [84] | |
| SIRT7 | 17 | nucleus | medium | – | H3K18, H3K36 | KO promotes hypertrophy and inflammatory cardiomyopathy [85] | |
| IV | HDAC11 | 3 | nucleus | – | NOTCH1 | H2A, H2B, H3, H4 | – |
| A | Gene Expression Relevant to Cardiac Epigenetics | ||||
| Gene | Fold change | Ref. | |||
| Writers | HATs | p300 (EP300) | – | ||
| pCAF (KAT2B) | – | ||||
| HMTs | SMYD1 | – | |||
| WHSC1 | 1.54 | [116] | |||
| Ezh2 | 3.95 | [117] | |||
| SUV39h | – | ||||
| DOT1L | 1.42 | [116] | |||
| Erasers | HDAC classes | I | HDAC1 | 1.88 | [117] |
| HDAC2 | 7.74 | [117] | |||
| 4.45 | [116] | ||||
| HDAC3 | 1.51 | [116] | |||
| HDAC8 | 1.72 | [116] | |||
| IIa | HDAC4 | – | |||
| HDAC5 | −1.28 | [117] | |||
| HDAC7 | 1.21 | [116] | |||
| HDAC9 | 1.55 | [116] | |||
| IIb | HDAC6 | – | |||
| HDAC10 | – | ||||
| III | SIRT1 | 2.24 | [116] | ||
| 1.79 | [117] | ||||
| SIRT2 | −1.96 | [117] | |||
| SIRT3 | 1.31 | [116] | |||
| SIRT4 | – | ||||
| SIRT5 | 1.4 | [116] | |||
| −4.75 | [117] | ||||
| SIRT6 | – | ||||
| SIRT7 | – | ||||
| IV | HDAC11 | – | |||
| HDMs | Jarid2 | – | |||
| Jmjd1 | 2.85 | [116] | |||
| Jmjd2 | 1.41 | [116] | |||
| Jmjd3 | – | ||||
| UTX | 2.68 | [116] | |||
| Readers | SWI/SNF | Brg1 (SMARCA4) | – | ||
| Baf60a (SMARCD1) | 1.99 | [117] | |||
| Baf180 (PBRM1) | 1.43 | [117] | |||
| 1.25 | [116] | ||||
| Baf250 (ARID1A) | – | ||||
| BETs | Brd4 | – | |||
| 14-3-3 (YWHAB) | – | ||||
| (DDR)-related readers | ZMYND8 (RACK7/PRKCBP1) | 3.01 | [117] | ||
| 1.4 | [116] | ||||
| B | Gene Expression Relevant to Cardiac AP | ||||
| Gene | Gene info | Fold change | Ref. | ||
| SCN5A | NaV1.5 → INa | 1.48 | [116] | ||
| CACNA1C | CaV1.2 → ICa,L | 1.39 | [116] | ||
| 1.64 | [117] | ||||
| CACNA1G | CaV3.1/3.2 → ICa,T | 1.41 | [116] | ||
| KCNH2 | KC11.1 (hERG) → Ikr | 1.51 | [116] | ||
| −4.15 | [117] | ||||
| KCNQ1 | KV7.1 → Iks | 1.51 | [116] | ||
| −1.8 | [117] | ||||
| KCNJ2 | Kir2.1 → IK1 | −4.24 | [117] | ||
| KCNJ12 | Kir2.1 → IK1 | 1.42 | [116] | ||
| KCND2 | KV 1.4/1.7/3.4 → Ito,s | 1.39 | [116] | ||
| KCND3 | KV 4.2/4.3 → Ito,f | 1.2 | [116] | ||
| KCNA4 | KV 1.4/1.7/3.4 → Ito,s | – | |||
| KCNA5 | KV1.5 → IKur | −1.97 | [116] | ||
| −3.37 | [117] | ||||
| KCNK1 | TWK-1/2 → IKP | −2.61 | [117] | ||
| −1.92 | [117] | ||||
| KCNK6 | TWK-1/2 → IKP | 1.6 | [116] | ||
| KCNK3 | TASK-1 → IKP | – | |||
| KCNK4 | TRAAK → IKP | 1.33 | [116] | ||
| KCNJ11 | Kir6.2 → IK,ATP | −1.6 | [117] | ||
| HCN2 | HCN2/4 → If | – | |||
| HCN4 | HCN2/4 → If | – | |||
| ATP1A1 | INaK | 2.47 | [116] | ||
| ATP1A2 | INaK | −7.15 | [116] | ||
| −10.2 | [117] | ||||
| ATP1A3 | INaK | −1.38 | [116] | ||
| −1.6 | [117] | ||||
| ATP1A4 | INaK | 1.46 | [116] | ||
| NCX1 | INCX | 2.03 | [116] | ||
| ATP2A2 | SERCA2 | 3.59 | [116] | ||
| −1.87 | [117] | ||||
| RYR2 | Ryanodine receptor 2 | −4.18 | [116] | ||
| −1.87 | [116] | ||||
| CALM1 | Calmodulin 1 | −2.36 | [117] | ||
| CALM2 | Calmodulin 2 | – | |||
| CALM3 | Calmodulin 3 | – | |||
| CASQ2 | Calsequestrin | −2.53 | [116] | ||
| −80.1 | [117] | ||||
| KCNIP2 | K+ channel interacting protein 2 | −1.63 | [116] | ||
| −1.44 | [117] | ||||
| KCNE1 | Auxiliary unit for IKs | −1.73 | [117] | ||
| KCNE2 | Auxiliary unit for IKs | – | |||
| GJA1 | Cx43 | – | |||
| GJC1 | Cx45 | 1.27 | [116] | ||
| 1.53 | [117] | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pozo, M.R.; Meredith, G.W.; Entcheva, E. Human iPSC-Cardiomyocytes as an Experimental Model to Study Epigenetic Modifiers of Electrophysiology. Cells 2022, 11, 200. https://doi.org/10.3390/cells11020200
Pozo MR, Meredith GW, Entcheva E. Human iPSC-Cardiomyocytes as an Experimental Model to Study Epigenetic Modifiers of Electrophysiology. Cells. 2022; 11(2):200. https://doi.org/10.3390/cells11020200
Chicago/Turabian StylePozo, Maria R., Gantt W. Meredith, and Emilia Entcheva. 2022. "Human iPSC-Cardiomyocytes as an Experimental Model to Study Epigenetic Modifiers of Electrophysiology" Cells 11, no. 2: 200. https://doi.org/10.3390/cells11020200
APA StylePozo, M. R., Meredith, G. W., & Entcheva, E. (2022). Human iPSC-Cardiomyocytes as an Experimental Model to Study Epigenetic Modifiers of Electrophysiology. Cells, 11(2), 200. https://doi.org/10.3390/cells11020200