
Accelerated Aging Characterizes the Early Stage of Alzheimer’s Disease

 ,
,  and
and 
Abstract
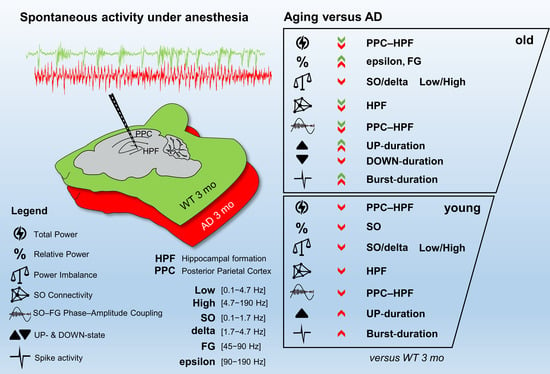
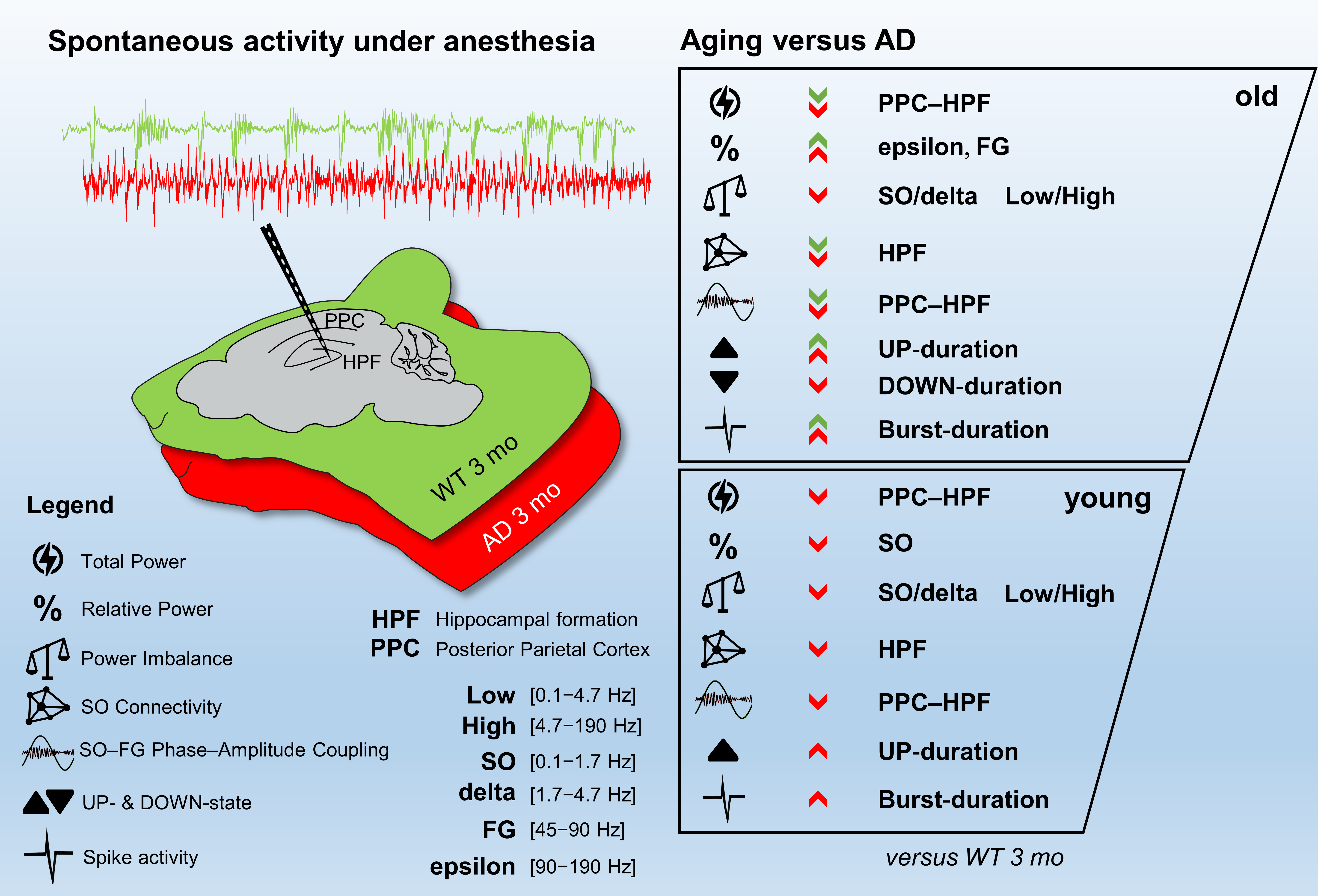
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Data Presentation and Statistical Analyses
3. Results
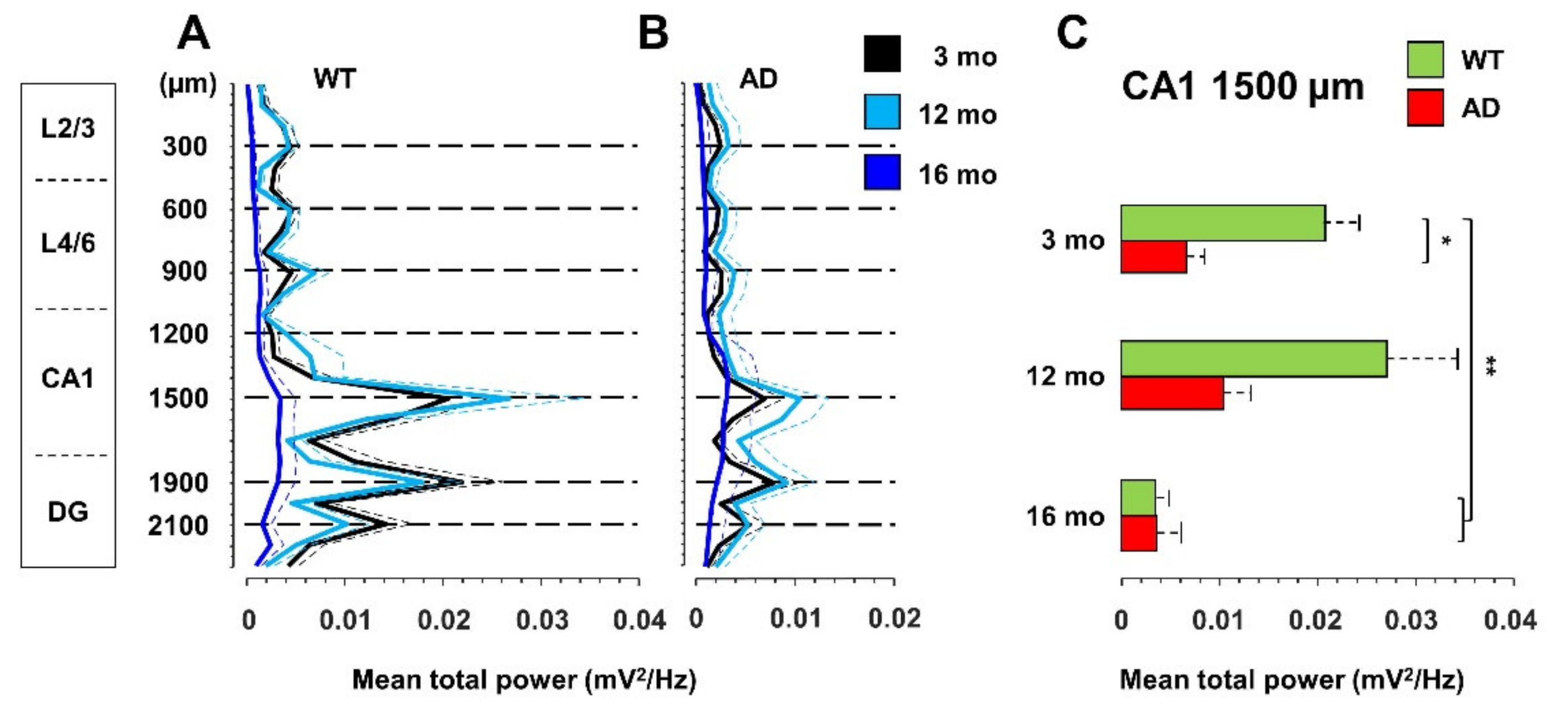
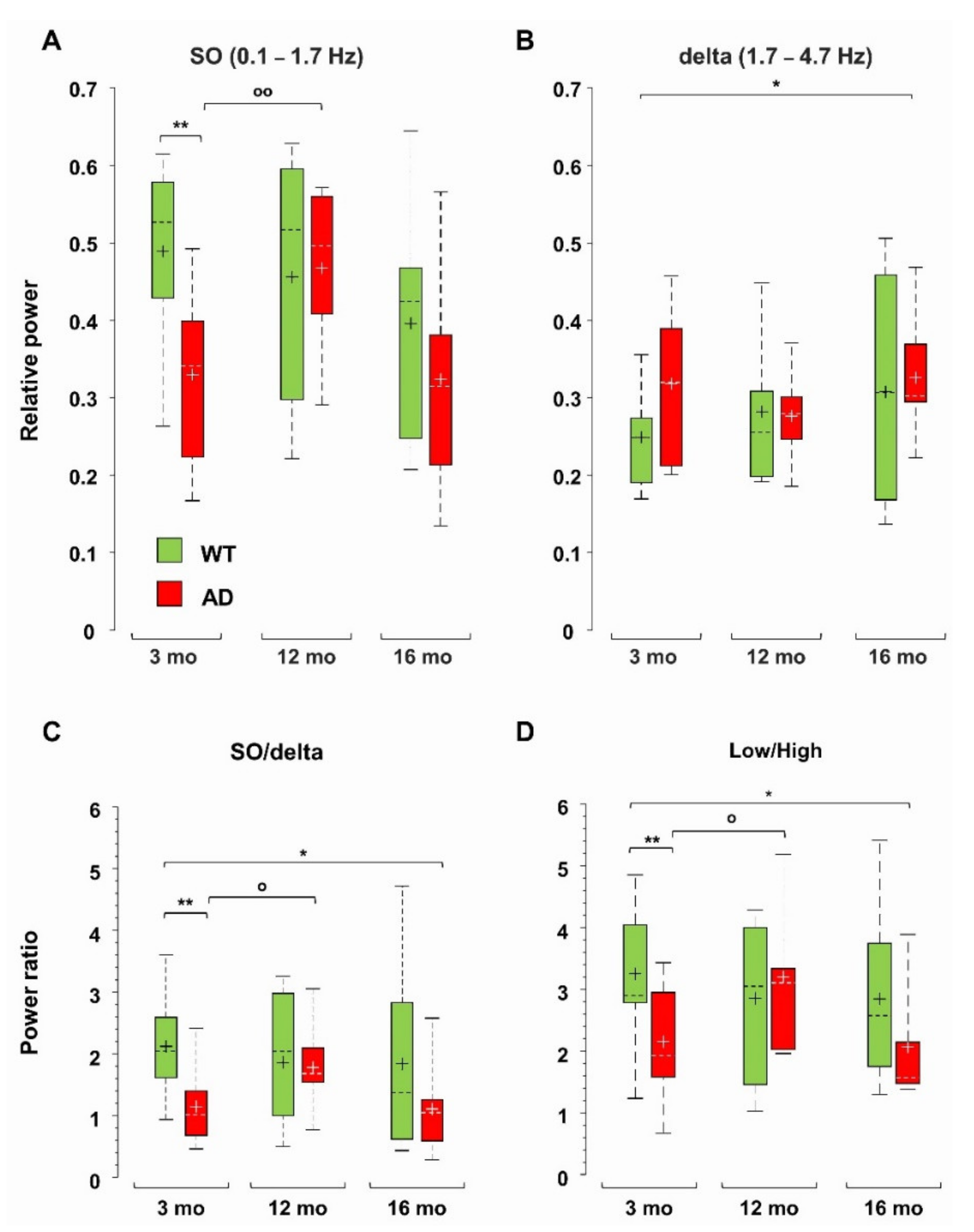
3.1. Total Power Is Reduced in Young AD and Old WT Mice
3.2. Power Imbalance in the Low Frequency Range Characterizes Young AD Mice
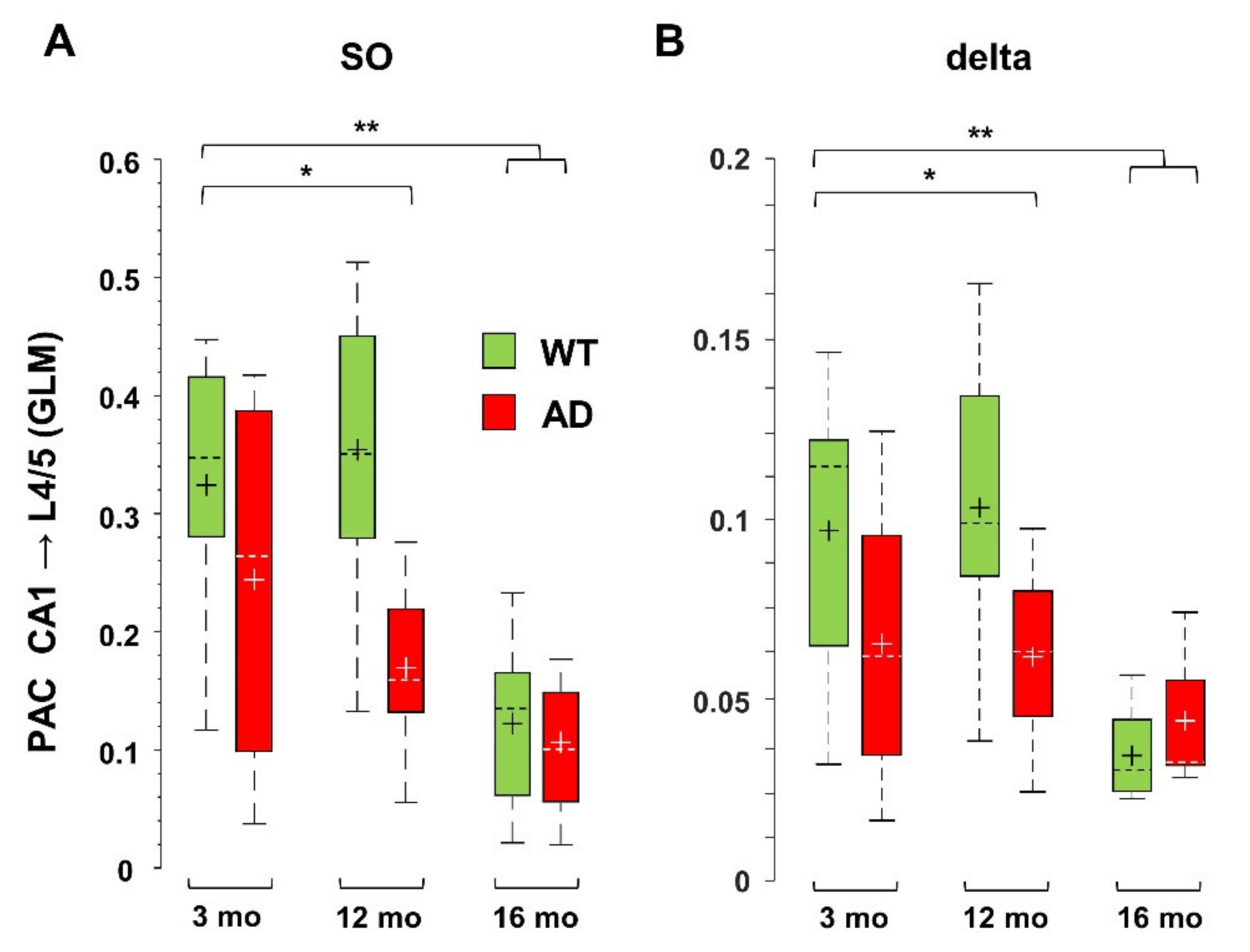
3.3. Impaired Coupling of SO to Higher Frequencies Is Shared between Young AD and Aged WT Mice
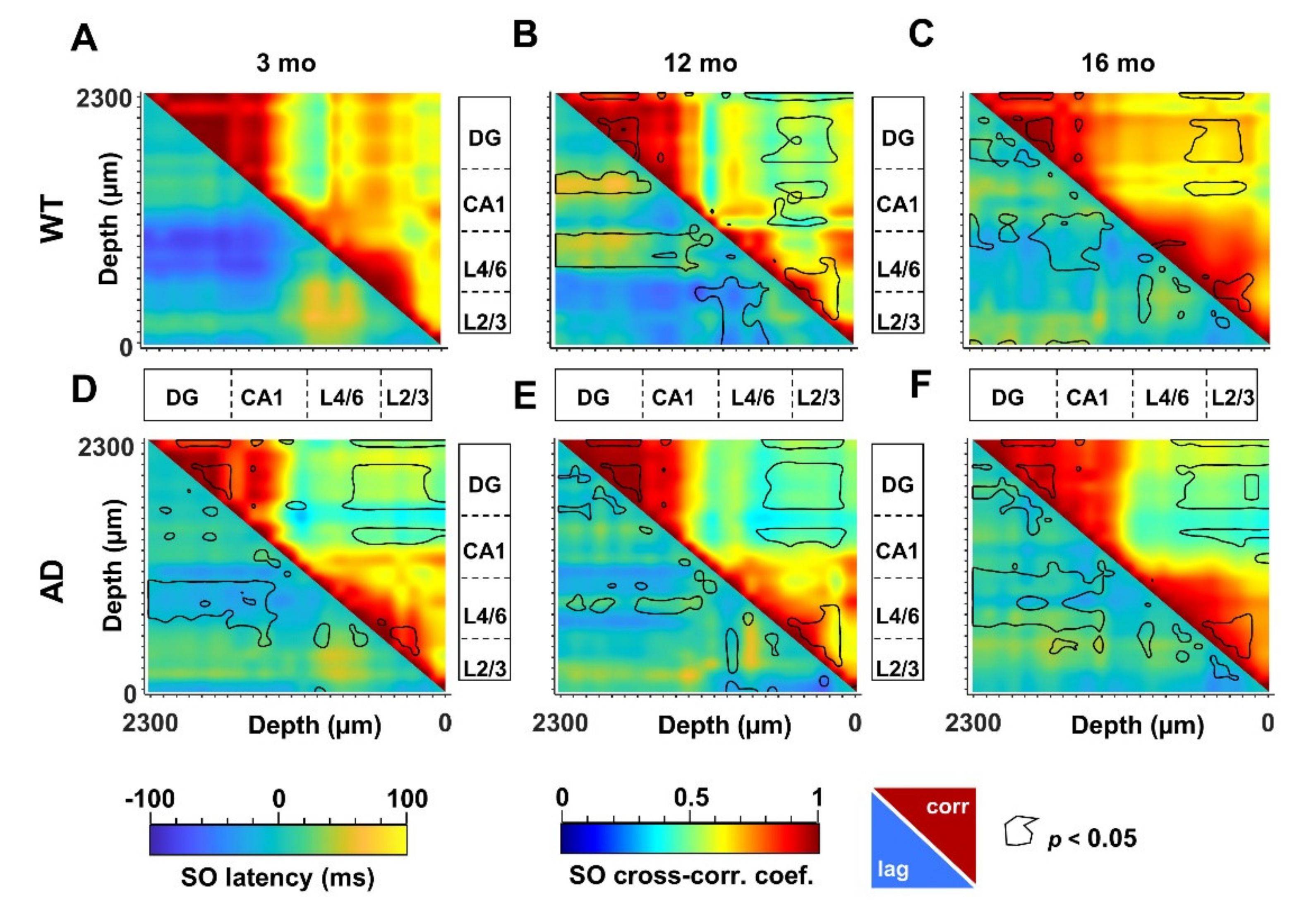
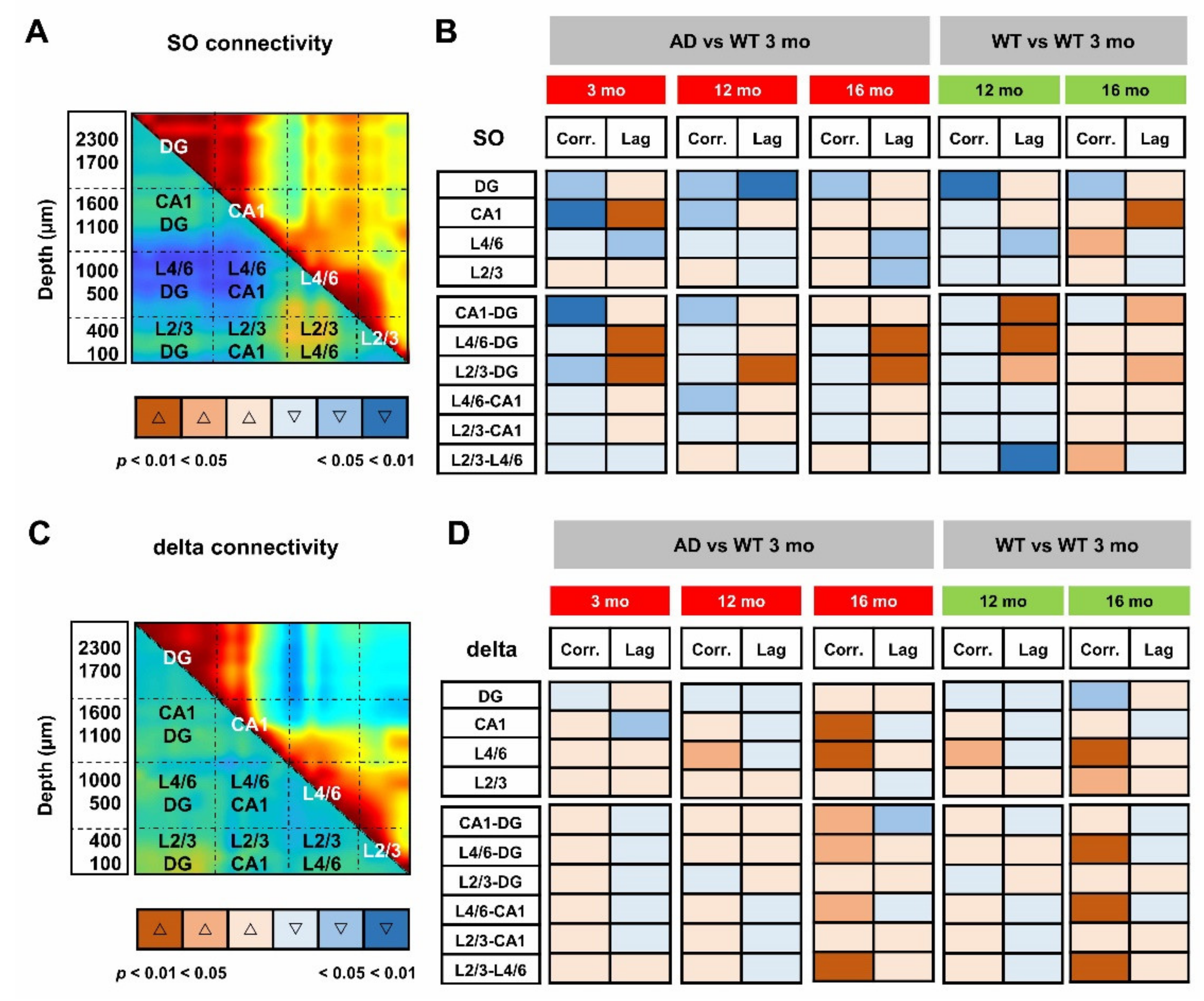
3.4. Defective SO Connectivity Anticipates the Aging Process in Young AD Mice
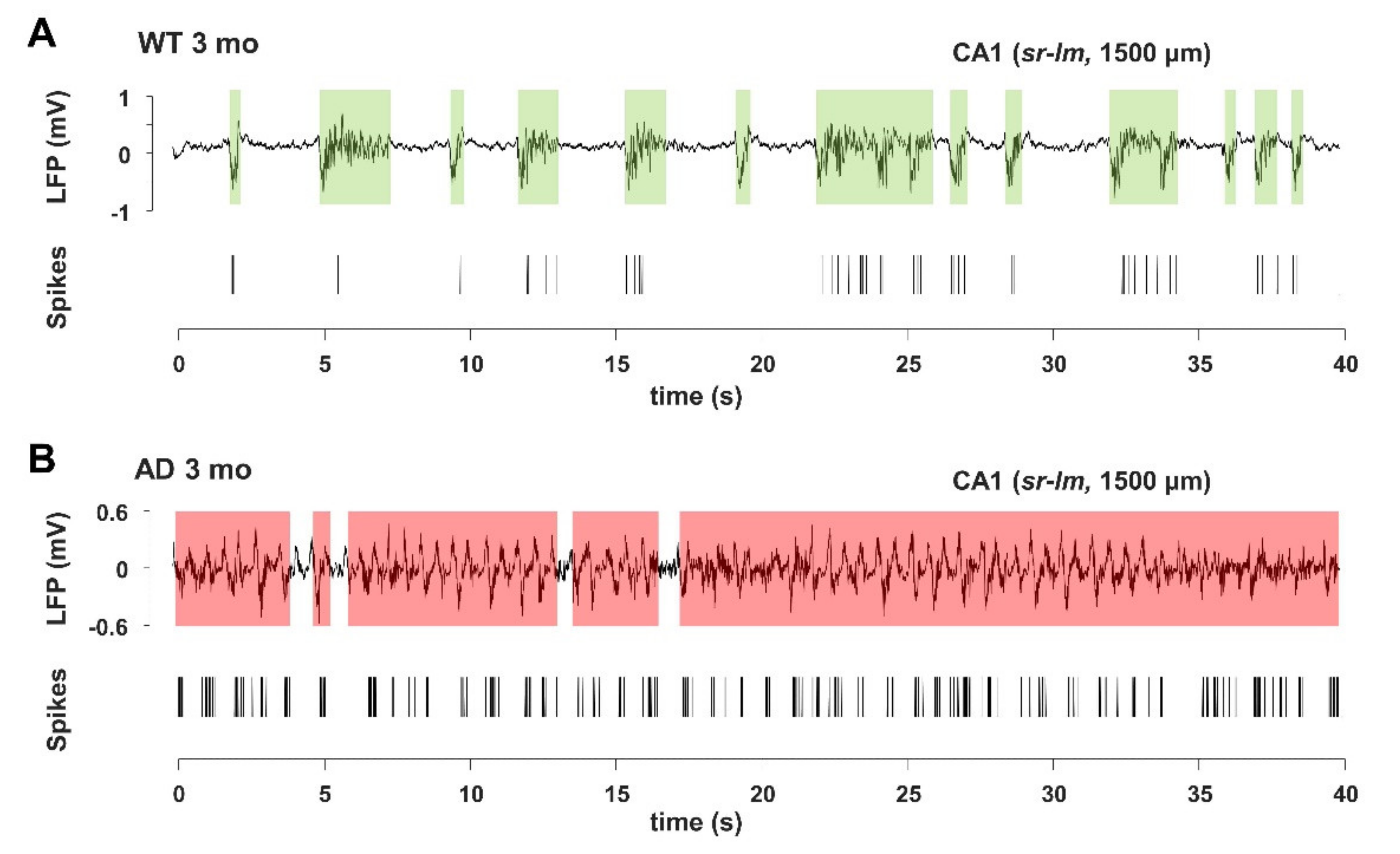
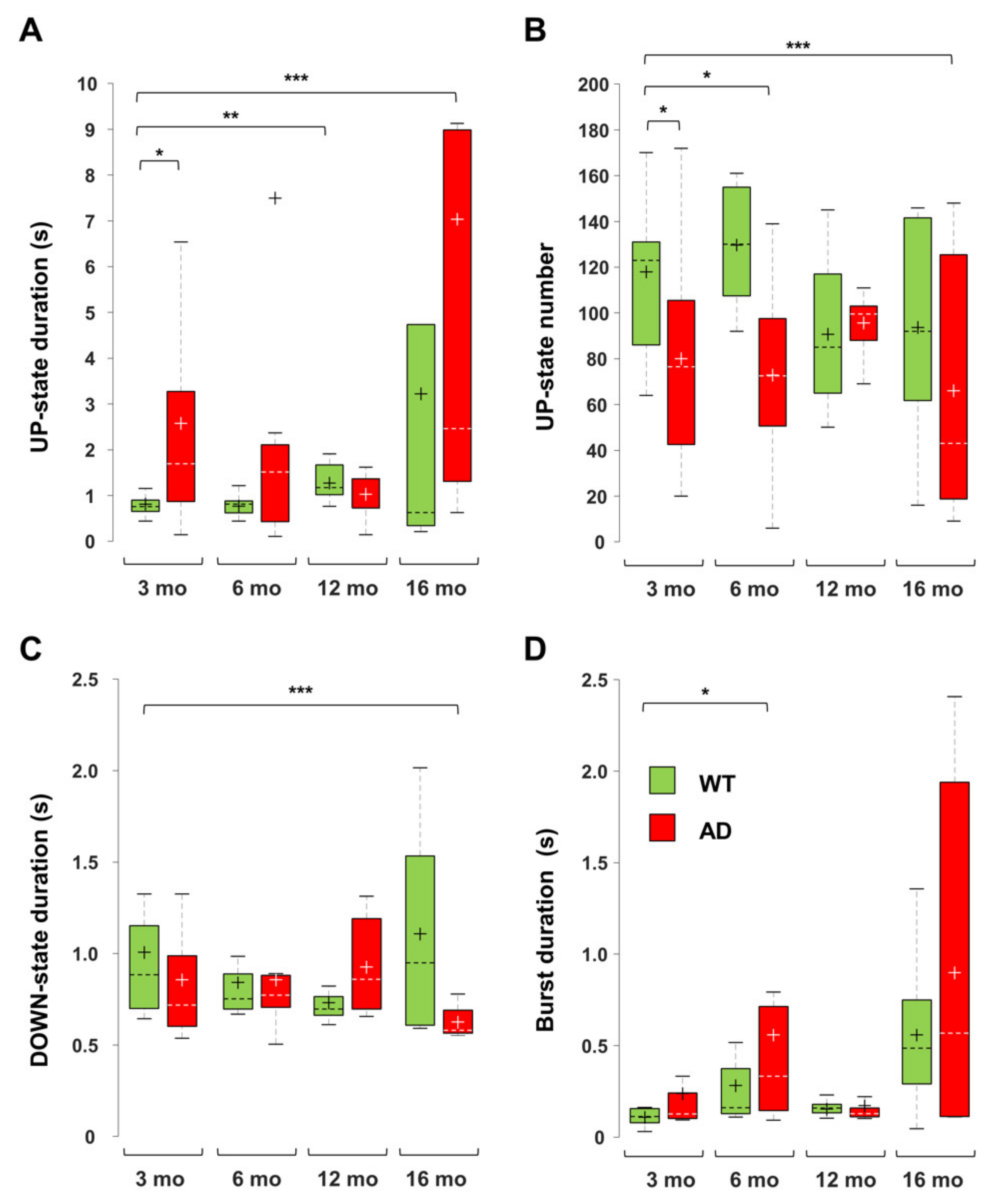
3.5. Imbalance in UP- and DOWN-States Marks AD Progression
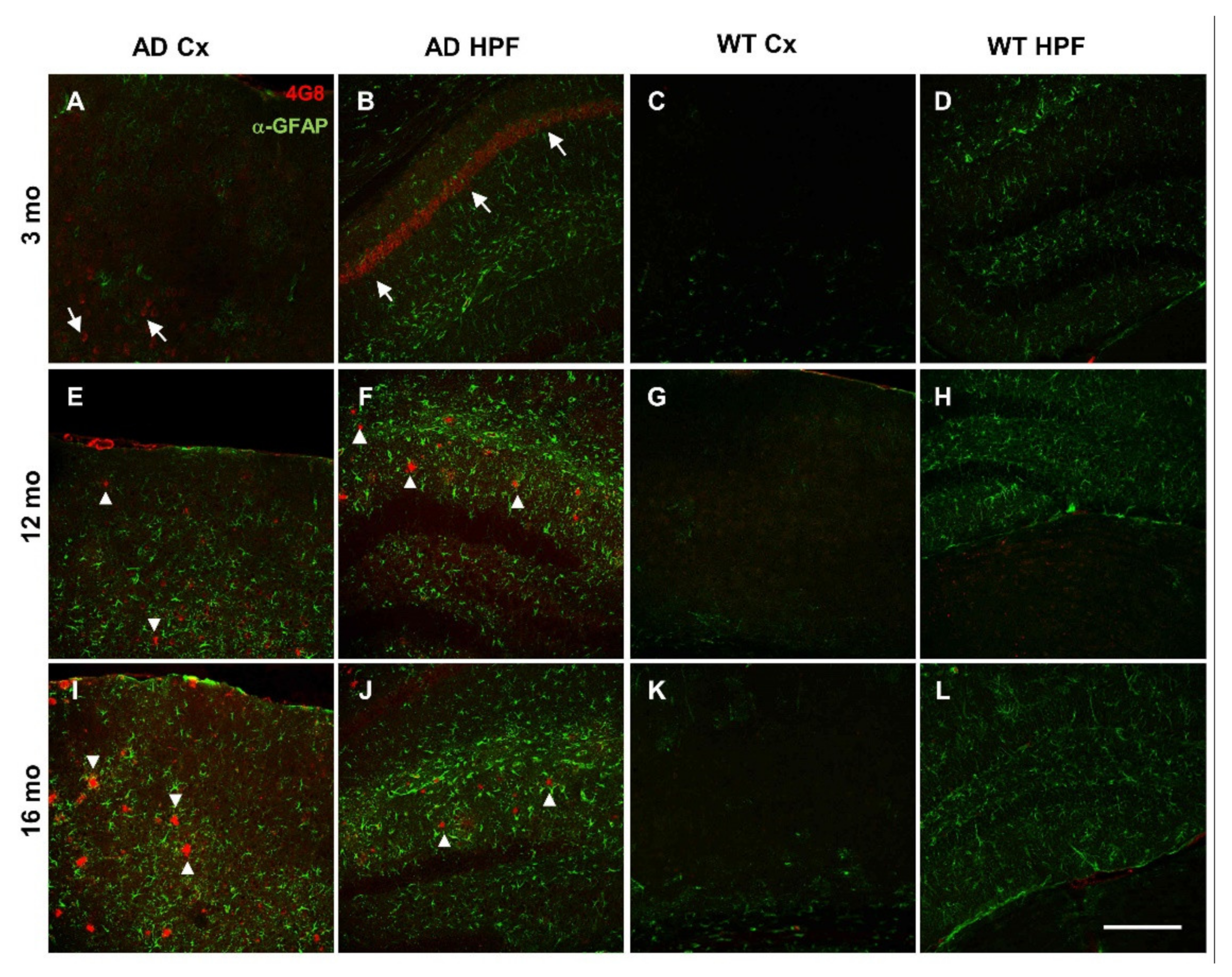
3.6. Amyloidosis and Inflammation in AD and Aged WT Mice
4. Discussion
4.1. Specific Markers of Brain Changes in Young AD Mice
4.2. Premature Aging Also Characterizes the Brain Alterations in AD Mice
4.3. AD Mice Show More Brain Changes
4.4. Mechanistic Insights into Brain Network Alterations
4.5. Study Relevance
4.6. Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Prim. 2021, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- D’Atri, A.; Scarpelli, S.; Gorgoni, M.; Truglia, I.; Lauri, G.; Cordone, S.; Ferrara, M.; Marra, C.; Rossini, P.M.; De Gennaro, L. EEG alterations during wake and sleep in mild cognitive impairment and Alzheimer’s disease. iScience 2021, 24, 102386. [Google Scholar] [CrossRef]
- Jelic, V.; Johansson, S.E.; Almkvist, O.; Shigeta, M.; Julin, P.; Nordberg, A.; Winblad, B.; Wahlund, L.O. Quantitative electroencephalography in mild cognitive impairment: Longitudinal changes and possible prediction of Alzheimer’s disease. Neurobiol. Aging 2000, 21, 533–540. [Google Scholar] [CrossRef]
- Hamm, V.; Héraud, C.; Cassel, J.-C.; Mathis, C.; Goutagny, R. Precocious Alterations of Brain Oscillatory Activity in Alzheimer’s Disease: A Window of Opportunity for Early Diagnosis and Treatment. Front. Cell. Neurosci. 2015, 9, 491. [Google Scholar] [CrossRef] [Green Version]
- Prichep, L.S.; John, E.R.; Ferris, S.H.; Rausch, L.; Fang, Z.; Cancro, R.; Torossian, C.; Reisberg, B. Prediction of longitudinal cognitive decline in normal elderly with subjective complaints using electrophysiological imaging. Neurobiol. Aging 2006, 27, 471–481. [Google Scholar] [CrossRef]
- Frere, S.; Slutsky, I. Alzheimer’s Disease: From Firing Instability to Homeostasis Network Collapse. Neuron 2018, 97, 32–58. [Google Scholar] [CrossRef] [Green Version]
- Palop, J.J.; Mucke, L. Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2016, 17, 777–792. [Google Scholar] [CrossRef]
- Šišková, Z.; Justus, D.; Kaneko, H.; Friedrichs, D.; Henneberg, N.; Beutel, T.; Pitsch, J.; Schoch, S.; Becker, A.; von der Kammer, H.; et al. Dendritic Structural Degeneration Is Functionally Linked to Cellular Hyperexcitability in a Mouse Model of Alzheimer’s Disease. Neuron 2014, 84, 1023–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Styr, B.; Slutsky, I. Imbalance between firing homeostasis and synaptic plasticity drives early-phase Alzheimer’s disease. Nat. Neurosci. 2018, 21, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Zott, B.; Busche, M.A.; Sperling, R.A.; Konnerth, A. What Happens with the Circuit in Alzheimer’s Disease in Mice and Humans? Annu. Rev. Neurosci. 2018, 41, 277–297. [Google Scholar] [CrossRef]
- De Risi, M.; Torromino, G.; Tufano, M.; Moriceau, S.; Pignataro, A.; Rivagorda, M.; Carrano, N.; Middei, S.; Settembre, C.; Ammassari-Teule, M.; et al. Mechanisms by which autophagy regulates memory capacity in ageing. Aging Cell 2020, 19, e13189. [Google Scholar] [CrossRef]
- Glatigny, M.; Moriceau, S.; Rivagorda, M.; Ramos-Brossier, M.; Nascimbeni, A.C.; Lante, F.; Shanley, M.R.; Boudarene, N.; Rousseaud, A.; Friedman, A.K.; et al. Autophagy Is Required for Memory Formation and Reverses Age-Related Memory Decline. Curr. Biol. 2019, 29, 435–448.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, S.; Hofer, S.J.; Zimmermann, A.; Pechlaner, R.; Dammbrueck, C.; Pendl, T.; Marcello, G.M.; Pogatschnigg, V.; Bergmann, M.; Müller, M.; et al. Dietary spermidine improves cognitive function. Cell Rep. 2021, 35, 108985. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, A.; Armato, U.; Hu, P.; Dal Prà, I. Danger-Sensing/Patten Recognition Receptors and Neuroinflammation in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 9036. [Google Scholar] [CrossRef] [PubMed]
- Grabert, K.; Michoel, T.; Karavolos, M.H.; Clohisey, S.; Baillie, J.K.; Stevens, M.P.; Freeman, T.C.; Summers, K.M.; McColl, B.W. Microglial brain region−dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 2016, 19, 504–516. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Huang, Y.; Cai, W.; Chen, X.; Men, X.; Lu, T.; Wu, A.; Lu, Z. Age-related cerebral small vessel disease and inflammaging. Cell Death Dis. 2020, 11, 932. [Google Scholar] [CrossRef]
- Yang, F.; Chu, X.; Yin, M.; Liu, X.; Yuan, H.; Niu, Y.; Fu, L. mTOR and autophagy in normal brain aging and caloric restriction ameliorating age-related cognition deficits. Behav. Brain Res. 2014, 264, 82–90. [Google Scholar] [CrossRef]
- Harris, S.S.; Wolf, F.; De Strooper, B.; Busche, M.A. Tipping the Scales: Peptide-Dependent Dysregulation of Neural Circuit Dynamics in Alzheimer’s Disease. Neuron 2020, 107, 417–435. [Google Scholar] [CrossRef]
- Walsh, D.M.; Selkoe, D.J. Amyloid β-protein and beyond: The path forward in Alzheimer’s disease. Curr. Opin. Neurobiol. 2020, 61, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Maestú, F.; de Haan, W.; Busche, M.A.; DeFelipe, J. Neuronal excitation/inhibition imbalance: Core element of a translational perspective on Alzheimer pathophysiology. Ageing Res. Rev. 2021, 69, 101372. [Google Scholar] [CrossRef]
- Wang, W. Brain network features based on theta-gamma cross-frequency coupling connections in EEG for emotion recognition. Neurosci. Lett. 2021, 761, 136106. [Google Scholar] [CrossRef]
- Busche, M.A.; Chen, X.; Henning, H.A.; Reichwald, J.; Staufenbiel, M.; Sakmann, B.; Konnerth, A. Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2012, 109, 8740–8745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, R.; Agostini, M.; Murana, E.; Mahmud, M.; Scremin, E.; Rubega, M.; Sparacino, G.; Vassanelli, S.; Fasolato, C. Early hippocampal hyperexcitability in PS2APP mice: Role of mutant PS2 and APP. Neurobiol. Aging 2017, 50, 64–76. [Google Scholar] [CrossRef]
- Goutagny, R.; Gu, N.; Cavanagh, C.; Jackson, J.; Chabot, J.-G.; Quirion, R.; Krantic, S.; Williams, S. Alterations in hippocampal network oscillations and theta-gamma coupling arise before Aβ overproduction in a mouse model of Alzheimer’s disease. Eur. J. Neurosci. 2013, 37, 1896–1902. [Google Scholar] [CrossRef] [PubMed]
- Bezzina, C.; Verret, L.; Juan, C.; Remaud, J.; Halley, H.; Rampon, C.; Dahan, L. Early Onset of Hypersynchronous Network Activity and Expression of a Marker of Chronic Seizures in the Tg2576 Mouse Model of Alzheimer’s Disease. PLoS ONE 2015, 10, e0119910. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.D.; Deck, G.; Goldman, A.; Eskandar, E.N.; Noebels, J.; Cole, A.J. Silent hippocampal seizures and spikes identified by foramen ovale electrodes in Alzheimer’s disease. Nat. Med. 2017, 23, 678–680. [Google Scholar] [CrossRef]
- Sanchez, P.E.; Zhu, L.; Verret, L.; Vossel, K.A.; Orr, A.G.; Cirrito, J.R.; Devidze, N.; Ho, K.; Yu, G.-Q.; Palop, J.J.; et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc. Natl. Acad. Sci. USA 2012, 109, E2895–E2903. [Google Scholar] [CrossRef] [Green Version]
- Verret, L.; Mann, E.O.; Hang, G.B.; Barth, A.M.I.; Cobos, I.; Ho, K.; Devidze, N.; Masliah, E.; Kreitzer, A.C.; Mody, I.; et al. Inhibitory Interneuron Deficit Links Altered Network Activity and Cognitive Dysfunction in Alzheimer Model. Cell 2012, 149, 708–721. [Google Scholar] [CrossRef] [Green Version]
- Gillespie, A.K.; Jones, E.A.; Lin, Y.-H.; Karlsson, M.P.; Kay, K.; Yoon, S.Y.; Tong, L.M.; Nova, P.; Carr, J.S.; Frank, L.M.; et al. Apolipoprotein E4 Causes Age-Dependent Disruption of Slow Gamma Oscillations during Hippocampal Sharp-Wave Ripples. Neuron 2016, 90, 740–751. [Google Scholar] [CrossRef] [Green Version]
- Iaccarino, H.F.; Singer, A.C.; Martorell, A.J.; Rudenko, A.; Gao, F.; Gillingham, T.Z.; Mathys, H.; Seo, J.; Kritskiy, O.; Abdurrob, F.; et al. Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature 2016, 540, 230–235. [Google Scholar] [CrossRef] [Green Version]
- Hyafil, A.; Giraud, A.-L.; Fontolan, L.; Gutkin, B. Neural Cross-Frequency Coupling: Connecting Architectures, Mechanisms, and Functions. Trends Neurosci. 2015, 38, 725–740. [Google Scholar] [CrossRef]
- Wang, D.X.; Schmitt, K.; Seger, S.; Davila, C.E.; Lega, B.C. Cross-regional phase amplitude coupling supports the encoding of episodic memories. Hippocampus 2021, 31, 481–492. [Google Scholar] [CrossRef]
- Abubaker, M.; Al Qasem, W.; Kvašňák, E. Working Memory and Cross-Frequency Coupling of Neuronal Oscillations. Front. Psychol. 2021, 12, 756661. [Google Scholar] [CrossRef] [PubMed]
- Riddle, J.; McFerren, A.; Frohlich, F. Causal role of cross-frequency coupling in distinct components of cognitive control. Prog. Neurobiol. 2021, 202, 102033. [Google Scholar] [CrossRef] [PubMed]
- Sweeney-Reed, C.M.; Zaehle, T.; Voges, J.; Schmitt, F.C.; Buentjen, L.; Kopitzki, K.; Esslinger, C.; Hinrichs, H.; Heinze, H.-J.; Knight, R.T.; et al. Corticothalamic phase synchrony and cross-frequency coupling predict human memory formation. eLife 2014, 3, e05352. [Google Scholar] [CrossRef] [PubMed]
- Lega, B.; Burke, J.; Jacobs, J.; Kahana, M.J. Slow-Theta-to-Gamma Phase-Amplitude Coupling in Human Hippocampus Supports the Formation of New Episodic Memories. Cereb. Cortex 2016, 26, 268–278. [Google Scholar] [CrossRef] [Green Version]
- Ahnaou, A.; Chave, L.; Manyakov, N.V.; Drinkenburg, W.H.I.M. Odour Retrieval Processing in Mice: Cholinergic Modulation of Oscillatory Coupling in Olfactory Bulb-Piriform Networks. Neuropsychobiology 2021, 80, 374–392. [Google Scholar] [CrossRef]
- Kastanenka, K.V.; Hou, S.S.; Shakerdge, N.; Logan, R.; Feng, D.; Wegmann, S.; Chopra, V.; Hawkes, J.M.; Chen, X.; Bacskai, B.J. Optogenetic Restoration of Disrupted Slow Oscillations Halts Amyloid Deposition and Restores Calcium Homeostasis in an Animal Model of Alzheimer’s Disease. PLoS ONE 2017, 12, e0170275. [Google Scholar] [CrossRef]
- Martorell, A.J.; Paulson, A.L.; Suk, H.-J.; Abdurrob, F.; Drummond, G.T.; Guan, W.; Young, J.Z.; Kim, D.N.-W.; Kritskiy, O.; Barker, S.J.; et al. Multi-sensory Gamma Stimulation Ameliorates Alzheimer’s-Associated Pathology and Improves Cognition. Cell 2019, 177, 256–271.e22. [Google Scholar] [CrossRef] [Green Version]
- Jafari, Z.; Kolb, B.E.; Mohajerani, M.H. Neural oscillations and brain stimulation in Alzheimer’s disease. Prog. Neurobiol. 2020, 194, 101878. [Google Scholar] [CrossRef]
- Koolschijn, R.S.; Emir, U.E.; Pantelides, A.C.; Nili, H.; Behrens, T.E.J.; Barron, H.C. The Hippocampus and Neocortical Inhibitory Engrams Protect against Memory Interference. Neuron 2019, 101, 528–541.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leparulo, A.; Mahmud, M.; Scremin, E.; Pozzan, T.; Vassanelli, S.; Fasolato, C. Dampened Slow Oscillation Connectivity Anticipates Amyloid Deposition in the PS2APP Mouse Model of Alzheimer’s Disease. Cells 2019, 9, 54. [Google Scholar] [CrossRef] [Green Version]
- Busche, M.A.; Kekuš, M.; Adelsberger, H.; Noda, T.; Förstl, H.; Nelken, I.; Konnerth, A. Rescue of long-range circuit dysfunction in Alzheimer’s disease models. Nat. Neurosci. 2015, 18, 1623–1630. [Google Scholar] [CrossRef] [PubMed]
- Ladenbauer, J.; Ladenbauer, J.; Külzow, N.; de Boor, R.; Avramova, E.; Grittner, U.; Flöel, A. Promoting Sleep Oscillations and Their Functional Coupling by Transcranial Stimulation Enhances Memory Consolidation in Mild Cognitive Impairment. J. Neurosci. 2017, 37, 7111–7124. [Google Scholar] [CrossRef] [Green Version]
- Mitra, A.; Snyder, A.Z.; Hacker, C.D.; Pahwa, M.; Tagliazucchi, E.; Laufs, H.; Leuthardt, E.C.; Raichle, M.E. Human cortical–hippocampal dialogue in wake and slow-wave sleep. Proc. Natl. Acad. Sci. USA 2016, 113, E6868–E6876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozmen, L.; Albientz, A.; Czech, C.; Jacobsen, H. Expression of Transgenic APP mRNA Is the Key Determinant for Beta-Amyloid Deposition in PS2APP Transgenic Mice. Neurodegener. Dis. 2009, 6, 29–36. [Google Scholar] [CrossRef]
- Gurevicius, K.; Lipponen, A.; Tanila, H. Increased Cortical and Thalamic Excitability in Freely Moving APPswe/PS1dE9 Mice Modeling Epileptic Activity Associated with Alzheimer’s Disease. Cereb. Cortex 2013, 23, 1148–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, J.G.; Higgins, G.A.; Ouagazzal, A.-M.M.; Ozmen, L.; Kew, J.N.C.; Bohrmann, B.; Malherbe, P.; Brockhaus, M.; Loetscher, H.; Czech, C.; et al. PS2APP transgenic mice, coexpressing hPS2mut and hAPPswe, show age-related cognitive deficits associated with discrete brain amyloid deposition and inflammation. J. Neurosci. 2003, 23, 8989–9003. [Google Scholar] [CrossRef] [Green Version]
- Chan, R.W.; Leong, A.T.L.; Ho, L.C.; Gao, P.P.; Wong, E.C.; Dong, C.M.; Wang, X.; He, J.; Chan, Y.-S.; Lim, L.W.; et al. Low-frequency hippocampal–cortical activity drives brain-wide resting-state functional MRI connectivity. Proc. Natl. Acad. Sci. USA 2017, 114, E6972–E6981. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Krishnan, G.P.; Bazhenov, M. Synaptic Mechanisms of Memory Consolidation during Sleep Slow Oscillations. J. Neurosci. 2016, 36, 4231–4247. [Google Scholar] [CrossRef] [Green Version]
- Born, J. Slow-wave sleep and the consolidation of long-term memory. World J. Biol. Psychiatry 2010, 11, 16–21. [Google Scholar] [CrossRef]
- Helfrich, R.F.; Mander, B.A.; Jagust, W.J.; Knight, R.T.; Walker, M.P. Old Brains Come Uncoupled in Sleep: Slow Wave-Spindle Synchrony, Brain Atrophy, and Forgetting. Neuron 2018, 97, 221–230.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalweit, A.N.; Yang, H.; Colitti-Klausnitzer, J.; Fülöp, L.; Bozsó, Z.; Penke, B.; Manahan-Vaughan, D. Acute intracerebral treatment with amyloid-beta (1-42) alters the profile of neuronal oscillations that accompany LTP induction and results in impaired LTP in freely behaving rats. Front. Behav. Neurosci. 2015, 9, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier-Umaña, C.; Muñoz-Cabrera, J.; Valderrama, M.; Múnera, A.; Nava-Mesa, M.O. Acute Effects of Two Different Species of Amyloid-β on Oscillatory Activity and Synaptic Plasticity in the Commissural CA3-CA1 Circuit of the Hippocampus. Neural Plast. 2020, 2020, 8869526. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Gulati, T.; Ganguly, K. Competing Roles of Slow Oscillations and Delta Waves in Memory Consolidation versus Forgetting. Cell 2019, 179, 514–526.e13. [Google Scholar] [CrossRef]
- Adhikari, A.; Sigurdsson, T.; Topiwala, M.A.; Gordon, J.A. Cross-correlation of instantaneous amplitudes of field potential oscillations: A straightforward method to estimate the directionality and lag between brain areas. J. Neurosci. Methods 2010, 191, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Stroh, A.; Adelsberger, H.; Groh, A.; Rühlmann, C.; Fischer, S.; Schierloh, A.; Deisseroth, K.; Konnerth, A. Making Waves: Initiation and Propagation of Corticothalamic Ca2+ Waves In Vivo. Neuron 2013, 77, 1136–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steriade, M.; Nunez, A.; Amzica, F. A novel slow (<1 Hz) oscillation of neocortical neurons in vivo: Depolarizing and hyperpolarizing components. J. Neurosci. 1993, 13, 3252–3265. [Google Scholar] [CrossRef] [Green Version]
- Stern, E.A.; Kincaid, A.E.; Wilson, C.J. Spontaneous Subthreshold Membrane Potential Fluctuations and Action Potential Variability of Rat Corticostriatal and Striatal Neurons In Vivo. J. Neurophysiol. 1997, 77, 1697–1715. [Google Scholar] [CrossRef]
- Cossart, R.; Aronov, D.; Yuste, R. Attractor dynamics of network UP states in the neocortex. Nature 2003, 423, 283–288. [Google Scholar] [CrossRef]
- Guo, Z.V.; Inagaki, H.K.; Daie, K.; Druckmann, S.; Gerfen, C.R.; Svoboda, K. Maintenance of persistent activity in a frontal thalamocortical loop. Nature 2017, 545, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Hahn, T.T.G.; Sakmann, B.; Mehta, M.R. Differential responses of hippocampal subfields to cortical up–down states. Proc. Natl. Acad. Sci. USA 2007, 104, 5169–5174. [Google Scholar] [CrossRef] [Green Version]
- Doi, A.; Mizuno, M.; Katafuchi, T.; Furue, H.; Koga, K.; Yoshimura, M. Slow oscillation of membrane currents mediated by glutamatergic inputs of rat somatosensory cortical neurons: In vivo patch-clamp analysis. Eur. J. Neurosci. 2007, 26, 2565–2575. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.A.; Buonomano, D. V Development and Plasticity of Spontaneous Activity and Up States in Cortical Organotypic Slices. J. Neurosci. 2007, 27, 5915–5925. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Vives, M.V.; McCormick, D.A. Cellular and network mechanisms of rhythmic recurrent activity in neocortex. Nat. Neurosci. 2000, 3, 1027–1034. [Google Scholar] [CrossRef]
- Shu, Y.; Hasenstaub, A.; Badoual, M.; Bal, T.; McCormick, D.A. Barrages of Synaptic Activity Control the Gain and Sensitivity of Cortical Neurons. J. Neurosci. 2003, 23, 10388–10401. [Google Scholar] [CrossRef]
- Lisman, J.E. Bursts as a unit of neural information: Making unreliable synapses reliable. Trends Neurosci. 1997, 20, 38–43. [Google Scholar] [CrossRef]
- Dolev, I.; Fogel, H.; Milshtein, H.; Berdichevsky, Y.; Lipstein, N.; Brose, N.; Gazit, N.; Slutsky, I. Spike bursts increase amyloid-β 40/42 ratio by inducing a presenilin-1 conformational change. Nat. Neurosci. 2013, 16, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Kipanyula, M.J.; Contreras, L.; Zampese, E.; Lazzari, C.; Wong, A.K.C.; Pizzo, P.; Fasolato, C.; Pozzan, T. Ca 2+ dysregulation in neurons from transgenic mice expressing mutant presenilin 2. Aging Cell 2012, 11, 885–893. [Google Scholar] [CrossRef] [Green Version]
- Fedeli, C.; Filadi, R.; Rossi, A.; Mammucari, C.; Pizzo, P. PSEN2 (presenilin 2) mutants linked to familial Alzheimer disease impair autophagy by altering Ca2+ homeostasis. Autophagy 2019, 15, 2044–2062. [Google Scholar] [CrossRef]
- Jayadev, S.; Leverenz, J.B.; Steinbart, E.; Stahl, J.; Klunk, W.; Yu, C.-E.E.; Bird, T.D. Alzheimer’s disease phenotypes and genotypes associated with mutations in presenilin 2. Brain 2010, 133, 1143–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iulita, M.F.; Allard, S.; Richter, L.; Munter, L.-M.; Ducatenzeiler, A.; Weise, C.; Do Carmo, S.; Klein, W.L.; Multhaup, G.; Cuello, A.C. Intracellular Aβ pathology and early cognitive impairments in a transgenic rat overexpressing human amyloid precursor protein: A multidimensional study. Acta Neuropathol. Commun. 2014, 2, 61. [Google Scholar] [CrossRef] [Green Version]
- Welikovitch, L.A.; Do Carmo, S.; Maglóczky, Z.; Szocsics, P.; Lőke, J.; Freund, T.; Cuello, A.C. Evidence of intraneuronal Aβ accumulation preceding tau pathology in the entorhinal cortex. Acta Neuropathol. 2018, 136, 901–917. [Google Scholar] [CrossRef] [PubMed]
- Focke, C.; Blume, T.; Zott, B.; Shi, Y.; Deussing, M.; Peters, F.; Schmidt, C.; Kleinberger, G.; Lindner, S.; Gildehaus, F.-J.; et al. Early and Longitudinal Microglial Activation but Not Amyloid Accumulation Predicts Cognitive Outcome in PS2APP Mice. J. Nucl. Med. 2019, 60, 548–554. [Google Scholar] [CrossRef]
- Meilandt, W.J.; Ngu, H.; Gogineni, A.; Lalehzadeh, G.; Lee, S.-H.; Srinivasan, K.; Imperio, J.; Wu, T.; Weber, M.; Kruse, A.J.; et al. Trem2 Deletion Reduces Late-Stage Amyloid Plaque Accumulation, Elevates the Aβ42:Aβ40 Ratio, and Exacerbates Axonal Dystrophy and Dendritic Spine Loss in the PS2APP Alzheimer’s Mouse Model. J. Neurosci. 2020, 40, 1956–1974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brendel, M.; Kleinberger, G.; Probst, F.; Jaworska, A.; Overhoff, F.; Blume, T.; Albert, N.L.; Carlsen, J.; Lindner, S.; Gildehaus, F.J.; et al. Increase of TREM2 during Aging of an Alzheimer’s Disease Mouse Model Is Paralleled by Microglial Activation and Amyloidosis. Front. Aging Neurosci. 2017, 9, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gui, B.; Slone, J.; Huang, T. Perspective: Is Random Monoallelic Expression a Contributor to Phenotypic Variability of Autosomal Dominant Disorders? Front. Genet. 2017, 8, 191. [Google Scholar] [CrossRef] [Green Version]
- Gonneaud, J.; Baria, A.T.; Pichet Binette, A.; Gordon, B.A.; Chhatwal, J.P.; Cruchaga, C.; Jucker, M.; Levin, J.; Salloway, S.; Farlow, M.; et al. Accelerated functional brain aging in pre-clinical familial Alzheimer’s disease. Nat. Commun. 2021, 12, 5346. [Google Scholar] [CrossRef]
- Khrimian, L.; Obri, A.; Ramos-Brossier, M.; Rousseaud, A.; Moriceau, S.; Nicot, A.-S.; Mera, P.; Kosmidis, S.; Karnavas, T.; Saudou, F.; et al. Gpr158 mediates osteocalcin’s regulation of cognition. J. Exp. Med. 2017, 214, 2859–2873. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Aron, L.; Zullo, J.; Pan, Y.; Kim, H.; Chen, Y.; Yang, T.-H.; Kim, H.-M.; Drake, D.; Liu, X.S.; et al. REST and stress resistance in ageing and Alzheimer’s disease. Nature 2014, 507, 448–454. [Google Scholar] [CrossRef] [Green Version]
- Kaja, S.; Sumien, N.; Shah, V.V.; Puthawala, I.; Maynard, A.N.; Khullar, N.; Payne, A.J.; Forster, M.J.; Koulen, P. Loss of Spatial Memory, Learning, and Motor Function During Normal Aging Is Accompanied by Changes in Brain Presenilin 1 and 2 Expression Levels. Mol. Neurobiol. 2015, 52, 545–554. [Google Scholar] [CrossRef] [Green Version]
- Pizzo, P.; Basso, E.; Filadi, R.; Greotti, E.; Leparulo, A.; Pendin, D.; Redolfi, N.; Rossini, M.; Vajente, N.; Pozzan, T.; et al. Presenilin-2 and Calcium Handling: Molecules, Organelles, Cells and Brain Networks. Cells 2020, 9, 2166. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Rodríguez-Arellano, J.J.; Parpura, V.; Zorec, R. Astroglial calcium signalling in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2017, 483, 1005–1012. [Google Scholar] [CrossRef] [Green Version]
- Poskanzer, K.E.; Yuste, R. Astrocytes regulate cortical state switching in vivo. Proc. Natl. Acad. Sci. USA 2016, 113, E2675–E2684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poskanzer, K.E.; Yuste, R. Astrocytic regulation of cortical UP states. Proc. Natl. Acad. Sci. USA 2011, 108, 18453–18458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greotti, E.; Capitanio, P.; Wong, A.; Pozzan, T.; Pizzo, P.; Pendin, D. Familial Alzheimer’s disease-linked presenilin mutants and intracellular Ca2+ handling: A single-organelle, FRET-based analysis. Cell Calcium 2019, 79, 44–56. [Google Scholar] [CrossRef]
- Zampese, E.; Fasolato, C.; Kipanyula, M.J.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. USA 2011, 108, 2777–2782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, S.; Tononi, G. Modeling Sleep and Wakefulness in the Thalamocortical System. J. Neurophysiol. 2005, 93, 1671–1698. [Google Scholar] [CrossRef]
- Neske, G.T. The Slow Oscillation in Cortical and Thalamic Networks: Mechanisms and Functions. Front. Neural Circuits 2016, 9, 88. [Google Scholar] [CrossRef] [Green Version]
- Rigotto, G.; Zentilin, L.; Pozzan, T.; Basso, E. Effects of Mild Excitotoxic Stimulus on Mitochondria Ca2+ Handling in Hippocampal Cultures of a Mouse Model of Alzheimer’s Disease. Cells 2021, 10, 2046. [Google Scholar] [CrossRef]
- Rossi, A.; Rigotto, G.; Valente, G.; Giorgio, V.; Basso, E.; Filadi, R.; Pizzo, P. Defective Mitochondrial Pyruvate Flux Affects Cell Bioenergetics in Alzheimer’s Disease-Related Models. Cell Rep. 2020, 30, 2332–2348.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agostini, M.; Fasolato, C. When, where and how? Focus on neuronal calcium dysfunctions in Alzheimer’s Disease. Cell Calcium 2016, 60, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Tong, B.C.-K.; Wu, A.J.; Li, M.; Cheung, K.-H. Calcium signaling in Alzheimer’s disease & therapies. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1745–1760. [Google Scholar] [PubMed]
- Hay, Y.A.; Deperrois, N.; Fuchsberger, T.; Quarrell, T.M.; Koerling, A.-L.; Paulsen, O. Thalamus mediates neocortical Down state transition via GABAB-receptor-targeting interneurons. Neuron 2021, 109, 2682–2690.e5. [Google Scholar] [CrossRef] [PubMed]
- Hazra, A.; Corbett, B.F.; You, J.C.; Aschmies, S.; Zhao, L.; Li, K.; Lepore, A.C.; Marsh, E.D.; Chin, J. Corticothalamic network dysfunction and behavioral deficits in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2016, 44, 96–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagirdar, R.; Chin, J. Corticothalamic network dysfunction and Alzheimer’s disease. Brain Res. 2019, 1702, 38–45. [Google Scholar] [CrossRef]
- Martín-Belmonte, A.; Aguado, C.; Alfaro-Ruíz, R.; Moreno-Martínez, A.E.; de la Ossa, L.; Martínez-Hernández, J.; Buisson, A.; Shigemoto, R.; Fukazawa, Y.; Luján, R. Density of GABAB Receptors Is Reduced in Granule Cells of the Hippocampus in a Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 2459. [Google Scholar] [CrossRef] [Green Version]
- Habib, N.; McCabe, C.; Medina, S.; Varshavsky, M.; Kitsberg, D.; Dvir-Szternfeld, R.; Green, G.; Dionne, D.; Nguyen, L.; Marshall, J.L.; et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci. 2020, 23, 701–706. [Google Scholar] [CrossRef]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Woolley, M.L.; Ballard, T.M. Age-related impairments in operant DMTP performance in the PS2APP mouse, a transgenic mouse model of Alzheimer’s disease. Behav. Brain Res. 2005, 161, 220–228. [Google Scholar] [CrossRef]
- Burrinha, T.; Martinsson, I.; Gomes, R.; Terrasso, A.P.; Gouras, G.K.; Almeida, C.G. Upregulation of APP endocytosis by neuronal aging drives amyloid-dependent synapse loss. J. Cell Sci. 2021, 134, jcs255752. [Google Scholar] [CrossRef] [PubMed]
- Born, H.A. Seizures in Alzheimer’s disease. Neuroscience 2015, 286, 251–263. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leparulo, A.; Bisio, M.; Redolfi, N.; Pozzan, T.; Vassanelli, S.; Fasolato, C. Accelerated Aging Characterizes the Early Stage of Alzheimer’s Disease. Cells 2022, 11, 238. https://doi.org/10.3390/cells11020238
Leparulo A, Bisio M, Redolfi N, Pozzan T, Vassanelli S, Fasolato C. Accelerated Aging Characterizes the Early Stage of Alzheimer’s Disease. Cells. 2022; 11(2):238. https://doi.org/10.3390/cells11020238
Chicago/Turabian StyleLeparulo, Alessandro, Marta Bisio, Nelly Redolfi, Tullio Pozzan, Stefano Vassanelli, and Cristina Fasolato. 2022. "Accelerated Aging Characterizes the Early Stage of Alzheimer’s Disease" Cells 11, no. 2: 238. https://doi.org/10.3390/cells11020238
APA StyleLeparulo, A., Bisio, M., Redolfi, N., Pozzan, T., Vassanelli, S., & Fasolato, C. (2022). Accelerated Aging Characterizes the Early Stage of Alzheimer’s Disease. Cells, 11(2), 238. https://doi.org/10.3390/cells11020238