
Emerging Role of cAMP/AMPK Signaling



{kind=link}
Abstract
:1. Current Model of cAMP Signaling
1.1. Structure
1.2. Regulation
2. AMPK
2.1. AMPK Structure
2.2. AMPK Regulation
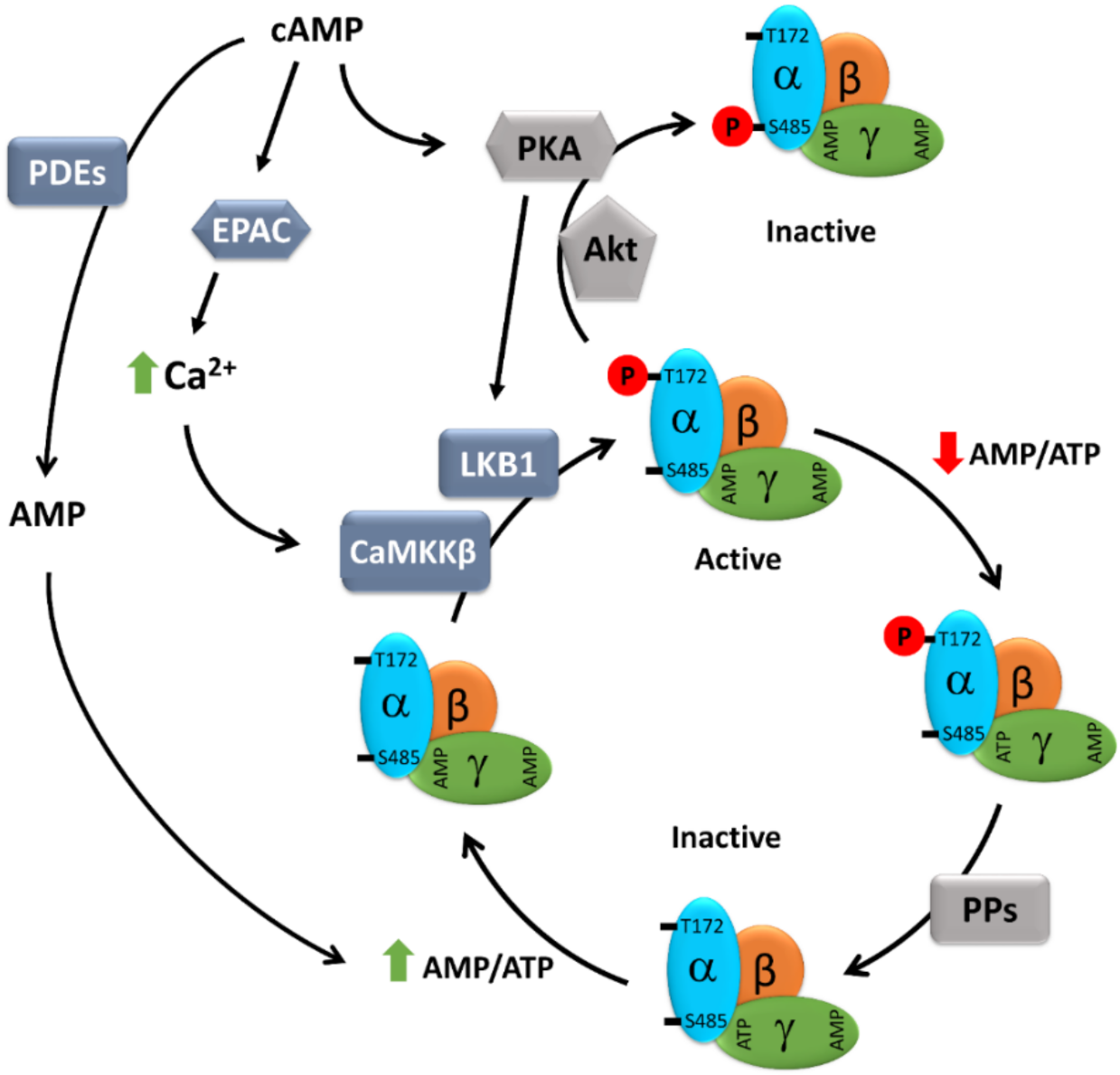
3. Regulation of AMPK Activity by cAMP Signaling
4. Functional and Translational Significance of the cAMP/AMPK Axis
4.1. Mitochondrial Biology
4.2. Lipid Metabolism
4.3. Ischemia
4.4. Inflammation
4.5. Type 2 Diabetes
4.6. Miscellaneous
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, Y.; Cann, M.J.; Litvin, T.N.; Iourgenko, V.; Sinclair, M.L.; Levin, L.R.; Buck, J. Soluble adenylyl cyclase as an evolutionarily conserved bicarbonate sensor. Science 2000, 289, 625–628. [Google Scholar] [CrossRef] [Green Version]
- Zippin, J.H.; Chen, Y.; Nahirney, P.; Kamenetsky, M.; Wuttke, M.S.; Fischman, D.A.; Levin, L.R.; Buck, J. Compartmentalization of bicarbonate-sensitive adenylyl cyclase in distinct signaling microdomains. FASEB J. 2003, 17, 82–84. [Google Scholar] [CrossRef] [PubMed]
- Boularan, C.; Gales, C. Cardiac cAMP: Production, hydrolysis, modulation and detection. Front. Pharmacol. 2015, 6, 203. [Google Scholar] [CrossRef] [Green Version]
- Zaccolo, M.; Zerio, A.; Lobo, M.J. Subcellular Organization of the cAMP Signaling Pathway. Pharm. Rev. 2021, 73, 278–309. [Google Scholar] [CrossRef]
- Godbole, A.; Lyga, S.; Lohse, M.J.; Calebiro, D. Internalized TSH receptors en route to the TGN induce local Gs-protein signaling and gene transcription. Nat. Commun. 2017, 8, 443. [Google Scholar] [CrossRef] [Green Version]
- Appukuttan, A.; Kasseckert, S.A.; Micoogullari, M.; Flacke, J.P.; Kumar, S.; Woste, A.; Abdallah, Y.; Pott, L.; Reusch, H.P.; Ladilov, Y. Type 10 adenylyl cyclase mediates mitochondrial Bax translocation and apoptosis of adult rat cardiomyocytes under simulated ischaemia/reperfusion. Cardiovasc. Res. 2012, 93, 340–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, R.L.; Buck, J.; Levin, L.R.; Winger, R.C.; Wang, J.; Arase, H.; Muller, W.A. Endothelial CD99 signals through soluble adenylyl cyclase and PKA to regulate leukocyte transendothelial migration. J. Exp. Med. 2015, 212, 1021–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appukuttan, A.; Kasseckert, S.A.; Kumar, S.; Reusch, H.P.; Ladilov, Y. Oxysterol-induced apoptosis of smooth muscle cells is under the control of a soluble adenylyl cyclase. Cardiovasc. Res. 2013, 99, 734–742. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Kostin, S.; Flacke, J.P.; Reusch, H.P.; Ladilov, Y. Soluble adenylyl cyclase controls mitochondria-dependent apoptosis in coronary endothelial cells. J. Biol. Chem. 2009, 284, 14760–14768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mika, D.; Leroy, J.; Vandecasteele, G.; Fischmeister, R. PDEs create local domains of cAMP signaling. J. Mol. Cell. Cardiol. 2012, 52, 323–329. [Google Scholar] [CrossRef]
- Zhang, L.; Bouadjel, K.; Manoury, B.; Vandecasteele, G.; Fischmeister, R.; Leblais, V. Cyclic nucleotide signalling compartmentation by PDEs in cultured vascular smooth muscle cells. Br. J. Pharm. 2019, 176, 1780–1792. [Google Scholar] [CrossRef]
- Baldwin, T.A.; Dessauer, C.W. Function of Adenylyl Cyclase in Heart: The AKAP Connection. J. Cardiovasc. Dev. Dis. 2018, 5, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrom, R.S.; Naugle, J.E.; Hase, M.; Gregorian, C.; Swaney, J.S.; Insel, P.A.; Brunton, L.L.; Meszaros, J.G. Angiotensin II enhances adenylyl cyclase signaling via Ca2+/calmodulin. Gq-Gs cross-talk regulates collagen production in cardiac fibroblasts. J. Biol. Chem. 2003, 278, 24461–24468. [Google Scholar] [CrossRef] [Green Version]
- Sadana, R.; Dessauer, C.W. Physiological roles for G protein-regulated adenylyl cyclase isoforms: Insights from knockout and overexpression studies. Neurosignals 2009, 17, 5–22. [Google Scholar] [CrossRef]
- Rossetti, T.; Jackvony, S.; Buck, J.; Levin, L.R. Bicarbonate, carbon dioxide and pH sensing via mammalian bicarbonate-regulated soluble adenylyl cyclase. Interface Focus 2021, 11, 20200034. [Google Scholar] [CrossRef]
- Zippin, J.H.; Chen, Y.; Straub, S.G.; Hess, K.C.; Diaz, A.; Lee, D.; Tso, P.; Holz, G.G.; Sharp, G.W.G.; Levin, L.R.; et al. CO2/HCO3(-)- and calcium-regulated soluble adenylyl cyclase as a physiological ATP sensor. J. Biol. Chem. 2013, 288, 33283–33291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litvin, T.N.; Kamenetsky, M.; Zarifyan, A.; Buck, J.; Levin, L.R. Kinetic properties of “soluble” adenylyl cyclase. Synergism between calcium and bicarbonate. J. Biol. Chem. 2003, 278, 15922–15926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, W.; Wang, Z.; Zhang, J.; Reed, B.Y.; Pak, C.Y.; Moe, O.W. Cloning and characterization of the human soluble adenylyl cyclase. Am. J. Physiol. Cell Physiol. 2005, 288, C1305–C1316. [Google Scholar] [CrossRef] [PubMed]
- Acin-Perez, R.; Salazar, E.; Kamenetsky, M.; Buck, J.; Levin, L.R.; Manfredi, G. Cyclic AMP produced inside mitochondria regulates oxidative phosphorylation. Cell Metab. 2009, 9, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Chagtoo, M.; George, N.; Pathak, N.; Tiwari, S.; Godbole, M.M.; Ladilov, Y. Inhibition of Intracellular Type 10 Adenylyl Cyclase Protects Cortical Neurons Against Reperfusion-Induced Mitochondrial Injury and Apoptosis. Mol. Neurobiol. 2018, 55, 2471–2482. [Google Scholar] [CrossRef] [PubMed]
- Flacke, J.P.; Flacke, H.; Appukuttan, A.; Palisaar, R.J.; Noldus, J.; Robinson, B.D.; Reusch, H.P.; Zippin, J.H.; Ladilov, Y. Type 10 soluble adenylyl cyclase is overexpressed in prostate carcinoma and controls proliferation of prostate cancer cells. J. Biol. Chem. 2013, 288, 3126–3135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Espiritu, L.; Diaz, A.; Nardin, C.; Saviola, A.J.; Shaw, F.; Plitt, T.; Yang, X.; Wolchok, J.; Pirog, E.C.; Desman, G.; et al. The metabolic/pH sensor soluble adenylyl cyclase is a tumor suppressor protein. Oncotarget 2016, 7, 45597–45607. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, I.; Bualeong, T.; Budde, H.; Cimiotti, D.; Appukuttan, A.; Klein, N.; Steinwascher, P.; Reusch, P.; Mugge, A.; Meyer, R.; et al. Soluble adenylyl cyclase: A novel player in cardiac hypertrophy induced by isoprenaline or pressure overload. PLoS ONE 2018, 13, e0192322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, Y.C.; Surdo, N.C.; Pantano, S.; Zaccolo, M. Imaging cAMP nanodomains in the heart. Biochem. Soc. Trans. 2019, 47, 1383–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baillie, G.S.; Tejeda, G.S.; Kelly, M.P. Therapeutic targeting of 3′,5′-cyclic nucleotide phosphodiesterases: Inhibition and beyond. Nat. Rev. Drug Discov. 2019, 18, 770–796. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G. AMP-activated protein kinase: Maintaining energy homeostasis at the cellular and whole-body levels. Annu. Rev. Nutr. 2014, 34, 31–55. [Google Scholar] [CrossRef] [Green Version]
- Stapleton, D.; Mitchelhill, K.I.; Gao, G.; Widmer, J.; Michell, B.J.; Teh, T.; House, C.M.; Fernandez, C.S.; Cox, T.; Witters, L.A.; et al. Mammalian AMP-activated protein kinase subfamily. J. Biol. Chem. 1996, 271, 611–614. [Google Scholar] [CrossRef] [Green Version]
- Ross, F.A.; MacKintosh, C.; Hardie, D.G. AMP-activated protein kinase: A cellular energy sensor that comes in 12 flavours. FEBS J. 2016, 283, 2987–3001. [Google Scholar] [CrossRef]
- Kim, M.; Shen, M.; Ngoy, S.; Karamanlidis, G.; Liao, R.; Tian, R. AMPK isoform expression in the normal and failing hearts. J. Mol. Cell Cardiol. 2012, 52, 1066–1073. [Google Scholar] [CrossRef] [Green Version]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Zhou, X.E.; Xu, H.E.; Melcher, K. Structure and Physiological Regulation of AMPK. Int. J. Mol. Sci. 2018, 19, 3534. [Google Scholar] [CrossRef] [Green Version]
- Woods, A.; Johnstone, S.R.; Dickerson, K.; Leiper, F.C.; Fryer, L.G.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 2003, 13, 2004–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, A.; Dickerson, K.; Heath, R.; Hong, S.-P.; Momcilovic, M.; Johnstone, S.R.; Carlson, M.; Carling, D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005, 2, 21–33. [Google Scholar] [CrossRef] [Green Version]
- Carling, D.; Sanders, M.J.; Woods, A. The regulation of AMP-activated protein kinase by upstream kinases. Int. J. Obes. 2008, 32 (Suppl. S4), S55–S59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBride, A.; Ghilagaber, S.; Nikolaev, A.; Hardie, D.G. The glycogen-binding domain on the AMPK beta subunit allows the kinase to act as a glycogen sensor. Cell Metab. 2009, 9, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Bultot, L.; Guigas, B.; Von Wilamowitz-Moellendorff, A.; Maisin, L.; Vertommen, D.; Hussain, N.; Beullens, M.; Guinovart, J.J.; Foretz, M.; Viollet, B.; et al. AMP-activated protein kinase phosphorylates and inactivates liver glycogen synthase. Biochem. J. 2012, 443, 193–203. [Google Scholar] [CrossRef] [Green Version]
- Xiao, B.; Sanders, M.J.; Carmena, D.; Bright, N.J.; Haire, L.F.; Underwood, E.; Patel, B.R.; Heath, R.B.; Walker, P.A.; Hallen, S.; et al. Structural basis of AMPK regulation by small molecule activators. Nat. Commun. 2013, 4, 3017. [Google Scholar] [CrossRef] [Green Version]
- Oakhill, J.S.; Chen, Z.-P.; Scott, J.; Steel, R.; Castelli, L.A.; Ling, N.; Macaulay, S.L.; Kemp, B.E. β-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK). Proc. Natl. Acad. Sci. USA 2010, 107, 19237–19241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Xu, Z.X.; Ding, Z.; Lu, Y.; Yu, Q.; Werle, K.D.; Zhou, G.; Park, Y.Y.; Peng, G.; Gambello, M.J.; et al. Myristoylation confers noncanonical AMPK functions in autophagy selectivity and mitochondrial surveillance. Nat. Commun. 2015, 6, 7926. [Google Scholar] [CrossRef]
- Scott, J.W.; Hawley, S.A.; Green, K.A.; Anis, M.; Stewart, G.; Scullion, G.A.; Norman, D.G.; Hardie, D.G. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J. Clin. Investig. 2004, 113, 274–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, B.; Heath, R.; Saiu, P.; Leiper, F.C.; Leone, P.; Jing, C.; Walker, P.A.; Haire, L.; Eccleston, J.F.; Davis, C.T.; et al. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 2007, 449, 496–500. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Carling, D.; Gamblin, S.J. AMP-activated protein kinase: Also regulated by ADP? Trends Biochem. Sci. 2011, 36, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Suter, M.; Riek, U.; Tuerk, R.; Schlattner, U.; Wallimann, T.; Neumann, D. Dissecting the role of 5′-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase. J. Biol. Chem. 2006, 281, 32207–32216. [Google Scholar] [CrossRef] [Green Version]
- Sanders, M.J.; Grondin, P.O.; Hegarty, B.D.; Snowden, M.A.; Carling, D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem. J. 2007, 403, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Gowans, G.J.; Hawley, S.A.; Ross, F.A.; Hardie, D.G. AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab. 2013, 18, 556–566. [Google Scholar] [CrossRef] [Green Version]
- Davies, S.P.; Helps, N.R.; Cohen, P.T.; Hardie, D.G. 5′-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS Lett. 1995, 377, 421–425. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, C.; Munoz, M.; Contreras, C.; Prieto, D. AMPK, metabolism, and vascular function. FEBS J. 2021, 288, 3746–3771. [Google Scholar] [CrossRef]
- Hawley, S.A.; Pan, D.A.; Mustard, K.J.; Ross, L.; Bain, J.; Edelman, A.M.; Frenguelli, B.G.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005, 2, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Fogarty, S.; Hawley, S.A.; Green, K.A.; Saner, N.; Mustard, K.J.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-beta activates AMPK without forming a stable complex: Synergistic effects of Ca2+ and AMP. Biochem. J. 2010, 426, 109–118. [Google Scholar] [CrossRef]
- Stahmann, N.; Woods, A.; Carling, D.; Heller, R. Thrombin activates AMP-activated protein kinase in endothelial cells via a pathway involving Ca2+/calmodulin-dependent protein kinase kinase beta. Mol. Cell Biol. 2006, 26, 5933–5945. [Google Scholar] [CrossRef] [Green Version]
- Hawley, S.A.; Ross, F.A.; Gowans, G.J.; Tibarewal, P.; Leslie, N.R.; Hardie, D.G. Phosphorylation by Akt within the ST loop of AMPK-alpha1 down-regulates its activation in tumour cells. Biochem. J. 2014, 459, 275–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopietz, F.; Rupar, K.; Berggreen, C.; Sall, J.; Vertommen, D.; Degerman, E.; Rider, M.H.; Göransson, O. Inhibition of AMPK activity in response to insulin in adipocytes: Involvement of AMPK pS485, PDEs, and cellular energy levels. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E459–E471. [Google Scholar] [CrossRef] [PubMed]
- Dagon, Y.; Hur, E.; Zheng, B.; Wellenstein, K.; Cantley, L.C.; Kahn, B.B. p70S6 kinase phosphorylates AMPK on serine 491 to mediate leptin’s effect on food intake. Cell Metab. 2012, 16, 104–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djouder, N.; Tuerk, R.D.; Suter, M.; Salvioni, P.; Thali, R.F.; Scholz, R.; Vaahtomeri, K.; Auchli, Y.; Rechsteiner, H.; Brunisholz, R.A.; et al. PKA phosphorylates and inactivates AMPKalpha to promote efficient lipolysis. EMBO J. 2010, 29, 469–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferretti, A.C.; Hidalgo, F.; Tonucci, F.M.; Almada, E.; Pariani, A.; Larocca, M.C.; Favre, C. Metformin and glucose starvation decrease the migratory ability of hepatocellular carcinoma cells: Targeting AMPK activation to control migration. Sci. Rep. 2019, 9, 2815. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Mu, J.; Birnbaum, M.J. Role of AMP-activated protein kinase in cyclic AMP-dependent lipolysis in 3T3-L1 adipocytes. J. Biol. Chem. 2003, 278, 43074–43080. [Google Scholar] [CrossRef] [Green Version]
- Omar, B.; Zmuda-Trzebiatowska, E.; Manganiello, V.; Goransson, O.; Degerman, E. Regulation of AMP-activated protein kinase by cAMP in adipocytes: Roles for phosphodiesterases, protein kinase B, protein kinase A, Epac and lipolysis. Cell Signal. 2009, 21, 760–766. [Google Scholar] [CrossRef] [Green Version]
- Fu, D.; Wakabayashi, Y.; Lippincott-Schwartz, J.; Arias, I.M. Bile acid stimulates hepatocyte polarization through a cAMP-Epac-MEK-LKB1-AMPK pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 1403–1408. [Google Scholar] [CrossRef] [Green Version]
- Park, S.J.; Ahmad, F.; Philp, A.; Baar, K.; Williams, T.; Luo, H.; Ke, H.; Rehmann, H.; Taussig, R.; Brown, A.L.; et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 2012, 148, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.L.; Yi, L.; Jin, X.; Liang, X.Y.; Zhou, Y.; Zhang, T.; Xie, Q.; Zhou, X.; Chang, H.; Fu, Y.J.; et al. Resveratrol attenuates vascular endothelial inflammation by inducing autophagy through the cAMP signaling pathway. Autophagy 2013, 9, 2033–2045. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.P.; Reoma, J.L.; Gamm, D.M.; Uhler, M.D. LKB1, a novel serine/threonine protein kinase and potential tumour suppressor, is phosphorylated by cAMP-dependent protein kinase (PKA) and prenylated in vivo. Biochem. J. 2000, 345, 673–680. [Google Scholar] [CrossRef]
- Kari, S.; Vasko, V.V.; Priya, S.; Kirschner, L.S. PKA Activates AMPK Through LKB1 Signaling in Follicular Thyroid Cancer. Front. Endocrinol. 2019, 10, 769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayarajan, V.; Appukuttan, A.; Aslam, M.; Reusch, P.; Regitz-Zagrosek, V.; Ladilov, Y. Regulation of AMPK activity by type 10 adenylyl cyclase: Contribution to the mitochondrial biology, cellular redox and energy homeostasis. Cell Mol. Life Sci. 2019, 76, 4945–4959. [Google Scholar] [CrossRef] [PubMed]
- Hurley, R.L.; Barre, L.K.; Wood, S.D.; Anderson, K.A.; Kemp, B.E.; Means, A.R.; Witters, L.A. Regulation of AMP-activated protein kinase by multisite phosphorylation in response to agents that elevate cellular cAMP. J. Biol. Chem. 2006, 281, 36662–36672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferretti, A.C.; Tonucci, F.M.; Hidalgo, F.; Almada, E.; Larocca, M.C.; Favre, C. AMPK and PKA interaction in the regulation of survival of liver cancer cells subjected to glucose starvation. Oncotarget 2016, 7, 17815–17828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Chi, M.M.; Moley, K.H.; Downs, S.M. cAMP pulsing of denuded mouse oocytes increases meiotic resumption via activation of AMP-activated protein kinase. Reproduction 2009, 138, 759–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johanns, M.; Lai, Y.C.; Hsu, M.F.; Jacobs, R.; Vertommen, D.; van Sande, J.; Dumont, J.E.; Woods, A.; Carling, D.; Hue, L.; et al. AMPK antagonizes hepatic glucagon-stimulated cyclic AMP signalling via phosphorylation-induced activation of cyclic nucleotide phosphodiesterase 4B. Nat. Commun. 2016, 7, 10856. [Google Scholar] [CrossRef]
- Garcia-Roves, P.M.; Osler, M.E.; Holmstrom, M.H.; Zierath, J.R. Gain-of-function R225Q mutation in AMP-activated protein kinase gamma3 subunit increases mitochondrial biogenesis in glycolytic skeletal muscle. J. Biol. Chem. 2008, 283, 35724–35734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [Green Version]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L.; Loson, O.C., Jr.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aslam, M.; Ladilov, Y. Regulation of Mitochondrial Homeostasis by sAC-Derived cAMP Pool: Basic and Translational Aspects. Cells 2021, 10, 473. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, G.; Gerbino, A.; Lefkimmiatis, K. Shaping mitochondrial dynamics: The role of cAMP signalling. Biochem. Biophys. Res. Commun. 2018, 500, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Hamidie, R.D.R.; Shibaguchi, T.; Yamada, T.; Koma, R.; Ishizawa, R.; Saito, Y.; Hosoi, T.; Masuda, K. Curcumin induces mitochondrial biogenesis by increasing cyclic AMP levels via phosphodiesterase 4A inhibition in skeletal muscle. Br. J. Nutr. 2021, 126, 1642–1650. [Google Scholar] [CrossRef]
- Burgin, A.B.; Magnusson, O.T.; Singh, J.; Witte, P.; Staker, B.L.; Bjornsson, J.M.; Thorsteinsdottir, M.; Hrafnsdottir, S.; Hagen, T.; Kiselyov, A.S.; et al. Design of phosphodiesterase 4D (PDE4D) allosteric modulators for enhancing cognition with improved safety. Nat. Biotechnol. 2010, 28, 63–70. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, S.; Zhai, A.; Zhang, B.; Tian, G. AMPK-Mediated Regulation of Lipid Metabolism by Phosphorylation. Biol. Pharm. Bull. 2018, 41, 985–993. [Google Scholar] [CrossRef] [Green Version]
- Ben-Shlomo, S.; Zvibel, I.; Shnell, M.; Shlomai, A.; Chepurko, E.; Halpern, Z.; Barzilai, N.; Oren, R.; Fishman, S. Glucagon-like peptide-1 reduces hepatic lipogenesis via activation of AMP-activated protein kinase. J. Hepatol. 2011, 54, 1214–1223. [Google Scholar] [CrossRef] [PubMed]
- Lv, S.; Qiu, X.; Li, J.; Liang, J.; Li, W.; Zhang, C.; Zhang, Z.N.; Luan, B. Glucagon-induced extracellular cAMP regulates hepatic lipid metabolism. J. Endocrinol. 2017, 234, 73–87. [Google Scholar] [CrossRef]
- Wang, Z.; Liang, Y.; Zhang, L.; Zhang, N.; Liu, Q.; Wang, Z. Phosphodiesterase 4 inhibitor activates AMPK-SIRT6 pathway to prevent aging-related adipose deposition induced by metabolic disorder. Aging 2018, 10, 2394–2406. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Chung, Y.W.; Ahmad, F.; Tang, Y.; Hockman, S.C.; Kee, H.J.; Berger, K.; Guirguis, E.; Choi, Y.H.; Schimel, D.M.; Aponte, A.M.; et al. White to beige conversion in PDE3B KO adipose tissue through activation of AMPK signaling and mitochondrial function. Sci. Rep. 2017, 7, 40445. [Google Scholar] [CrossRef] [Green Version]
- Wan, D.; Zhou, Y.; Wang, K.; Hou, Y.; Hou, R.; Ye, X. Resveratrol provides neuroprotection by inhibiting phosphodiesterases and regulating the cAMP/AMPK/SIRT1 pathway after stroke in rats. Brain Res. Bull. 2016, 121, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Qian, T.; Wang, W. CTRP3 Activates the AMPK/SIRT1-PGC-1alpha Pathway to Protect Mitochondrial Biogenesis and Functions in Cerebral Ischemic Stroke. Neurochem. Res. 2020, 45, 3045–3058. [Google Scholar] [CrossRef]
- Yang, B.; Wang, S.; Yu, S.; Chen, Y.; Li, L.; Zhang, H.; Zhao, Y. C1q/tumor necrosis factor-related protein 3 inhibits oxidative stress during intracerebral hemorrhage via PKA signaling. Brain Res. 2017, 1657, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X.L.; Zhao, J.; Wang, Y.J.; Lau, W.B.; Yuan, Y.X.; Gao, E.H.; Koch, W.J.; Ma, X.L. Adiponectin inhibits oxidative/nitrative stress during myocardial ischemia and reperfusion via PKA signaling. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E1436–E1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Tao, L.; Yuan, Y.; Lau, W.B.; Li, R.; Lopez, B.L.; Christopher, T.A.; Tian, R.; Ma, X.L. Cardioprotective effect of adiponectin is partially mediated by its AMPK-independent antinitrative action. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E384–E391. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.T.; He, P.C.; Li, A.Q.; Cao, K.X.; Yan, J.W.; Guo, S.; Jiang, L.; Yao, L.; Dai, X.Y.; Feng, D.; et al. Caffeine promotes angiogenesis through modulating endothelial mitochondrial dynamics. Acta Pharmacol. Sin. 2021, 42, 2033–2045. [Google Scholar] [CrossRef] [PubMed]
- Tseng, S.Y.; Chao, T.H.; Li, Y.H.; Liu, P.Y.; Lee, C.H.; Cho, C.L.; Wu, H.L.; Chen, J.H. Cilostazol improves high glucose-induced impaired angiogenesis in human endothelial progenitor cells and vascular endothelial cells as well as enhances vasculoangiogenesis in hyperglycemic mice mediated by the adenosine monophosphate-activated protein kinase pathway. J. Vasc. Surg. 2016, 63, 1051–1062.e3. [Google Scholar] [CrossRef] [Green Version]
- Feehan, K.T.; Gilroy, D.W. Is Resolution the End of Inflammation? Trends Mol. Med. 2019, 25, 198–214. [Google Scholar] [CrossRef]
- Galkina, E.; Ley, K. Immune and inflammatory mechanisms of atherosclerosis. Annu. Rev. Immunol. 2009, 27, 165–197. [Google Scholar] [CrossRef] [Green Version]
- Joshi, N.V.; Toor, I.; Shah, A.S.; Carruthers, K.; Vesey, A.T.; Alam, S.R.; Sills, A.; Hoo, T.Y.; Melville, A.J.; Langlands, S.P.; et al. Systemic Atherosclerotic Inflammation Following Acute Myocardial Infarction: Myocardial Infarction Begets Myocardial Infarction. J. Am. Heart Assoc. 2015, 4, e001956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavares, L.P.; Negreiros-Lima, G.L.; Lima, K.M.; Silva, P.M.E.; Pinho, V.; Teixeira, M.M.; Sousa, L.P. Blame the signaling: Role of cAMP for the resolution of inflammation. Pharmacol. Res. 2020, 159, 105030. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Hyttinen, J.M.; Kaarniranta, K. AMP-activated protein kinase inhibits NF-kappaB signaling and inflammation: Impact on healthspan and lifespan. J. Mol. Med. 2011, 89, 667–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mounier, R.; Theret, M.; Arnold, L.; Cuvellier, S.; Bultot, L.; Goransson, O.; Sanz, N.; Ferry, A.; Sakamoto, K.; Foretz, M.; et al. AMPKalpha1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab. 2013, 18, 251–264. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [Green Version]
- Neumann, M.; Grieshammer, T.; Chuvpilo, S.; Kneitz, B.; Lohoff, M.; Schimpl, A.; Franza, B.R., Jr.; Serfling, E. RelA/p65 is a molecular target for the immunosuppressive action of protein kinase A. EMBO J. 1995, 14, 1991–2004. [Google Scholar] [CrossRef]
- Kamthong, P.J.; Wu, M. Inhibitor of nuclear factor-kappaB induction by cAMP antagonizes interleukin-1-induced human macrophage-colony-stimulating-factor expression. Biochem. J. 2001, 356, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Wall, E.A.; Zavzavadjian, J.R.; Chang, M.S.; Randhawa, B.; Zhu, X.; Hsueh, R.C.; Liu, J.; Driver, A.; Bao, X.R.; Sternweis, P.C.; et al. Suppression of LPS-induced TNF-alpha production in macrophages by cAMP is mediated by PKA-AKAP95-p105. Sci. Signal. 2009, 2, ra28. [Google Scholar] [CrossRef] [Green Version]
- Scheibner, K.A.; Boodoo, S.; Collins, S.; Black, K.E.; Chan-Li, Y.; Zarek, P.; Powell, J.D.; Horton, M.R. The adenosine a2a receptor inhibits matrix-induced inflammation in a novel fashion. Am. J. Respir. Cell Mol. Biol. 2009, 40, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zheng, Y.; Liu, L.; Lin, C.; Liao, C.; Xin, L.; Zhong, S.; Cheng, Q.; Zhang, L. Adiponectin Inhibits TNF-alpha-Activated PAI-1 Expression Via the cAMP-PKA-AMPK-NF-kappaB Axis in Human Umbilical Vein Endothelial Cells. Cell Physiol. Biochem. 2017, 42, 2342–2352. [Google Scholar] [CrossRef]
- Hu, F.; Dong, X.; Li, W.; Lv, J.; Lin, F.; Song, G.; Hou, G.; Li, R. miR3515p aggravates lipopolysaccharideinduced acute lung injury via inhibiting AMPK. Mol. Med. Rep. 2021, 24, 689. [Google Scholar] [CrossRef]
- Salehi, M.; Aulinger, B.A.; D’Alessio, D.A. Targeting beta-cell mass in type 2 diabetes: Promise and limitations of new drugs based on incretins. Endocr. Rev. 2008, 29, 367–379. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Jun, H.S. Anti-Inflammatory Effects of GLP-1-Based Therapies beyond Glucose Control. Mediators Inflamm. 2016, 2016, 3094642. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Fan, S.; Xiong, Q.; Niu, Y.; Zhang, X.; Qin, J.; Shi, Y.; Zhang, L. Glucagon-like peptide-1 attenuates cardiac hypertrophy via the AngII/AT1R/ACE2 and AMPK/mTOR/p70S6K pathways. Acta Biochim. Biophys. Sin. 2021, 53, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Hou, N.; Liu, Y.; Huang, N.; Pan, R.; Zhang, X.; Mao, E.; Sun, X. Liraglutide improves vascular dysfunction by regulating a cAMP-independent PKA-AMPK pathway in perivascular adipose tissue in obese mice. Biomed. Pharmacother. 2019, 120, 109537. [Google Scholar] [CrossRef]
- Jones, B. The therapeutic potential of GLP-1 receptor biased agonism. Br. J. Pharmacol. 2021. [Google Scholar] [CrossRef]
- Wei, R.; Ma, S.; Wang, C.; Ke, J.; Yang, J.; Li, W.; Liu, Y.; Hou, W.; Feng, X.; Wang, G.; et al. Exenatide exerts direct protective effects on endothelial cells through the AMPK/Akt/eNOS pathway in a GLP-1 receptor-dependent manner. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E947–E957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holz, G.G. Epac: A new cAMP-binding protein in support of glucagon-like peptide-1 receptor-mediated signal transduction in the pancreatic beta-cell. Diabetes 2004, 53, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.G.; Yuan, Y.P.; Xu, S.C.; Wei, W.Y.; Xu, C.R.; Zhang, X.; Wu, Q.Q.; Liao, H.H.; Ni, J.; Tang, Q.Z. CTRP3 attenuates cardiac dysfunction, inflammation, oxidative stress and cell death in diabetic cardiomyopathy in rats. Diabetologia 2017, 60, 1126–1137. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Huo, J.; Ding, X.; Yang, M.; Li, L.; Dai, J.; Hosoe, K.; Kubo, H.; Mori, M.; Higuchi, K.; et al. Coenzyme Q10 Improves Lipid Metabolism and Ameliorates Obesity by Regulating CaMKII-Mediated PDE4 Inhibition. Sci. Rep. 2017, 7, 8253. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Steinle, J.J. Epac1 Requires AMPK Phosphorylation to Regulate HMGB1 in the Retinal Vasculature. Invest. Ophthalmol. Vis. Sci. 2020, 61, 33. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Chang, E.; Peng, J.; An, H.; McMillin, S.M.; Radovick, S.; Stratakis, C.A.; Wondisford, F.E. Activation of the cAMP-PKA pathway Antagonizes Metformin Suppression of Hepatic Glucose Production. J. Biol. Chem. 2016, 291, 10562–10570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wang, S.; Yang, X.; Chu, H. miR1423p targets AC9 to regulate sciatic nerve injuryinduced neuropathic pain by regulating the cAMP/AMPK signalling pathway. Int. J. Mol. Med. 2021, 47, 561–572. [Google Scholar] [CrossRef]
- De Llera, A.H.; Martin-Hidalgo, D.; Gil, M.C.; Garcia-Marin, L.J.; Bragado, M.J. The calcium/CaMKKalpha/beta and the cAMP/PKA pathways are essential upstream regulators of AMPK activity in boar spermatozoa. Biol. Reprod. 2014, 90, 29. [Google Scholar] [CrossRef]
- Langley, E.; Pearson, M.; Faretta, M.; Bauer, U.M.; Frye, R.A.; Minucci, S.; Pelicci, P.G.; Kouzarides, T. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 2002, 21, 2383–2396. [Google Scholar] [CrossRef] [Green Version]
- Giannakou, M.E.; Partridge, L. The interaction between FOXO and SIRT1: Tipping the balance towards survival. Trends Cell Biol. 2004, 14, 408–412. [Google Scholar] [CrossRef]
- Wang, Y.; Liang, Y.; Vanhoutte, P.M. SIRT1 and AMPK in regulating mammalian senescence: A critical review and a working model. FEBS Lett. 2011, 585, 986–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerhart-Hines, Z.; Dominy, J.E., Jr.; Blattler, S.M.; Jedrychowski, M.P.; Banks, A.S.; Lim, J.H.; Chim, H.; Gygi, S.P.; Puigserver, P. The cAMP/PKA pathway rapidly activates SIRT1 to promote fatty acid oxidation independently of changes in NAD(+). Mol. Cell 2011, 44, 851–863. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Zhao, H.; Cai, Y.; Xiong, J.; Mohan, A.; Lou, D.; Shi, H.; Zhang, Y.; Long, X.; Wang, J.; et al. Cyclic nucleotide phosphodiesterase 1C contributes to abdominal aortic aneurysm. Proc. Natl. Acad. Sci. USA 2021, 118, e2107898118. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, L.S.; Ren, J.L.; Zhang, Y.R.; Wu, N.; Jia, M.Z.; Yu, Y.R.; Ning, Z.P.; Tang, C.S.; Qi, Y.F. Intermedin1-53 attenuates aging-associated vascular calcification in rats by upregulating sirtuin 1. Aging 2020, 12, 5651–5674. [Google Scholar] [CrossRef]
- Han, X.; Tai, H.; Wang, X.; Wang, Z.; Zhou, J.; Wei, X.; Ding, Y.; Gong, H.; Mo, C.; Zhang, J.; et al. AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD(+) elevation. Aging Cell 2016, 15, 416–427. [Google Scholar] [CrossRef] [Green Version]
- Ford, R.J.; Desjardins, E.M.; Steinberg, G.R. Are SIRT1 activators another indirect method to increase AMPK for beneficial effects on aging and the metabolic syndrome? EBioMedicine 2017, 19, 16–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.J.; Ahmad, F.; Um, J.H.; Brown, A.L.; Xu, X.; Kang, H.; Ke, H.; Feng, X.; Ryall, J.; Philp, A.; et al. Specific Sirt1 Activator-mediated Improvement in Glucose Homeostasis Requires Sirt1-Independent Activation of AMPK. EBioMedicine 2017, 18, 128–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, J.Y.; Kim, S.G.; Cho, D.H.; Kim, J.R.; Choi, H.C. SRT1720-induced activation of SIRT1 alleviates vascular smooth muscle cell senescence through PKA-dependent phosphorylation of AMPKalpha at Ser485. FEBS Open Bio 2020, 10, 1316–1325. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aslam, M.; Ladilov, Y. Emerging Role of cAMP/AMPK Signaling. Cells 2022, 11, 308. https://doi.org/10.3390/cells11020308
Aslam M, Ladilov Y. Emerging Role of cAMP/AMPK Signaling. Cells. 2022; 11(2):308. https://doi.org/10.3390/cells11020308
Chicago/Turabian StyleAslam, Muhammad, and Yury Ladilov. 2022. "Emerging Role of cAMP/AMPK Signaling" Cells 11, no. 2: 308. https://doi.org/10.3390/cells11020308
APA StyleAslam, M., & Ladilov, Y. (2022). Emerging Role of cAMP/AMPK Signaling. Cells, 11(2), 308. https://doi.org/10.3390/cells11020308