1. Introduction
Myelinating oligodendrocytes (OLs) are essential for saltatory nerve conduction in the central nervous system and are involved in the metabolic support of neurons and modulation of neuronal excitability [
1,
2]. OLs are derived from oligodendrocyte precursor cells (OPCs) and both cell types are considered heterogeneous populations with regional specifications and functionally different states [
3,
4,
5,
6]. Moreover, OL dysfunction is associated with several major neurological diseases, e.g., multiple sclerosis, leukodystrophy, stroke, and schizophrenia [
1,
7]. The advent of human-induced pluripotent stem cells (hiPSCs) paved the way for generating hiPSC-derived OPCs (hiPSC OPCs) and hiPSC-derived OLs (hiPSC OLs) that improved our understanding of human biology and enabled dissection of pathophysiological mechanisms, including cell-based therapies [
8,
9].
Initial protocols applied small molecules and peptides but took 75 to 200 days to generate hiPSC OPCs and hiPSC OLs [
8,
9], thus limiting larger-scaled studies such as compound screens or large cohort investigations [
10]. A subsequent, more rapid strategy used overexpression of lineage-specific transcription factors (TFs), which forces oligodendroglial differentiation and allows generation of induced OPCs (iOPCs) and iOLs within 20 to 30 days [
8,
9]. Most protocols identified overexpression of SRY-box 10 (SOX10) as sufficient for iOL generation [
11,
12,
13] and overexpression of SOX9 alone was also recently shown to be an efficient approach [
14]. Garcia-León et al., suggested that SOX10 (referred to as S) alone is most efficient for iOL generation [
13]. In contrast, Ehrlich et al., showed that the combination SON–consisting of S, oligodendrocyte transcription factor 2 (OLIG2, referred to as O), and the NK6 homeobox 2 (NKX6.2, referred to as N)–enriched the yield of iOLs [
11] and allowed direct reprogramming of human fibroblasts [
15]. In addition, Pawlowski et al., applied the combination SO to generate iOLs from embryonic stem cells [
12]. Nonetheless, iOLs displayed transcriptional similarities to primary OLs and allowed axonal myelination in vitro and in vivo [
11,
13]. Despite these functional and morphological similarities between studies, several experimental conditions beyond the different TF combinations varied, preventing direct comparison of the findings. First, the neural patterning strategies applied before initiating TF overexpression differed: Garcia et al., performed neural induction on a monolayer [
13], whereas Ehrlich et al., performed free-floating embryoid body formation with neural patterning and applied different combinations of small molecules [
11]. Second, different configurations of the TF-expressing lentivirus constructs were applied: Ehrlich et al., expressed all TFs as multicistronic units, however, Garcia et al., used combinations of individual TFs expressing lentiviral constructs, which likely reduced the efficacy of multi-TF combinations in generating iOLs compared with that of single TFs. Moreover, to date, no studies have addressed the fate and heterogeneity of individual iOPCs and iOL populations.
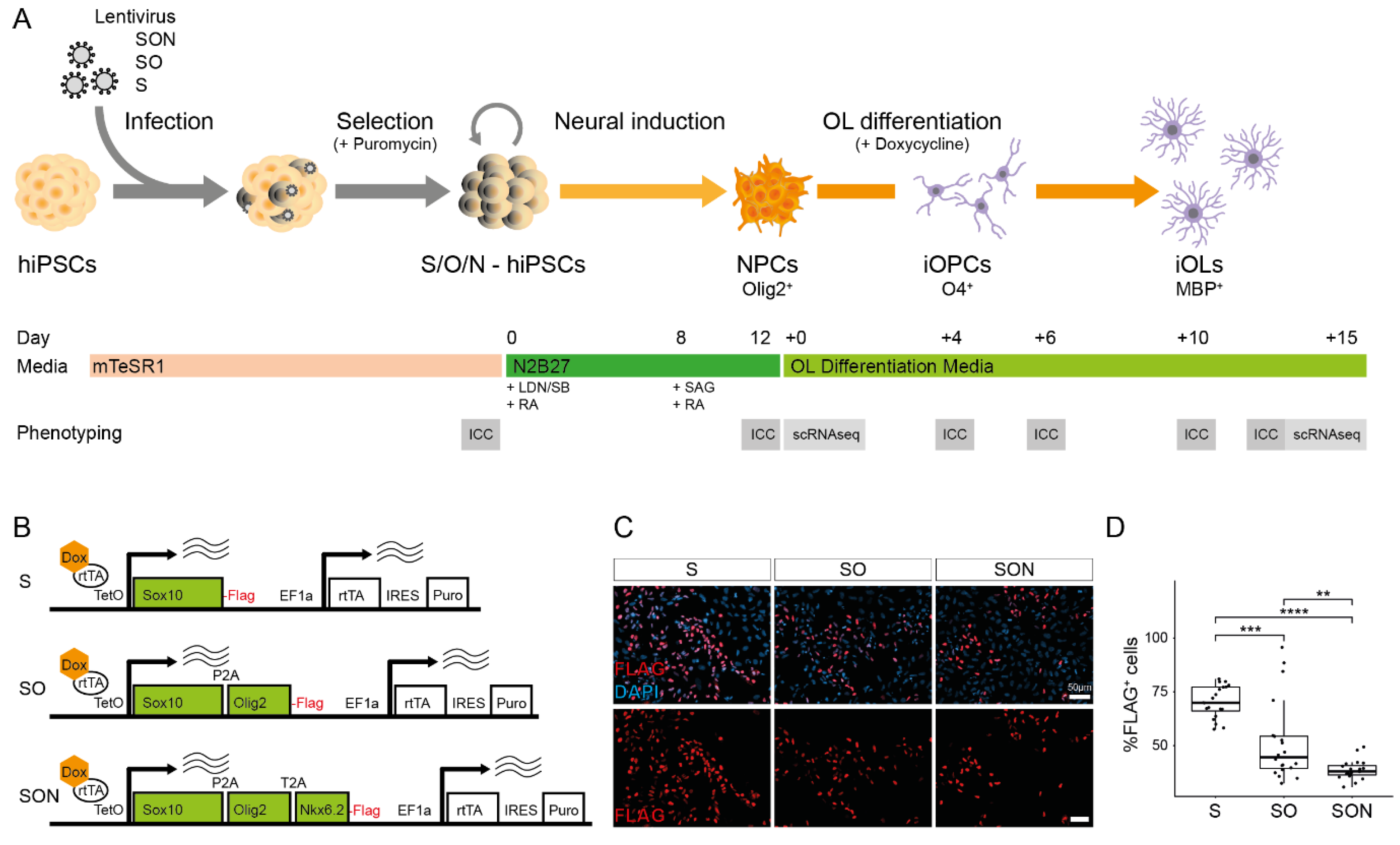
Therefore, we systematically compared S-, SO- and SON-directed differentiation of individual oligodendroglial lineage cells by using a streamlined protocol in which all TF combinations were expressed from an identical lentivirus backbone and all cell culture conditions were standardized. We show that S-, SO- and SON-directed differentiation were all sufficient to generate high yields of O4+ iOPCs, however, SON provided significant more yield than SO and S. Further investigations with S and SON show that SON allows an earlier generation of MBP+ iOLs with higher yields and more complex morphology. Subsequent scRNAseq experiments including RNA velocity analysis reveals a fastened directed oligodendroglial differentiation using SON and higher maturation stages of SON-iOLs compared to S-iOLs. We show that scRNAseq including RNA analysis is not limited to dissecting a static stage but allows to dissect the time-dependent dynamics of directed differentiation and highlights that SON-directed differentiation might be better suited for research with a focus on more mature iOL.
2. Materials and Methods
2.1. Lentiviral Vectors
The cDNA sequences of human SOX10 (S), SOX10-P2A-OLIG2 (SO) and SOX10-P2A-OLIG2-T2A-NKX6.2 (SON) were synthesized as plasmids (GenScript, Piscataway, NJ, USA), with each open reading frame flanked by attB1 and attB2 sites for Gateway recombination cloning. Synthesized genes were recombined into pDONR/Zeo (Thermo Fisher Scientific, Waltham, MA, USA, #12535035) to yield Entry clones. Entry clones were finally recombined into pINDUCER21-puro_Gateway-3xFLAG (Addgene, Watertown, MA, USA, plasmid #172981). HEK293 cells (ATCC) were used for lentivirus production. S-, SO- and SON-lentiviral vectors (12 µg of each) were transfected with packaging plasmids psPAX2 (Addgene plasmid #12260, 9 µg) and pMD2.G (Addgene plasmid #12259, 4 µg) with 4 µg polyethylenimine per 1 µg plasmid-DNA (Polysciences, Warrington, FL, USA, #9002-98-6). Then, 48 h after lentiviral transfection, the culture medium was collected, filtered through a 0.45 µM PVDF filter, precipitated with PEG-it (System Biosciences, Palo Alto, CA, USA, #LV810A-1) according to the manufacturer’s instructions, resuspended in DPBS, and stored at −80 °C for further use.
2.2. HiPSC Lines, HiPSC Cultivation and Lentiviral Transfection
hiPSC were derived from the hiPSC biobank at the Department of Psychiatry and Psychotherapy, University Hospital, LMU Munich, Munich, Germany. hiPSC were generated from PBMCs according to a previous protocol [
16]. Conventional hiPSC verification included validation of pluripotency by immunocytochemistry (Tra1-60, NANOG, OCT4, SOX2), genomic integrity by digital karyotyping [
17] (pipeline available at
https://gitlab.mpcdf.mpg.de/luciat/cnv_detection.git, last accessed 28 June 2021), and successful differentiation into all three germ layers [
18]. hiPSC were tested negative for HIV, HCV, CMV (by Synlab, Munich, Germany) and free of Mycoplasma infections (by Eurofins, Ebersberg, Germany). All used hiPSC lines and passaging numbers are annotated in
Table S1. Technical experiments were performed with line PSYLMUi001-A, SON-directed oligodendroglial differentiation efficiency was replicated with six independent hiPSC lines as annotated in
Table S1.
hiPSCs were cultivated in feeder-free conditions with iPS-Brew (Miltenyi Biotec, Bergisch Gladbach, Germany, #130-104-368) on Vitronectin (Thermo Fisher Scientific, Waltham, MA, USA, #A14700). Lentiviral transfection was performed in iPS-Brew, supplemented with 1 µM Y-27632 (Rock-Inhibitor, Selleckchem, Houston, USA, #S 1049) and plated with 5 × 10
4 cells/cm
2. Selection with 1 ug/mL puromycin (Thermo Fisher Scientific, Waltham, MA, USA, #A1113803) was performed 48 h after transfection. We performed the first passage after transfection with Accutase (Sigma-Aldrich, St. Louis, USA, #A6964) and single cells. Subsequent passaging for expansion or maintenance was performed as hiPSC clump passaging with 0.5 µM EDTA (Thermo Fisher Scientific, Waltham, MA, USA, #15575-020). An overview of used materials is annotated in
Table S2. Independent experiments were based on independent lentiviral infections.
2.3. Neural Induction
Neural induction and oligodendroglial differentiation were performed according to published protocols [
13,
19] with the modification that lentiviral transduction was performed before and not after neural induction (
Figure 1A). At least one passage before neural induction, the hiPSC medium was changed to mTeSR1 (StemCell, Vancouver, Canada, #85850). Two days before neural induction, hiPSCs were singularized with Accutase, resuspended in mTeSR1, supplemented with 1× RevitaCell (Thermo Fisher Scientific, Waltham, MA, USA, #A2644501), and plated at a cell density of 2 × 10
4 cells/cm
2 on 12- and 24-well plates (Corning, New York, NJ, USA, #CORN3513 and #CORN3526) coated with Matrigel (BD Bioscience, San Jose, CA, USA, #354277). Two days after cultivation in mTESR1, neural induction was initiated by medium exchange to N2B27, which consists of DMEM/F-12 with GlutaMAX™ (Gibco, brand of Thermo Fisher Scientific, Waltham, MA, USA, #31331028), 1× N2 (Gibco, #17502048), 1× NEAA (Gibco, #11140035), 50 µM Mercaptoethanol (Gibco, #21985023) and 25 µg/mL Insulin (Sigma, #I9278), supplemented with 10 µM SB431542 (StemCell, Vancouver, Canada, #72232), 1 µM LDN193189 (StemCell, Vancouver, Canada, #72147) and 0.1 µM retinoic acid (RA)(Sigma-Aldrich, St. Louis, MO, USA, #R2625-50MG). A daily full media change of 0.5 mL per 24 wells and 1 mL per 12 wells was performed and media volume was doubled after day 4 of neural induction. From day 8 after neural induction, daily media change was performed with N2B27 supplemented with 0.1 µM RA and 1 µM SAG (Millipore, Burlington, MA, USA, #566660) until day 12.
2.4. Oligodendroglial Differentiation
Twelve- or twenty-four-well plates were coated with PLO/Laminin. First, 50 µg/mL poly-L-ornithine (Sigma, P4957) in DPBS (ThermoFisher Scientific, Waltham, MA, USA, #1419009400) was coated overnight at 37 °C. Then, 3× washing steps were performed with DPBS, and plates were incubated overnight with 10 µg/mL mouse laminin (Sigma, #L2020) at 37 °C. Day 12 NPCs were passaged as single cells upon Accutase treatment and were seeded at a density of 150,000 cells/cm2 in N2B27 supplemented with 0.1 µM RA, 1 µM SAG (Millipore, #566660), and 1× RevitaCell. The next day, directed OL differentiation was initiated by adding OL differentiation medium (OL-DM), which consists of N2B27 supplemented with 10 ng/mL PDGF-AA (PeproTech, brand of Thermo Fisher Scientific, Waltham, MA, USA, #100-13A), 10 ng/mL IGF1 (PeproTech, #100-11), 5 ng/mL HGF (PeproTech, #100-39), 10 ng/mL NT3 (Peprotech, #AF-450-03), 0.1 ng/mL Biotin (Sigma, #B4639), 1 mM dbcAMP (Sigma, #D0627), 60 ng/mL T3 (Sigma, #T6397) and 1 µg/mL doxycycline (Clontech, brand of Takara Bio, Shiga, Japan, #NC0424034). Full media change was performed every second day and puromycin selection was performed from Day + 2 to Day + 4 by supplementing media with 1 µg puromycin (ThermoFisher Scientific, Waltham, MA, USA, #A1113803). Cells were passaged at Day + 10 or cryopreserved for further use. Singularized cells were resuspended in OL-DM, supplemented with 1× RevitaCell and mixed 1:1 with cooled ProFreeze CDM (Lonza, Basel, Switzerland, #BEBP12-769E) for freezing purposes. Cells were stored overnight in a freezing container (Nalgene Mr. Frosty) at −80 °C and subsequently placed in liquid nitrogen.
2.5. O4 Microbeads Purification
To select O4+ cells, we performed microbead purification with anti-O4 microbeads (Miltenyi Biotec, #130-096-670) according to the manufacturer’s instructions.
2.6. Immunofluorescence Staining and Imaging Analysis
Before fixation, cells were washed with PBS. Then, they were fixed with 4% PFA for 10 min at room temperature (RT). Subsequently, 3× washing with PBS was performed. Permeabilization and blocking were performed with 0.1% Triton and 5% goat serum in PBS for 60 min at RT. Triton was omitted for O4 staining. Primary antibodies were added in PBS supplemented with 0.1% Triton and 1% goat serum and incubated overnight at 4 °C. On the next day, the primary antibody solution was removed by 3× PBS washing. The respective secondary antibodies in PBS were added and the cells were incubated for 60 min at RT. The secondary antibody solution was removed by 3× PBS washing and nuclei were counterstained with DAPI, which was added during the second washing step. Finally, samples were washed 1× with water and mounted onto microscope slides. Imaging was performed on an Axio Observer.Z1 (Zeiss) inverted microscope and fields of view (FOV) were taken at randomly defined but fixed positions for each well at 20× magnification. Image analysis was performed with the Fiji [
20] and its plugin Simple Neurite Tracer [
21]. Average values per FOV were used for subsequent statistical analysis.
2.7. Single-Cell RNA Sequencing and Data Analysis
2.7.1. Sample Processing
Cells were detached and singularized with Accutase supplemented with DNAse1 (Merck, Darmstadt, Germany, #DN25-1G), cell aggregates were removed with 30 µm Pre-Separation Filters (Miltenyi Biotec, Bergisch-Gladbach, Germany #130-041-407), and cells were kept on ice in cold PBS with 1% BSA. Small aliquots of the samples were stained with trypan blue and cell density, viability, and multiplet rate were determined in an improved Neumann counting chamber. The density was adjusted to 2500 cells/µL. The multiplet rate was less than 7% in all samples.
2.7.2. Library Preparation
The single-cell libraries were prepared with the SureCell WTA 3′ Kit (Illumina, San Diego, CA, USA) on a ddSEQ Single-Cell Isolator (Bio-Rad, Hercules, USA) in accordance with the manufacturer’s protocol. Library quality and yield were evaluated with a high-sensitivity DNA kit on a Bioanalyzer 2100 (Agilent, Santa Clara, CA, USA) according to the manufacturer’s specifications.
2.7.3. Sequencing
Libraries for scRNAseq experiments were sequenced on a NextSeq550 (Illumina) in paired-end mode with 69 cycles in Read1 and 80 cycles in Read2. Subsequently, the sequencing data were demultiplexed with bcl2fastq (Illumina).
2.7.4. Upstream Analysis of scRNAseq Data
Reads were quality-checked with FastQC (v0.11.9). Barcodes in Read1 were identified, added to the Read2 BAM file as an XC tag, and quality-checked with the ddSeeker tool [
22], which found that 91% of barcodes were good quality in all samples. The tagged BAM files were further processed with Drop-seq tools (v2.3.0). A metabundle was created with the Ensembl annotation release 100 of the GRCh38 human genome (
create_Drop-seq_reference_metadata). A non-human fragment (3′LTR and WPRE) and the 3xFLAG-Tag region of the expression constructs were added to the genome FASTA and GTF to determine the construct expression levels. The data were filtered for rejected barcodes. The reads were sorted by query name, and the BAM files were converted back to FASTQ for mapping. Read2 was mapped by using the STAR aligner (v2.7.5) with default settings. Aligned data were merged with the unaligned but barcode-tagged data. Functional annotation was added and any remaining substitution and synthesis errors in the cell barcodes were repaired to create the final BAM file. Digital gene expression data were extracted (
Digital Expression) for the 2000 most abundant cell barcodes in each sample. The threshold was chosen to be well beyond the cut-off suggested by the knee plot generated by ddSeeker’s
make_graphs.R.
In the RNA velocity analysis pipeline, to acquire separate count matrices for spliced and unspliced reads we estimated counts with DropEst (v0.8.6) instead of Drop-seq’s
Digital Expression. The following command was used, in accordance with the suggestions of the developers of Velocyto [
23]:
dropest -m -V -b -f -g%GTF file from Drop-seq metabundle% -L eiEIBA -m -c %FILEPATH%/dropEst/configs/drop_seq_velocyto.xml. Note that the barcode tag was changed from
BC to
XC in
drop_seq_velocyto.xml to make the Drop-seq output compatible with Dropest.
2.7.5. Downstream Analysis of scRNAseq Data
The scRNAseq data were further processed with the R (v4.0.4) package
Seurat (v4.0.1) [
24]. The UMI count tables were imported in R, and Seurat objects were created for each sample. We filtered for features expressed in at least three cells in each dataset and for cells with at least 200 features in the first step. Mitochondrial RNA content was determined by grepping for “^
MT-” (
PercentageFeatureSet). We filtered the cells further for barcodes with less than 5% mitochondrial RNA content, at least 1700 UMI counts in the NPC and SON sample and 1000 counts in the S sample, and a minimum of 1000 detected features. After these filtering steps, 1194 cells were left in total.
2.7.6. Normalization of scRNAseq Data
All datasets were merged into one object. To check for artifacts potentially introduced by merging, we additionally performed all analyses on each sample dataset individually. The data were normalized by the single-cell transform procedure (
SCTransform) [
25], which regressed out the percentage of mitochondrial transcripts.
2.7.7. Dimension Reduction and Clustering of scRNAseq Data
Dimension reduction was performed as a principal component analysis (
RunPCA). The previously identified 3000 most variable features (
FindVariableFeatures) were used as input. The first 30 principal components were computed. Based on an elbow plot, as suggested in Macosko et al., 2015, we decided to use the first 15 principal components for downstream analysis. We performed SNN graph-based clustering with a resolution of 0.23 (
FindNeighbors, FindClusters); this clustering returned 5 distinct clusters of cells. Next, we computed a UMAP embedding (
RunUMAP; wrapper function for integration of the
Python library
umap-learn; [
26]). Later in the project, we revisited the clustering and increased the resolution to 0.28, which split the iOPC cluster into two subclusters, which we then used for differential expression analysis.
2.7.8. Integration with Bulk RNAseq Data
To integrate our data with publicly available bulk RNA-seq data, we followed the procedure from Ng et al. [
14]. We used a primary cell dataset from mouse brain tissue because human datasets are commonly based on postmortem samples. The dataset is available on the Gene Expression Omnibus website under the accession number GSE52564 [
27]. Briefly, we created an average expression table and excluded features that were expressed in less than 10% of all cells in our dataset. The filtering step was introduced to avoid artifacts resulting from bad coverage inherent to single-cell RNA-seq data and low expression. The resulting list was then filtered for variably expressed genes in our dataset; such genes were computed as differential expression with a minimum log2-fold change of 0.25 (
FindAllMarkers). This step improved the signal-to-noise ratio and reduced potential batch effects. Next, we filtered the reference dataset and our dataset for mouse/human homologs. Finally, a list of about 3000 features was entered into the analysis. From our datasets, we excluded all iOPC cells in the NI Day + 0 sample because of their low number and their high probability of being misclustered NPCs. From the reference dataset neurons, we retained myelinating oligodendrocytes (MO), newly formed oligodendrocytes (NFO), and oligodendrocyte progenitor cells (OPC). First, we calculated feature variability (
rowVars) with the
DESeq2 R package [
28] for variance stabilization (
VarianceStabilizingTransformation; vst). Then, we used the 2000 most variable features for the downstream analysis. A principal component analysis (PCA) was performed to check for batch effects and other artifacts, and Manhattan distances were calculated between samples and features. These distances were used for hierarchical clustering and shown in heatmaps. Additionally, Spearman correlation coefficients were calculated to ensure that our data correlated appropriately with the reference data.
2.7.9. Differential Expression Analysis
Differential gene expression was analyzed with
Seurat v3 [
29]. Genes were selected for expression in a minimum of 10% of all cells included in the analysis and for variability between groups. The data used were normalized but not integrated because the integration procedure smoothens expression differences, which leads to the underestimation of transcriptional differences. The results were tested in a Wilcoxon rank-sum test, and
p values were adjusted with the Bonferroni procedure. Marker genes for characterization of the clusters were identified with a minimum log2-fold change in average expression (avg_log2FC) of 0.25 between the respective cluster and background (
FindAllMarkers()). In pairwise comparisons between clusters, an avg_log2FC threshold of 1 was chosen because the lower threshold used for marker identification produced a high level of noise in downstream analyses.
2.7.10. Analysis of Maturation Marker Load and Construct Expression
Construct counts based on the 3′LTR-WPRE sequence were included in the
sctransform regression model for normalization (see
Section 2.7.6.) and used for construct expression equivalent. The FLAG tag region was not detected in the sequencing data.
For the maturation marker load, a set of genes with increasing levels of expression during oligodendrocyte-lineage differentiation [
9] and that were detected with scRNAseq was chosen, specifically:
CNP,
CD9,
CLDN11,
GALC,
PLP1,
MAG,
MAL,
MBP,
MOBP, and
MYRF. The normalized expression level of these genes was then summarized for each cell. A Spearman correlation factor between construct expression level and marker load was computed and tested for S and SON Day + 15 samples.
2.7.11. Hypergeometric Gene Ontology Term Enrichment Analysis
For hypergeometric enrichment analyses of gene ontology (GO) terms, we used the GOrilla online tool [
30]. Unranked gene lists were compared, of which one comprised all genes that entered the respective differential expression analysis (background) and the other identified differentially expressed genes (target). Only process terms went into the analysis. Enriched terms were filtered with an FDR-adjusted q-value threshold of 10
−3 and a B value of less than 500.
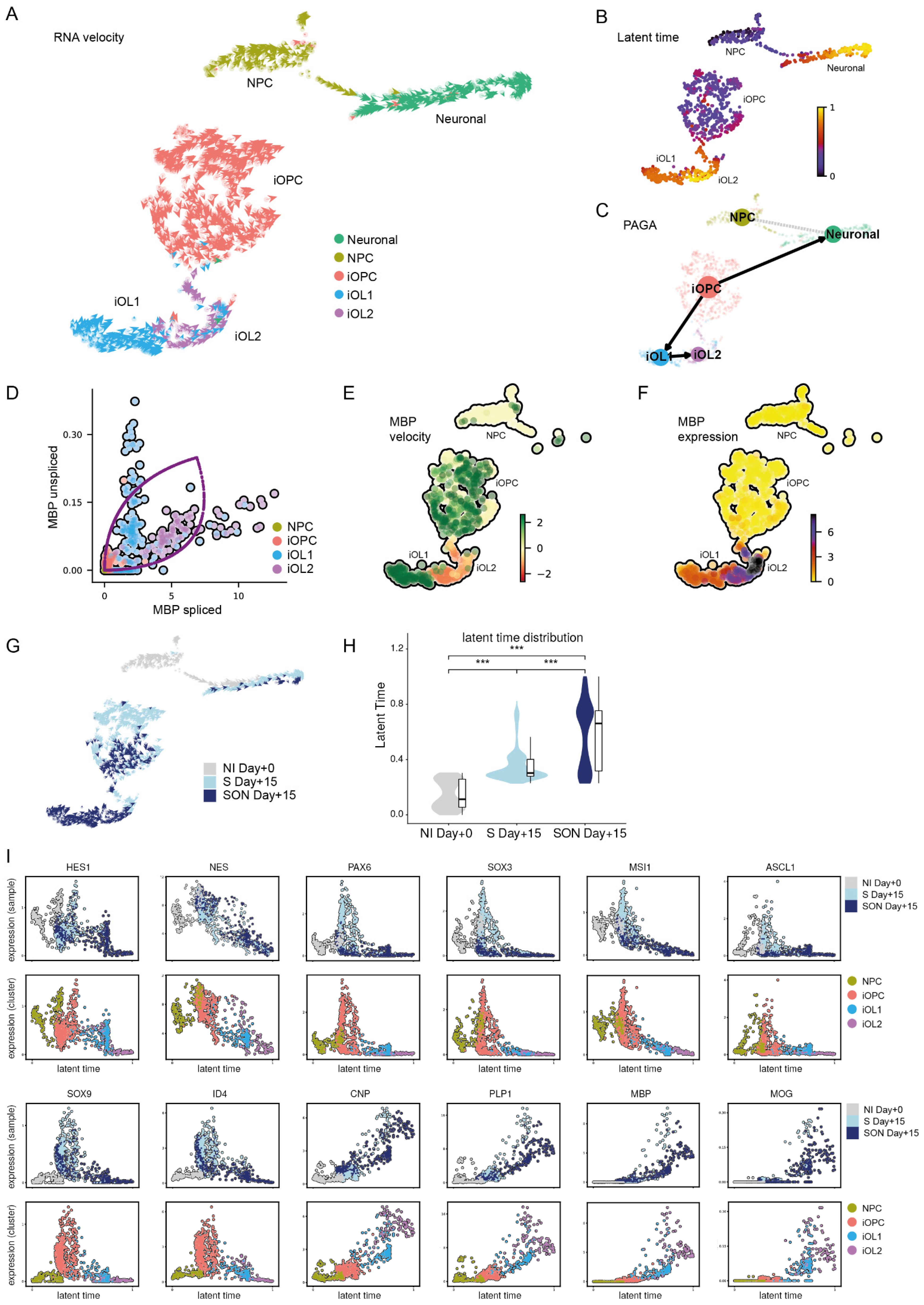
2.7.12. RNA Velocity Analysis
For RNA velocity analysis, we first created a loom file for each sample from the Dropest output by using the command line
Python implementation of the
velocyto (v0.17.17) [
23]. Next, we combined these loom files for downstream analysis. Additionally, we analyzed each sample independently to make sure that merging the samples had not introduced major artifacts. We used the
scVelo Python library [
31] for the rest of the pipeline. We imported the combined loom file as anndata with the
anndata library [
32] and imported the cellIDs, UMAP coordinates, and cluster assignment from the
Seurat pipeline. The cells were filtered for these IDs and their UMAP coordinates, and clusters were assigned. Further, the cells were filtered and normalized by using the default parameters with
pp.filter_and_normalize(). Briefly, highly variable genes were selected, and the data were normalized by total library size and subsequently transformed to a logarithmic scale. For quality control, the fraction of unspliced transcripts was determined and found to be 13%, which is well within the lower range of 10% to 25% that is typically found in scRNAseq data. A fraction of this size is to be expected from a 3′ UTR-enriched RNA-seq dataset and is sufficient for RNA velocity estimation [
31,
33]. Moments were calculated and a full dynamic model was fitted to the data [
31] (
tl.recover_dynamics(), tl.velocity(mode = “dynamical”). This model was used to compute the velocity graph (
tl.velocity_graph()).
2.7.13. Latent Time and PAGA Analysis
In addition, the gene-shared latent time was computed (
tl.latent_time()) to create a pseudo-timeline that was used to inform cell-to-cell transition inference. A PAGA analysis was computed to find cluster-to-cluster transitions (
tl.paga_plot()) [
34]. Latent times were reimported in R to be used as a measure for maturity and were tested between samples with a Wilcoxon rank-sum test.
2.8. Statistics
Data were first tested for normal distribution with a Shapiro–Wilk test. We used both parametric (Student’s t-test, ANOVA) and nonparametric analyses (two-sample Wilcoxon’s rank-sum test, Kruskal–Wallis test, chi-squared test, Fisher’s exact test), depending on the specific distribution properties of the data. Significance was set at * p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001.
4. Discussion
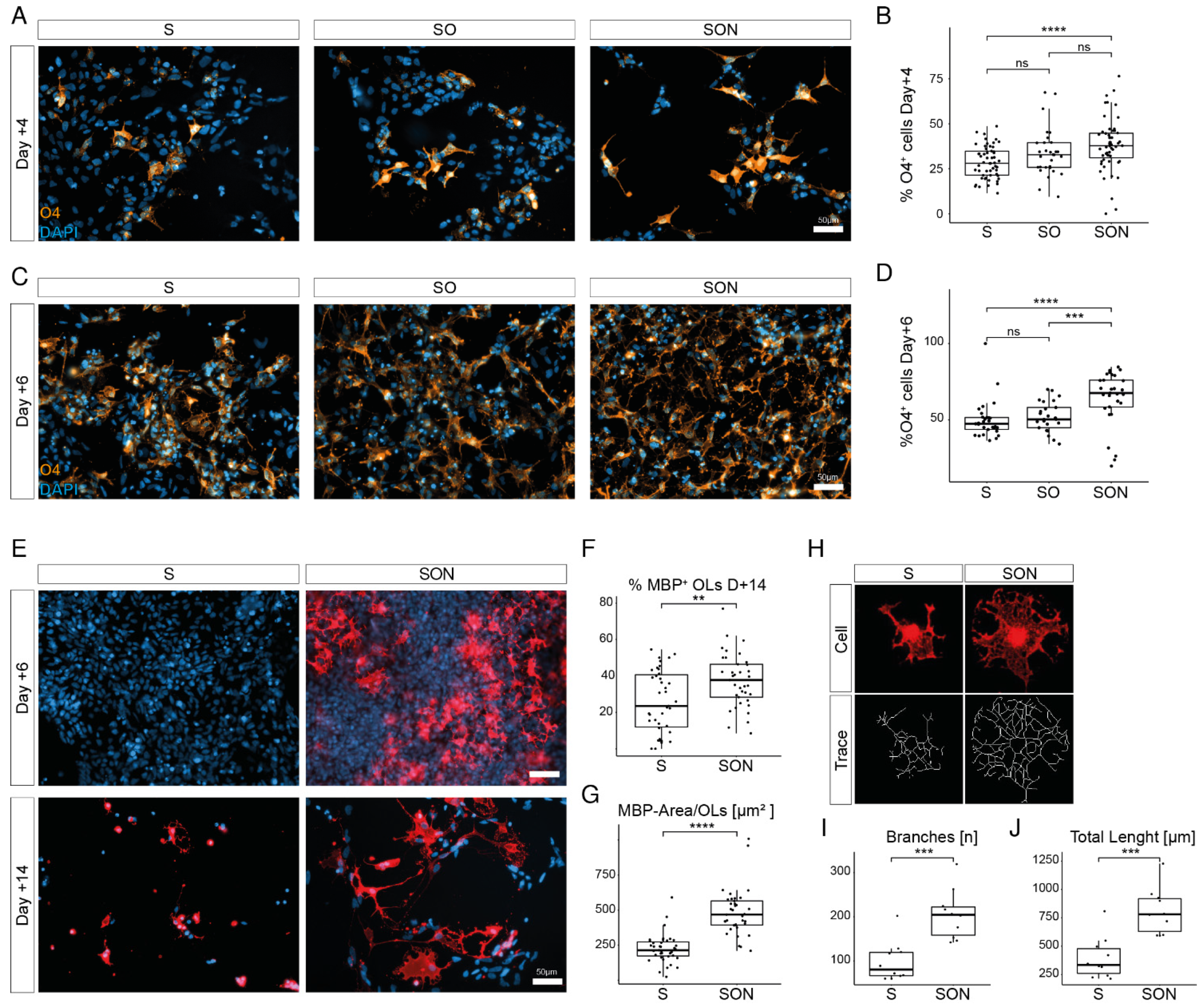
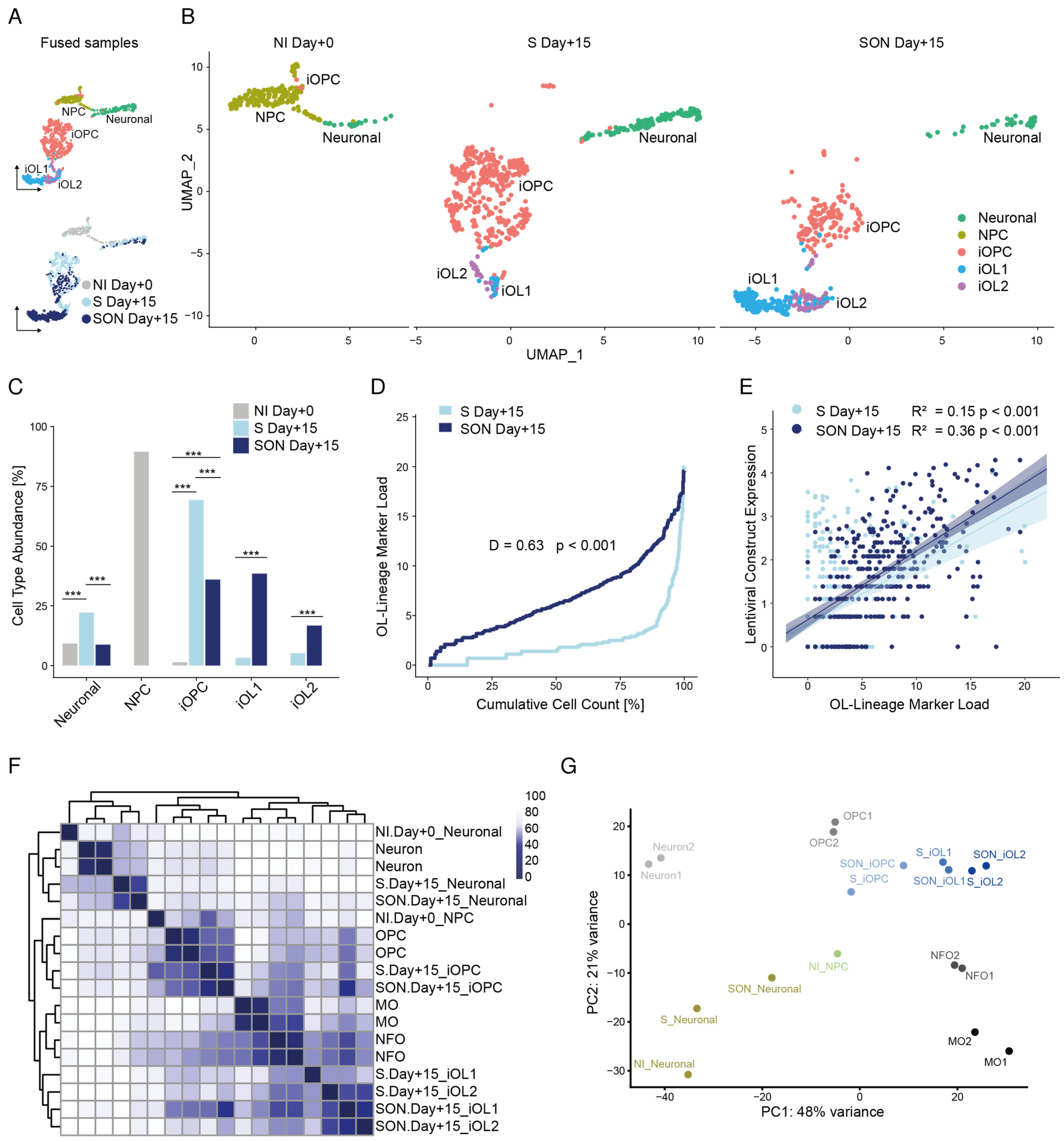
In our comparative study, we confirmed that S-, SO- and SON-directed conversion are all sufficient to rapidly generate oligodendroglial lineage cells from hiPSCs. As a result of the side-by-side analysis at the single-cell resolution, we showed that the application of SON generated more O4
+ late-stage iOPCs and more mature and complex MBP
+ iOLs than the application of S alone, supporting previous observations [
11]. S alone was most effective in generating a mixed population of iOPCs (see below). In agreement with previous studies [
36,
37], we observed a higher level of expression of smaller-sized constructs. However, an intermediate selection step performed in our protocol normalized these differences and allowed us to compare TF combinations irrespective of construct size. As mentioned above, a more recent study that used a systematic screen for lineage-converting TFs found that SOX9 was capable of inducing oligodendroglial cells on its own [
14]. Thus, similar to other SOX family members, both SOX9 and SOX10 can be considered as pioneering TFs in the oligodendroglial lineage that prime critical steps essential for stem cell conversion [
38]. SOX9 operates genetically upstream of SOX10 in the oligodendroglial lineage and—despite the fact that SOX9 can generate myelinating iOLs in a 3D system [
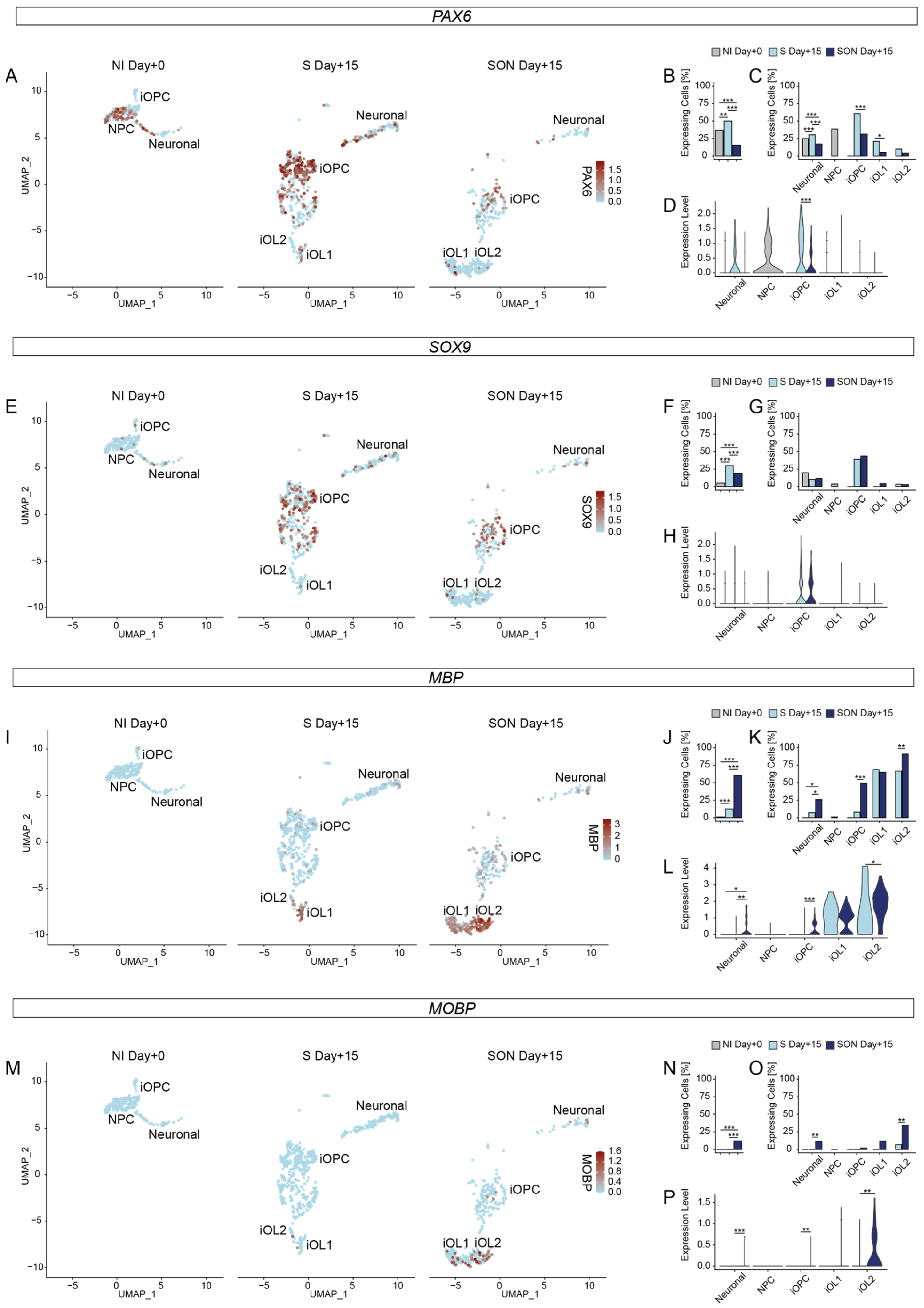
14]—it remains an open question whether SOX9 might generate more earlier stage iOPCs than SOX10 in a cell-autonomous setting such as the one applied in this study. We observed substantial differences in iOPC and iOL heterogeneity and dynamics of differentiation between S- and SON-directed oligodendroglial differentiation. Despite the finding that OPC marker genes such as
SOX9,
ST8SIA1,
CNP,
ID2 and
ID4 reached similar levels in S- and SON-iOPCs, the expression level of oligodendroglial maturation markers such as
GALC and
MOBP was higher in SON-iOLs than in S-iOLs under the applied conditions. Moreover, the cell cluster composition differed dramatically: Mature iOLs, that express
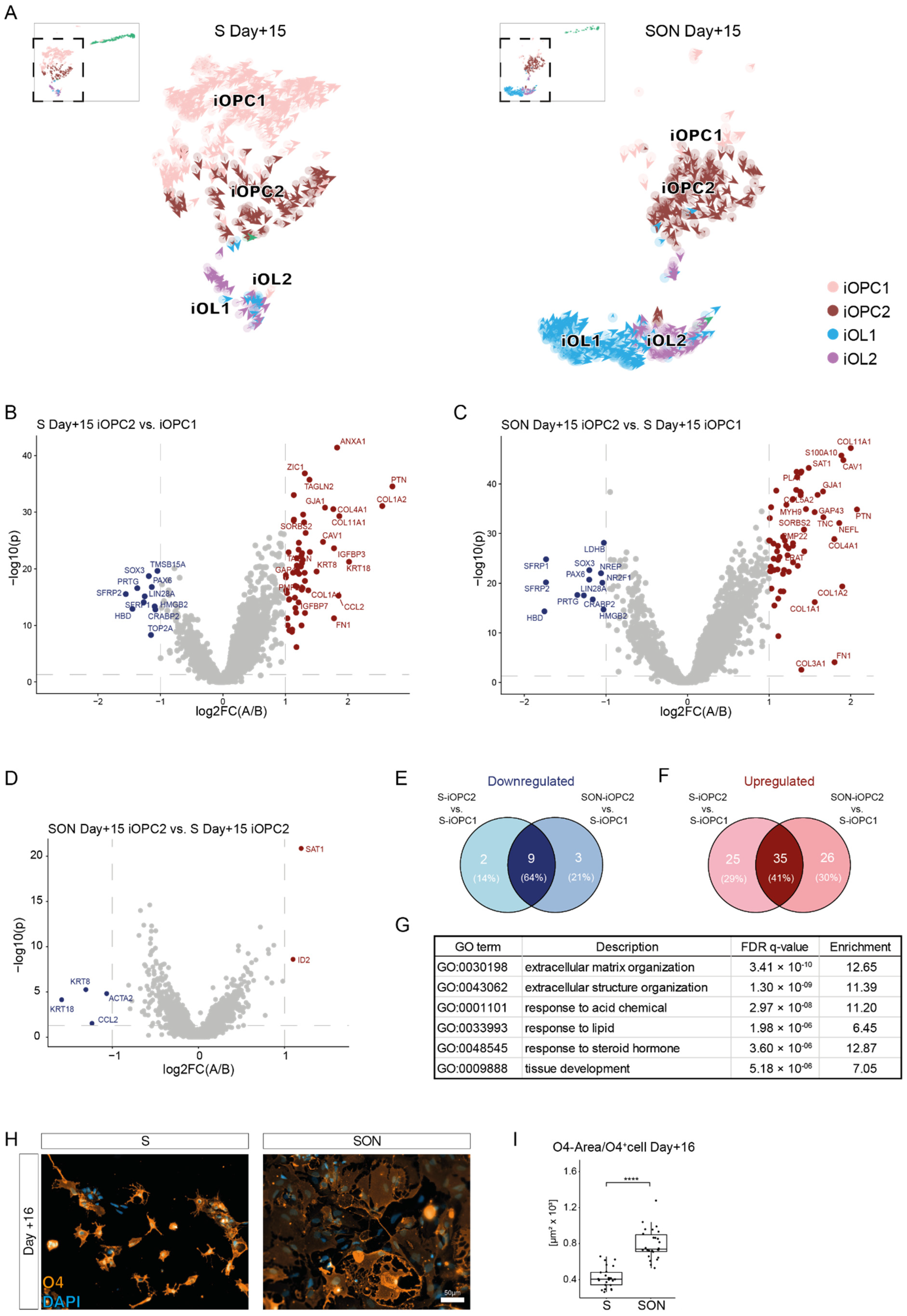
PLP1, GALC, MBP, MOBP, were far more abundant upon SON-directed differentiation but the iOPC cluster was larger and more diverse in the S-directed condition (see below).
To improve purity and scalability, we introduced the lentivirus-encoded lineage transcription factors at the level of iPSC cultivation followed by two puromycin selection steps, one before and one after initiation of iOPC/iOL differentiation; this protocol was robust for different iPSC lines obtained from six different human donors. The advantage of this approach is that less lentivirus is needed and only infected S/O/N-hiPSCs are proliferative and can be amplified, frozen, banked, and recovered en masse at the time needed. In the future, this approach will be particularly suited to enable larger-scaled case-control comparisons and may allow also genetic and compound screenings requiring high numbers of dedicated cells. Nonetheless, epigenetic lentivirus silencing is a confounding factor [
39]. The drop of FLAG
+ hiPSCs after doxycycline induction is likely due to the loss of cleavage efficiencies by single versus double P2A/T2A element-containing constructs [
40].
In both conditions (S and SON), the lentiviral construct expression level was associated with a higher level of OL-lineage marker load. However, SON-directed differentiation caused a much higher marker load underlining a more maturating oligodendroglial differentiation compared to S-directed differentiation in line with previous work [
11].
On the iOPC/iOL level, lentiviral construct transcriptome levels within the clusters were comparable between S and SON indicating that the expression levels are unlikely to cause cluster diversity and that this diversity is most likely attributed to the different TF combinations. However, expression levels were higher in iOL clusters compared to iOPC clusters and the neuronal cluster. Thus, it might be possible that higher expression levels sustain the directed differentiation. Thus, for genetic and chemical screens [
41,
42], safe harbor strategies may be a better choice to express OL-lineage-converting TFs and previous work could highlight that safe harbor strategies using SOX10 were sufficient to generate high yields of homogenous MBP
+ iOLs [
19].
However, safe harbourstrategies will be complicated to apply in case-control comparisons, where many subjects/cell lines are to be compared. Nonetheless, a safe harbor-mediated CRISPR/Cas9-based genetic screen has been described for hiPSC-derived NGN2-directed iNeurons [
41], showing the potential of this approach also for other cell types. Safe harbor strategies that apply CRISPR/Cas9 technology could also be applied to modify directed differentiation. In contrast to exogenous overexpression of TFs for directed differentiation, a recently published study applied CRISPR/Cas9-mediated activation of endogenous
Sox10,
Olig2, and
Nkx6.2 by specific gRNAs to differentiate mouse neural stem cells and mouse embryonic fibroblasts to O4
+ and MBP
+ oligodendroglial cells [
43].
In this study and previous studies that used the same neuronal patterning approach [
13,
19], the expression of early-stage OPCs markers, such as
CSPG4 (
NG2) and
PDGFRA, was barely detectable in iOPCs. Thus, with both SON and S this directed differentiation approach can be assumed to bypass early OPC stages. Chemical differentiation approaches may therefore be preferable for examining early-stage OPCs over a longer time period [
44,
45]. A study that used such an approach applied scRNAseq on PDGFRA-enriched, chemically transformed OL-lineage cells and uncovered several stages of OPCs [
46]. The scRNAseq analysis of our study revealed that directed oligodendroglial-lineage conversion also reflects several aspects of known OPC and OL development [
9,
47]. For example, we observed an upregulation of
ST8STIA1 (A2B5),
ID2,
ID4, and
SOX9 in iOPCs. In addition, we found increased expression of the oligodendroglial markers
CNP and
PLP1 during differentiation. In addition, in more mature iOLs we found an enhanced expression of marker genes for OL differentiation, such as
MBP and
MYRF that are crucial for myelination [
47].
To investigate hiPSC-based oligodendroglial lineage differentiation, we applied RNA velocity analysis, a technique that was developed for investigating differentiation processes in scRNAseq datasets to infer temporal changes without needing to obtain samples at several time points [
23]. Thereby, we described the transcriptional dynamics during fate decisions in induced oligodendroglial differentiation and identified
MBP, for example, as a velocity gene. Nevertheless, our data are based on 3′UTR-enriched single-cell transcriptome libraries, which limits their information content in RNA velocity analysis. Full-length scRNAseq protocols, which cover more splicing sites, would detect a higher variety of splicing variants and thus increase the number of potential velocity genes and might be beneficial to improve velocity analysis [
31]. Nonetheless, the RNA velocity-based analyses in this study provided additional insights. With the help of graph-based analysis methods such as PAGA [
34], we could visualize the oligodendroglial differentiation stream from iOPC to iOL1 and iOL2, which could be validated independently in S- and SON-directed differentiation and which would not have been identified with standard scRNAseq analysis. To generate a pseudo-timeline, we used gene-shared latent time instead of common similarity-based Markovian diffusion models, as suggested by Bergen et al., (2020). This approach improved the resulting timeline dramatically because the diffusion-based pseudo-timeline forced a unidirectional ‘time flow’ onto the dataset, which clearly did not fit the existence of two distinct lineages in one dataset. We used latent time analysis to highlight the ‘time’-dependent up- and downregulation of several oligodendroglial stage markers and to estimate the maturation state of a single cell within the whole span of oligodendroglial differentiation. Hereby, we observed a higher latent time distribution with SON than with S, indicating that oligodendroglial differentiation was faster in the SON- than in the S-directed differentiation. RNA velocity also uncovered a second neuronal leakage stream that was more prominent in S-directed differentiation. This leakage stream indicated that not only NPCs with low construct expression but also parts of the S-iOPC1s differentiated towards a neuronal state. Thus, we found evidence that S-directed differentiation might allow some iOPCs to ‘escape’ towards the neuronal lineage during the differentiation process, and that SON-directed differentiation seems to be less volatile and more efficient regarding OL differentiation under the applied conditions.
Differential gene expression analysis revealed that S-iOPC2 and SON-iOPC2 were quite similar and showed a substantial overlap in their differentially expressed genes when compared with iOPC1. This finding hints at a similar differentiation path with S and SON, although efficiency was higher with SON, as reflected by the abundance of more mature oligodendroglial cells, i.e., iOPC2, iOL1, and iOL2. On the other hand, iOPC1 constitutes a population of ‘escaping’ cells, giving us the opportunity to investigate the transcriptional networks and signaling machinery necessary for different steps of the differentiation process. Pathway analysis indicated greater expression of extracellular matrix components in iOPC2 cells than in iOPC1 cells; this difference may reflect a maturation step, as indicated by the change in direction of the velocity vectors towards the OL stage, or may represent a slight deflection towards other glial lineages [
46].
Oligodendroglial-lineage cells represent a heterogeneous cell population with a high spatio-temporal diversity [
3,
6]. A limitation of directed protocols is that they most likely generate only a subset of OPC/OL sub-lineages, depending on which TFs or combination of TFs they use. An advantage of directed conversion, however, is that human cells from a defined differentiation state can be generated in virtually unlimited amounts for various applications.
Our work highlights that applied TFs for generating iOPCs or iOLs should be chosen depending on the intended application or research question. Our analysis suggests that differentiation directed by S offers a useful tool for research on OPC biology and earlier stages of oligodendroglial differentiation, including research that addresses the effects of external cues promoting OPC and OL differentiation, i.e., in a 3D/spheroid culture system; the same might be true for SOX9 [
14]. On the other hand, applications, and research with a focus on more mature iOLs might benefit from the use of SON. TF combinations such as SON are likely to be better suited to studying OL biology and myelination-associated aspects, given the much higher conversion rate and easier access to much higher numbers of mature OLs with such combinations. Further investigations are certainly needed to unleash the potential of future cell therapies with engineered iOLs in myelin diseases, but a better understanding of the nature of induced cells via different TF-directed protocols will certainly be helpful to achieve this goal. We believe that the molecular and analysis tools applied in this study to force and dissect human oligodendrocyte development may support the understanding of dynamic aspects of cell-autonomous versus non-cell-autonomous contributions in in vivo disease models in the future, i.e., via grafting of iOLs generated from normal and diseased donors into mouse brains. The paradigmatic study on human iPSC-derived glia/mouse chimeras already revealed oligodendroglia-cell-autonomous contributions to childhood-onset schizophrenia [
48]. More such in vivo studies are needed that combine patient-derived oligodendoglial cell types with sophisticated single-cell level analyses to further increase our knowledge of disorders with myelination deficits.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





