
Fibrosis Is a Basement Membrane-Related Disease in the Cornea: Injury and Defective Regeneration of Basement Membranes May Underlie Fibrosis in Other Organs


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. BM-Related Corneal Fibrosis
2.1. Normal Cornea Structure and Transparency
2.2. The Corneal Wound Healing Response to Injury
2.3. Minor Injuries and Non-Fibrotic Healing in the Anterior Cornea
2.4. Major Injuries and Fibrotic Healing in the Anterior Cornea
2.5. Injuries and Fibrotic Healing in the Posterior Cornea
3. Other Candidate Organs Where BM Injury Can Be Associated with Fibrosis
3.1. Skin
3.2. Lung
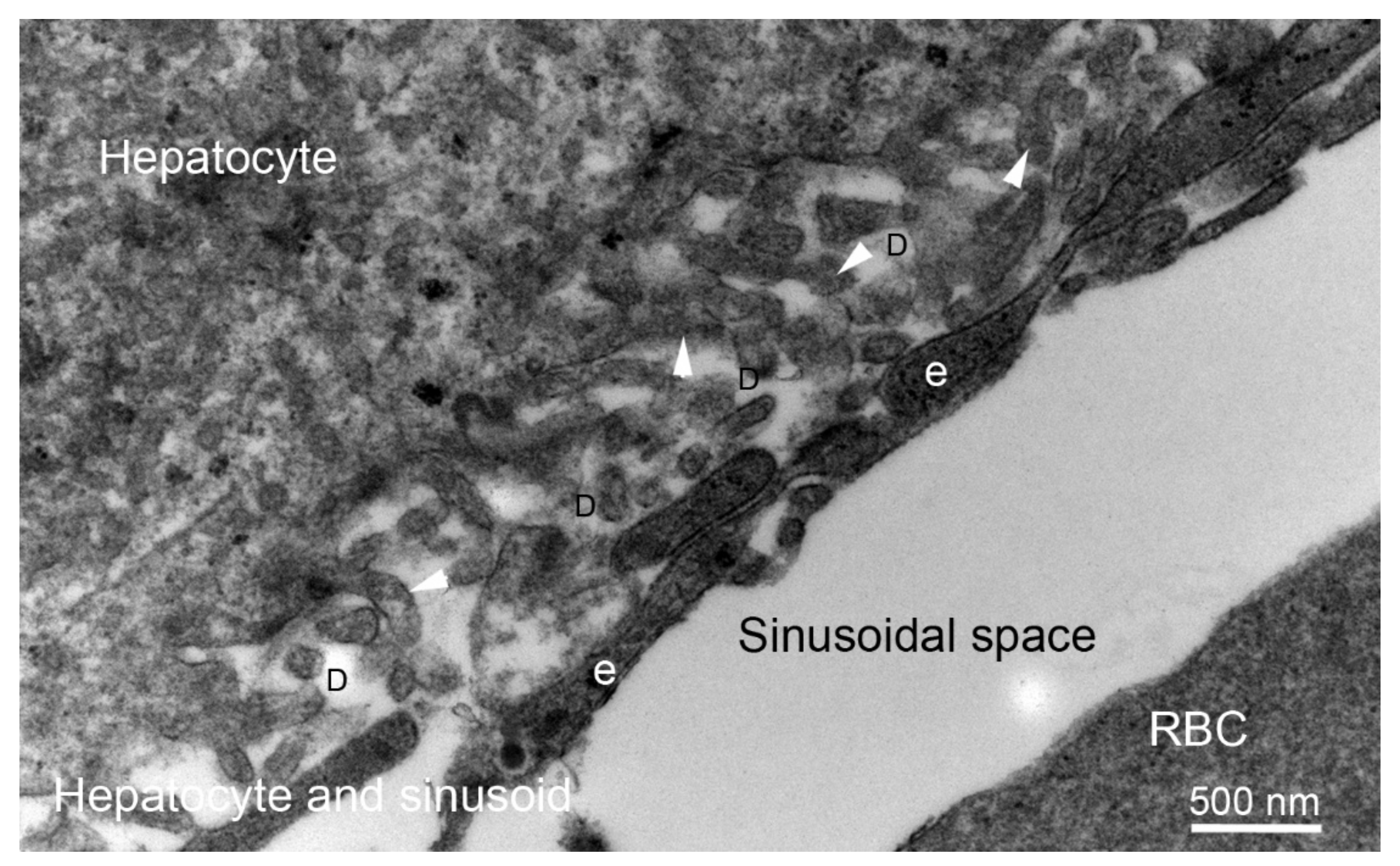
4. Liver Fibrosis: Capillarization of Hepatic Sinusoids Associated with the Generation of Endothelial BMs
5. Conclusions
Funding
Conflicts of Interest
References
- Meyer, K.C. Pulmonary fibrosis, part I: Epidemiology, pathogenesis, and diagnosis. Expert Rev. Respir. Med. 2017, 11, 343–359. [Google Scholar] [CrossRef]
- Wilson, S.E. TGF beta -1, -2 and -3 in the modulation of fibrosis in the cornea and other organs. Exp. Eye Res. 2021, 207, 108594. [Google Scholar] [CrossRef] [PubMed]
- Lipshitz, I.; Loewenstein, A.; Varssano, D.; Lazar, M. Late onset corneal haze after photorefractive keratectomy for moderate and high myopia. Ophthalmology 1997, 104, 369–373. [Google Scholar] [CrossRef]
- Marino, G.K.; Santhiago, M.R.; Santhanam, A.; Lassance, L.; Thangavadivel, S.; Medeiros, C.S.; Torricelli, A.A.M.; Wilson, S.E. Regeneration of defective epithelial basement membrane and restoration of corneal transparency. J. Ref. Surg. 2017, 33, 337–346. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira, R.C.; Tye, G.; Sampaio, L.P.; Shiju, T.M.; Dedreu, J.; Menko, A.S.; Santhiago, M.R.; Wilson, S.E. TGFβ1 and TGFβ2 proteins in corneas with and without stromal fibrosis: Delayed regeneration of epithelial barrier function and the epithelial basement membrane in corneas with stromal fibrosis. Exp. Eye Res. 2021, 202, 108325. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, C.S.; Saikia, P.; de Oliveira, R.C.; Lassance, L.; Santhiago, M.R.; Wilson, S.E. Descemet’s membrane modulation of posterior corneal fibrosis. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1010–1020. [Google Scholar] [CrossRef] [Green Version]
- Sampaio, L.P.; Shiju, T.M.; Hilgert, G.S.L.; de Oliveira, R.C.; DeDreu, J.; Menko, A.S.; Santhiago, M.R.; Wilson, S.E. Descemet’s membrane injury and regeneration, and posterior corneal fibrosis in rabbits. Exp. Eye Res. 2021, 213, 108803. [Google Scholar] [CrossRef] [PubMed]
- Espana, E.M.; Birk, D.E. Composition, structure and function of the corneal stroma. Exp. Eye Res. 2020, 198, 108137. [Google Scholar] [CrossRef]
- Wilson, S.E. Bowman’s layer in the cornea-structure and function and regeneration. Exp. Eye Res. 2020, 195, 108033. [Google Scholar] [CrossRef]
- Joyce, N.C. Proliferative capacity of the corneal endothelium. Prog. Retin. Eye Res. 2003, 22, 359–389. [Google Scholar] [CrossRef]
- De Oliveira, R.C.; Wilson, S.E. Descemet’s membrane development, structure, function and regeneration. Exp. Eye Res. 2020, 197, 108090. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, F.L.; Chaurasia, S.S.; Cutler, A.; Asosingh, K.; Kaur, H.; de Medeiros, F.W.; Agrawal, V.; Wilson, S.E. Corneal myofibroblast generation from bone marrow-derived cells. Exp. Eye Res. 2010, 91, 92–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, L.J.; Marfurt, C.F.; Kruse, F.; Tervo, T.M. Corneal nerves: Structure, contents and function. Exp. Eye Res. 2003, 76, 521–542. [Google Scholar] [CrossRef]
- Mousa, H.M.; Saban, D.R.; Perez, V.L. The cornea IV immunology, infection, neovascularization, and surgery chapter 1: Corneal immunology. Exp. Eye Res. 2021, 205, 108502. [Google Scholar] [CrossRef]
- Wilson, S.E.; He, Y.-G.; Weng, J.; Li, Q.; McDowall, A.W.; Vital, M.; Chwang, E.L. Epithelial injury induces keratocyte apoptosis: Hypothesized role for the interleukin-1 system in the modulation of corneal tissue organization and wound healing. Exp. Eye Res. 1996, 62, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, C.S.; Lassance, L.; Saikia, P.; Wilson, S.E. Posterior stromal keratocyte apoptosis triggered by mechanical endothelial injury and nidogen-1 production in the cornea. Exp. Eye Res. 2018, 172, 30–35. [Google Scholar] [CrossRef]
- Wilson, S.E.; Li, Q.; Weng, J.; Barry-Lane, P.A.; Jester, J.V.; Liang, Q.; Wordinger, R.J. The Fas/Fas ligand system and other modulators of apoptosis in the cornea. Investig. Ophthalmol. Vis. Sci. 1996, 37, 1582–1592. [Google Scholar]
- Mohan, R.R.; Liang, Q.; Kim, W.-J.; Helena, M.C.; Baerveldt, F.; Wilson, S.E. Apoptosis in the cornea: Further characterization of Fas-Fas ligand system. Exp. Eye Res. 1997, 65, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Mohan, R.R.; Hutcheon, A.E.K.; Choi, R.; Hong, J.-W.; Lee, J.-S.; Mohan, R.R.; Ambrósio, R.; Zieske, J.D.; Wilson, S.E. Apoptosis, necrosis, proliferation, and myofibroblast generation in the stroma following LASIK and PRK. Exp. Eye Res. 2003, 76, 71–87. [Google Scholar] [CrossRef]
- Wilson, S.E.; Pedroza, L.; Beuerman, R.; Hill, J.M. Herpes simplex virus type-1 infection of corneal epithelial cells induces apoptosis of the underlying keratocytes. Exp. Eye Res. 1997, 64, 775–779. [Google Scholar] [CrossRef]
- Lassance, L.; Marino, G.K.; Medeiros, C.S.; Thangavadivel, S.; Wilson, S.E. Fibrocyte migration, differentiation and apoptosis during the corneal wound healing response to injury. Exp. Eye Res. 2018, 170, 177–187. [Google Scholar] [CrossRef]
- Wilson, S.E. Corneal myofibroblasts and fibrosis. Exp. Eye Res. 2020, 201, 108272. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.E.; Medeiros, C.S.; Santhiago, M.R. Pathophysiology of corneal scarring in persistent epithelial defects after PRK and other corneal injuries. J. Ref. Surg. 2018, 34, 59–64. [Google Scholar] [CrossRef]
- Yurchenco, P.D.; Tsilibary, E.C.; Charonis, A.S.; Furthmayr, H. Models for the self-assembly of basement membrane. J. Histochem. Cytochem. 1986, 34, 93–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Oliveira, R.C.; Sampaio, L.P.; Shiju, T.M.; Santhiago, M.R.; Wilson, S.E. Epithelial basement membrane regeneration after PRK-induced epithelial-stromal injury in rabbits: Fibrotic vs. non-fibrotic corneal healing. J. Ref. Surg. 2022, 38, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, R.V.; Zoeller, J.J.; Nyström, A. Basement membrane proteoglycans: Modulators Par Excellence of cancer growth and angiogenesis. Mol. Cells 2009, 27, 503–513. [Google Scholar] [CrossRef]
- Paralkar, V.M.; Vukicevic, S.; Reddi, A.H. Transforming growth factor beta type 1 binds to collagen IV of basement membrane matrix: Implications for development. Dev. Biol. 1991, 143, 303–308. [Google Scholar] [CrossRef]
- Shibuya, H.; Okamoto, O.; Fujiwara, S. The bioactivity of transforming growth factor-beta1 can be regulated via binding to dermal collagens in mink lung epithelial cells. J. Dermatol. Sci. 2006, 41, 187–195. [Google Scholar] [CrossRef]
- Göhring, W.; Sasaki, T.; Heldin, C.H.; Timpl, R. Mapping of the binding of platelet-derived growth factor to distinct domains of the basement membrane proteins BM-40 and perlecan and distinction from the BM-40 collagen-binding epitope. Eur. J. Biochem. 1998, 255, 60–66. [Google Scholar] [CrossRef]
- Jester, J.V.; Huang, J.; Petroll, W.M.; Cavanagh, H.D. TGF beta induced myofibroblast differentiation of rabbit keratocytes requires synergistic TGF beta, PDGF and integrin signaling. Exp. Eye Res. 2002, 75, 645–657. [Google Scholar] [CrossRef]
- Lande, M.A.; Birk, D.E.; Nagpal, M.L.; Rader, R.L. Phagocytic properties of human keratocyte cultures. Investig. Ophthalmol. Vis. Sci. 1981, 20, 481–489. [Google Scholar]
- Marino, G.K.; Santhiago, M.R.; Santhanam, A.; Lassance, L.; Thangavadivel, S.; Medeiros, C.S.; Bose, K.; Tam, K.P.; Wilson, S.E. Epithelial basement membrane injury and regeneration modulates corneal fibrosis after pseudomonas corneal ulcers in rabbits. Exp. Eye Res. 2017, 161, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.E.; Marino, G.K.; Torricelli, A.A.M.; Medeiros, C.S. Injury and defective regeneration of the epithelial basement membrane in corneal fibrosis: A paradigm for fibrosis in other organs? Matrix Biol. 2017, 64, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Netto, M.V.; Mohan, R.R.; Sinha, S.; Sharma, A.; Dupps, W.; Wilson, S.E. Stromal haze, myofibroblasts, and surface irregularity after PRK. Exp. Eye Res. 2006, 82, 788–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torricelli, A.A.M.; Singh, V.; Agrawal, V.; Santhiago, M.R.; Wilson, S.E. Transmission electron microscopy analysis of epithelial basement membrane repair in rabbit corneas with haze. Investig. Ophthalmol. Vis. Sci. 2013, 54, 4026–4033. [Google Scholar] [CrossRef] [PubMed]
- Cintron, C.; Kublin, C.L. Regeneration of corneal tissue. Dev. Biol. 1977, 61, 346–357. [Google Scholar] [CrossRef]
- Wilson, S.E.; Chaurasia, S.S.; Medeiros, F.W. Apoptosis in the initiation, modulation and termination of the corneal wound healing response. Exp. Eye Res. 2007, 85, 305–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinz, B. Formation and function of the myofibroblast during tissue repair. J. Investig. Dermatol. 2007, 127, 526–537. [Google Scholar] [CrossRef]
- Chapman, H.A. Epithelial–mesenchymal interactions in pulmonary fibrosis. Annu. Rev. Physiol. 2011, 73, 413–435. [Google Scholar] [CrossRef] [PubMed]
- Piera-Velazquez, S.; Li, Z.; Jimenez, S.A. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am. J. Pathol. 2011, 179, 1074–1080. [Google Scholar] [CrossRef]
- Singh, V.; Agrawal, V.; Santhiago, M.R.; Wilson, S.E. Stromal fibroblast-bone marrow-derived cell interactions: Implications for myofibroblast development in the cornea. Exp. Eye Res. 2012, 98, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Saikia, P.; Crabb, J.S.; Dibbin, L.L.; Juszczak, M.J.; Willard, B.; Jang, G.-F.; Shiju, M.T.; Crabb, J.W.; Wilson, S.E. Quantitative proteomic comparison of myofibroblasts derived from bone marrow or locally from the cornea. Sci. Rep. 2020, 10, 16717. [Google Scholar] [CrossRef] [PubMed]
- Saikia, P.; Medeiros, C.S.; Thangavadivel, S.; Wilson, S.E. Basement membranes in the cornea and other organs that commonly develop fibrosis. Cell Tissue Res. 2018, 374, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.; Hardin, C.; Corkins, C.J.; Jiwani, A.Z.; Fletcher, J.; Carlsson, A.; Chan, R. Pathophysiologic mechanisms and current treatments for cutaneous sequelae of burn wounds. Compr. Physiol. 2017, 8, 371–405. [Google Scholar]
- Gabrielli, A.; Avvedimento, E.V.; Krieg, T. Scleroderma. N. Engl. J. Med. 2009, 360, 1989–2003. [Google Scholar] [CrossRef]
- Yang, L.; Hashimoto, K.; Tohyama, M.; Okazaki, H.; Dai, X.; Hanakawa, Y.; Sayama, K.; Shirakata, Y. Interactions between myofibroblast differentiation and epidermogenesis in constructing human living skin equivalents. J. Dermatol. Sci. 2012, 65, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Liarte, S.; Bernabé-García, Á.; Nicolás, F.J. Role of TGF-β in skin chronic wounds: A keratinocyte perspective. Cells 2020, 9, 306. [Google Scholar] [CrossRef] [Green Version]
- Wataya-Kaneda, M.; Hashimoto, K.; Kato, M.; Miyazono, K.; Yoshikawa, K. Differential localization of TGF-beta-precursor isotypes in psoriatic human skin. J. Dermatol. Sci. 1996, 11, 183–188. [Google Scholar] [CrossRef]
- Thompson, E.A.; Darrah, P.A.; Foulds, K.E.; Hoffer, E.; Caffrey-Carr, A.; Norenstedt, S.; Perbeck, L.; Seder, R.A.; Kedl, R.M.; Loré, K. Monocytes acquire the ability to prime tissue-resident T cells via IL-10-mediated TGF-β release. Cell Rep. 2019, 28, 1127–1135.e4. [Google Scholar] [CrossRef] [Green Version]
- Needleman, B.W.; Choi, J.; Burrows-Mezu, A.; Fontana, J.A. Secretion and binding of transforming growth factor beta by scleroderma and normal dermal fibroblasts. Arthritis Rheum. 1990, 33, 650–665. [Google Scholar] [CrossRef]
- Zhao, B.; Guan, H.; Liu, J.Q.; Zheng, Z.; Zhou, Q.; Zhang, J.; Su, L.L.; Hu, D.H. Hypoxia drives the transition of human dermal fibroblasts to a myofibroblast-like phenotype via the TGF-β1/Smad3 pathway. Int. J. Mol. Med. 2017, 39, 153–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, M.; Tokura, Y. Epithelial-mesenchymal transition in the skin. J. Dermatol. Sci. 2011, 61, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.C.; Pastar, I.; Ojeh, N.; Chen, V.; Liu, S.; Garzon, K.I.; Tomic-Canic, M. Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. 2016, 365, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Kruglikov, I.L.; Scherer, P.E. Adipocyte-myofibroblast transition as a possible pathophysiological step in androgenetic alopecia. Exp. Dermatol. 2017, 26, 522–523. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, V.S.; Howell, K.; Csiszar, K.; Denton, C.P.; Black, C.M.; Abraham, D.J. Shared expression of phenotypic markers in systemic sclerosis indicates a convergence of pericytes and fibroblasts to a myofibroblast lineage in fibrosis. Arthritis Res. Ther. 2005, 7, R1113–R1123. [Google Scholar] [CrossRef] [Green Version]
- Quan, T.E.; Cowper, S.E.; Bucala, R. The role of circulating fibrocytes in fibrosis. Curr. Rheumatol. Rep. 2006, 8, 145–150. [Google Scholar] [CrossRef]
- Basset, F.; Ferrans, V.J.; Soler, P.; Takemura, T.; Fukuda, Y.; Crystal, R.G. Intraluminal fibrosis in interstitial lung disorders. Am. J. Pathol. 1986, 122, 443–461. [Google Scholar]
- Parra, E.R.; Pincelli, M.S.; Teodoro, W.R.; Velosa, A.P.; Martins, V.; Rangel, M.P.; Barbas-Filho, J.V.; Capelozzi, V.L. Modeling pulmonary fibrosis by abnormal expression of telomerase/apoptosis/collagen V in experimental usual interstitial pneumonia. Braz. J. Med. Biol. Res. 2014, 47, 567–575. [Google Scholar] [CrossRef] [Green Version]
- Bowden, D.H. Alveolar response to injury. Thorax 1981, 36, 801–804. [Google Scholar] [CrossRef] [Green Version]
- Furuyama, A.; Mochitate, K. Assembly of the exogenous extracellular matrix during basement membrane formation by alveolar epithelial cells in vitro. J. Cell Sci. 2000, 113, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, O.; Tamura, A.; Ohdama, S.; Mark, E.J. Alveolar basement membrane breaks down in diffuse alveolar damage: An immunohistochemical study. Pathol. Int. 1995, 45, 473–482. [Google Scholar] [CrossRef]
- Willis, B.C.; Liebler, J.M.; Luby-Phelps, K.; Nicholson, A.G.; Crandall, E.D.; du Bois, R.M.; Borok, Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: Potential role in idiopathic pulmonary fibrosis. Am. J. Pathol. 2005, 166, 1321–1332. [Google Scholar] [CrossRef]
- Kasai, H.; Allen, J.T.; Mason, R.M.; Kamimura, T.; Zhang, Z. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir. Res. 2005, 6, 56. [Google Scholar] [CrossRef] [Green Version]
- Yao, H.W.; Xie, Q.M.; Chen, J.Q.; Deng, Y.M.; Tang, H.F. TGF-beta1 induces alveolar epithelial to mesenchymal transition in vitro. Life Sci. 2004, 76, 29–37. [Google Scholar] [CrossRef]
- Goldmann, T.; Zissel, G.; Watz, H.; Drömann, D.; Reck, M.; Kugler, C.; Rabe, K.F.; Marwitz, S. Human alveolar epithelial cells type II are capable of TGFβ-dependent epithelial-mesenchymal-transition and collagen-synthesis. Respir. Res. 2018, 19, 138. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Kugler, M.C.; Wolters, P.J.; Robillard, L.; Galvez, M.G.; Brumwell, A.N.; Sheppard, D.; Chapman, H.A. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by extracellular matrix. Proc. Natl. Acad. Sci. USA 2006, 103, 13180–13185. [Google Scholar] [CrossRef] [Green Version]
- Rock, J.R.; Barkauskas, C.E.; Cronce, M.J.; Xue, Y.; Harris, J.R.; Liang, J.; Noble, P.W.; Hogan, B.L. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc. Natl. Acad. Sci. USA 2011, 108, E1475–E1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kage, H.; Borok, Z. EMT and interstitial lung disease: A mysterious relationship. Curr. Opin. Pulm. Med. 2012, 18, 517–523. [Google Scholar] [CrossRef]
- Humphreys, B.D.; Lin, S.L.; Kobayashi, A.; Hudson, T.E.; Nowlin, B.T.; Bonventre, J.V.; Valerius, M.T.; McMahon, A.P.; Duffield, J.S. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 2010, 176, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Heukels, P.; van Hulst, J.A.C.; van Nimwegen, M.; Boorsma, C.E.; Melgert, B.N.; van den Toorn, L.M.; Boomars, K.A.T.; Wijsenbeek, M.S.; Hoogsteden, H.; von der Thüsen, J.H.; et al. Fibrocytes are increased in lung and peripheral blood of patients with idiopathic pulmonary fibrosis. Respir. Res. 2018, 19, 90. [Google Scholar] [CrossRef] [PubMed]
- Alhamad, E.H.; Shakoor, Z.; Al-Kassimi, F.A.; Almogren, A.; Gad ElRab, M.O.; Maharaj, S.; Kolb, M. Rapid detection of circulating fibrocytes by flowcytometry in idiopathic pulmonary fibrosis. Ann. Thorac. Med. 2015, 10, 279–283. [Google Scholar] [CrossRef]
- Moeller, A.; Gilpin, S.E.; Ask, K.; Cox, G.; Cook, D.; Gauldie, J.; Margetts, P.J.; Farkas, L.; Dobranowski, J.; Boylan, C.; et al. Circulating fibrocytes are an indicator of poor prognosis in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2009, 179, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Andersson-Sjöland, A.; de Alba, C.G.; Nihlberg, K.; Becerril, C.; Ramírez, R.; Pardo, A.; Westergren-Thorsson, G.; Selman, M. Fibrocytes are a potential source of lung fibroblasts in idiopathic pulmonary fibrosis. Int. J. Biochem. Cell Biol. 2008, 40, 2129–2140. [Google Scholar] [CrossRef]
- Vyalov, S.L.; Gabbiani, G.; Kapanci, Y. Rat alveolar myofibroblasts acquire alpha-smooth muscle actin expression during bleomycin-induced pulmonary fibrosis. Am. J. Pathol. 1993, 143, 1754–1765. [Google Scholar] [PubMed]
- Piera-Velazquez, S.; Mendoza, F.A.; Jimenez, S.A. Endothelial to mesenchymal transition (EndoMT) in the pathogenesis of human fibrotic diseases. J. Clin. Med. 2016, 5, 45. [Google Scholar] [CrossRef]
- Wilson, M.S.; Wynn, T.A. Pulmonary fibrosis: Pathogenesis, etiology and regulation. Mucosal. Immunol. 2009, 2, 103–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagome, K.; Dohi, M.; Okunishi, K.; Tanaka, R.; Miyazaki, J.; Yamamoto, K. In vivo IL-10 gene delivery attenuates bleomycin induced pulmonary fibrosis by inhibiting the production and activation of TGF-beta in the lung. Thorax 2006, 61, 886–894. [Google Scholar] [CrossRef] [Green Version]
- Park, H.R.; Jo, S.K.; Jung, U. Ionizing radiation promotes epithelial-to-mesenchymal transition in lung epithelial cells by TGF-β-producing M2 macrophages. In Vivo 2019, 33, 1773–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannella, K.M.; Wynn, T.A. Mechanisms of organ injury and repair by macrophages. Annu. Rev. Physiol. 2017, 79, 593–617. [Google Scholar] [CrossRef]
- Grotendorst, G.R.; Smale, G.; Pencev, D. Production of transforming growth factor beta by human peripheral blood monocytes and neutrophils. J. Cell Physiol. 1989, 140, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Flanders, K.C.; Phan, S.H. Cellular localization of transforming growth factor-beta expression in bleomycin-induced pulmonary fibrosis. Am. J. Pathol. 1995, 147, 352–361. [Google Scholar] [PubMed]
- Camelo, A.; Dunmore, R.; Sleeman, M.A.; Clarke, D.L. The epithelium in idiopathic pulmonary fibrosis: Breaking the barrier. Front. Pharmacol. 2014, 4, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, L.H.; Yamauchi, K.; Uzuki, M.; Nakanishi, T.; Takigawa, M.; Inoue, H.; Sawai, T. Type II alveolar epithelial cells and interstitial fibroblasts express connective tissue growth factor in IPF. Eur. Respir. J. 2001, 17, 1220–1227. [Google Scholar] [CrossRef] [Green Version]
- Saito, A.; Horie, M.; Nagase, T. TGF-b signaling in lung health and disease. Int. J. Mol. Sci. 2018, 19, 2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaccaro, C.A.; Brody, J.S.; Snider, G.L. Alveolar wall basement membranes in bleomycin-induced pulmonary fibrosis. Am. Rev. Respir. Dis. 1985, 132, 905–912. [Google Scholar]
- Furuyama, A.; Kimata, K.; Mochitate, K. Assembly of basement membrane in vitro by cooperation between alveolar epithelial cells and pulmonary fibroblasts. Cell Struct. Funct. 1997, 22, 603–614. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, N.M.; Bai, Y.; Mochitate, K.; Senior, R.M. Laminin alpha-chain expression and basement membrane formation by MLE-15 respiratory epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L1004–L1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskams, T.; Rosenbaum, J.; De Vos, R.; David, G.; Desmet, V. Heparan sulfate proteoglycan expression in chronic cholestatic human liver diseases. Hepatology 1996, 24, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Spatz, J.P.; Häussinger, D. Laminin-521 promotes quiescence in isolated stellate cells from rat liver. Biomaterials 2018, 180, 36–51. [Google Scholar]
- Huang, Y.; Deng, X.; Liang, J. Modulation of hepatic stellate cells and reversibility of hepatic fibrosis. Exp. Cell Res. 2017, 352, 420–426. [Google Scholar] [CrossRef]
- Tomte, L.T.; Annatshah, Y.; Schlüter, N.K.; Miosge, N.; Herken, R.; Quondamatteo, F. Hematopoietic cells are a source of nidogen-1 and nidogen-2 during mouse liver development. J. Histochem. Cytochem. 2006, 54, 593–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karsdal, M.A.; Detlefsen, S.; Daniels, S.J.; Nielsen, M.J.; Krag, A.; Schuppan, D. Is the Total Amount as Important as Localization and Type of Collagen in Liver Fibrosis Attributable to Steatohepatitis? Hepatology 2020, 71, 346–351. [Google Scholar] [CrossRef]
- Mak, K.M.; Mei, R. Basement membrane type IV collagen and laminin: An overview of their biology and value as fibrosis biomarkers of liver disease. Anat. Rec. 2017, 300, 1371–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Hernandez, A.; Martinez, J. The role of capillarization in hepatic failure: Studies in carbon tetrachloride-induced cirrhosis. Hepatology 1991, 14, 864–874. [Google Scholar] [CrossRef]
- Jia, J.D.; Bauer, M.; Sedlaczek, N.; Herbst, H.; Ruehl, M.; Hahn, E.G.; Riecken, E.O.; Schuppan, D. Modulation of collagen XVIII/endostatin expression in lobular and biliary rat liver fibrogenesis. J. Hepatol. 2001, 35, 386–391. [Google Scholar] [CrossRef]
- Mak, K.M.; Chen, L.L.; Lee, T.F. Codistribution of collagen type IV and laminin in liver fibrosis of elderly cadavers: Immunohistochemical marker of perisinusoidal basement membrane formation. Anat. Rec. 2013, 296, 953–964. [Google Scholar] [CrossRef] [PubMed]
- McLean, A.J.; Cogger, V.C.; Chong, G.C.; Warren, A.; Makus, A.M.A.; Dahlstrom, E.; Le Couteur, D.G. Age-related pseudocapillarization of the human liver. J. Pathol. 2003, 200, 112–117. [Google Scholar] [CrossRef]
- Le Couteur, D.G.; Warren, A.; Cogger, V.C.; Smedsrød, B.; Sørensen, K.K.; De Cabo, R.; Fraser, R.; McCuskey, R.S. Old age and the hepatic sinusoid. Anat. Rec. 2008, 291, 672–683. [Google Scholar] [CrossRef]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, N.; Corne, S.; Whitman, C.; Yacyshyn, H. Plasmin regulates the activation of cell-associated latent TGFβ1 secreted by rat alveolar macrophages after in vivo bleomycin injury. Am. J. Respir. Cell Mol. Biol. 1996, 15, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Schon, H.T.; Weiskirchen, R. Immunomodulatory effects of transforming growth factor-β in the liver. Hepatobiliary Surg. Nutr. 2014, 3, 386–406. [Google Scholar] [PubMed]
- Ghafoory, S.; Varshney, R.; Robison, T.; Kouzbari, K.; Woolington, S.; Murphy, B.; Xia, L.; Ahamed, J. Platelet TGF-β1 deficiency decreases liver fibrosis in a mouse model of liver injury. Blood Adv. 2018, 2, 470–480. [Google Scholar] [CrossRef] [Green Version]
- Puche, J.E.; Saiman, Y.; Friedman, S.L. Hepatic stellate cells and liver fibrosis. Compr. Physiol. 2013, 3, 1473–1492. [Google Scholar]
- Mederacke, I.; Hsu, C.C.; Troeger, J.S.; Huebener, P.; Mu, X.; Dapito, D.H.; Pradere, J.P.; Schwabe, R.F. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 2013, 4, 2823. [Google Scholar] [CrossRef] [Green Version]
- Direkze, N.C.; Forbes, S.J.; Brittan, M.; Hunt, T.; Jeffery, R.; Preston, S.L.; Poulsom, R.; Hodivala-Dilke, K.; Alison, M.R.; Wright, N. Multiple organ engraftment by bone-marrow-derived myofibroblasts and fibroblasts in bone-marrow-transplanted mice. Stem Cells 2003, 21, 514–520. [Google Scholar] [CrossRef]
- Forbes, S.J.; Russo, F.P.; Rey, V.; Burra, P.; Rugge, M.; Wright, N.A.; Allison, M.R. A significant proportion of myofibroblasts are of bone marrow origin in human liver fibrosis. Gastroenterology 2004, 126, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Taura, K.; Iwaisako, K.; Hatano, E.; Uemoto, S. Controversies over the epithelial-to-mesenchymal transition in liver fibrosis. J. Clin. Med. 2016, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Lafoz, E.; Ruart, M.; Anton, A.; Oncins, A.; Hernández-Gea, V. The endothelium as a driver of liver fibrosis and regeneration. Cells 2020, 9, 929. [Google Scholar] [CrossRef] [PubMed] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilson, S.E. Fibrosis Is a Basement Membrane-Related Disease in the Cornea: Injury and Defective Regeneration of Basement Membranes May Underlie Fibrosis in Other Organs. Cells 2022, 11, 309. https://doi.org/10.3390/cells11020309
Wilson SE. Fibrosis Is a Basement Membrane-Related Disease in the Cornea: Injury and Defective Regeneration of Basement Membranes May Underlie Fibrosis in Other Organs. Cells. 2022; 11(2):309. https://doi.org/10.3390/cells11020309
Chicago/Turabian StyleWilson, Steven E. 2022. "Fibrosis Is a Basement Membrane-Related Disease in the Cornea: Injury and Defective Regeneration of Basement Membranes May Underlie Fibrosis in Other Organs" Cells 11, no. 2: 309. https://doi.org/10.3390/cells11020309
APA StyleWilson, S. E. (2022). Fibrosis Is a Basement Membrane-Related Disease in the Cornea: Injury and Defective Regeneration of Basement Membranes May Underlie Fibrosis in Other Organs. Cells, 11(2), 309. https://doi.org/10.3390/cells11020309