



Deficient Sarcolemma Repair in ALS: A Novel Mechanism with Therapeutic Potential

 ,
, 

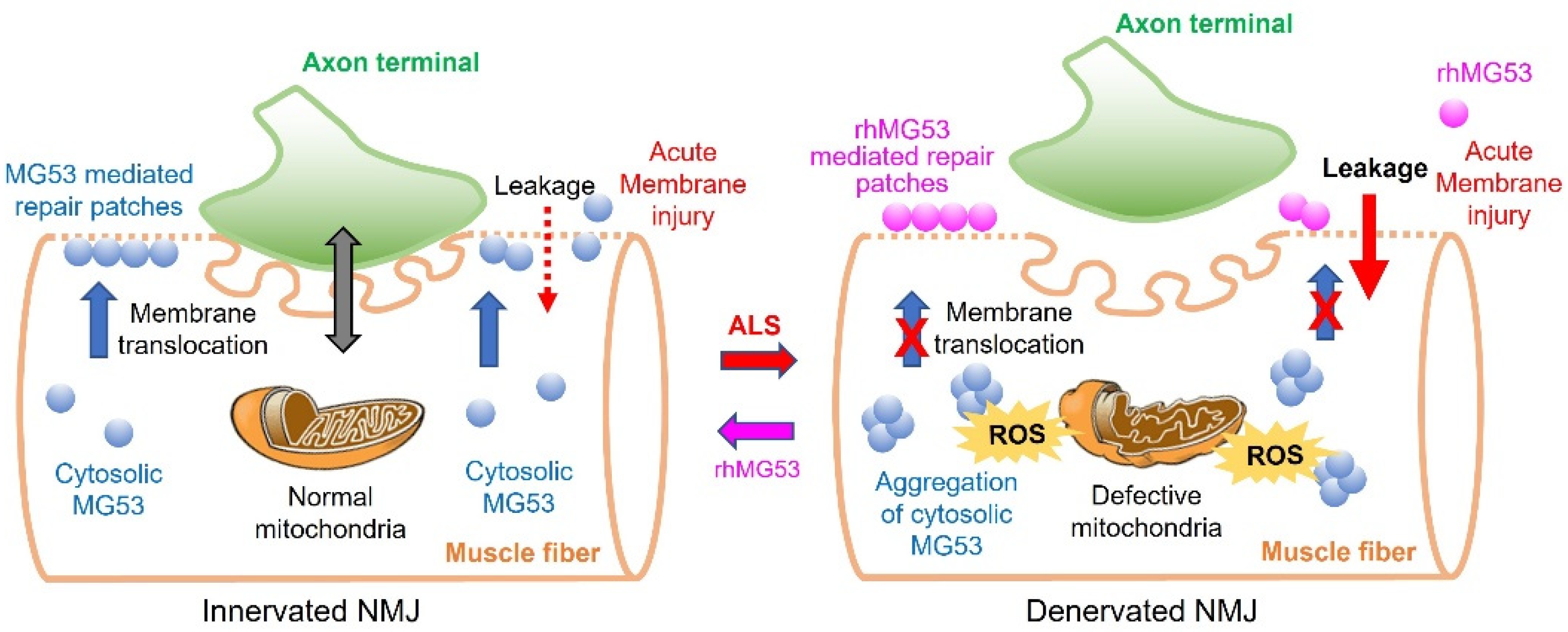{kind=link}
Abstract
:1. Introduction
2. Mitochondrial Dysfunction and Oxidative Stress in ALS Skeletal Muscle
3. ALS Mice Exhibit Sarcolemma Fragility and Mitochondrial Dysfunction in Proximity to NMJs Prior to Symptom Onset
4. MG53-Mediated Membrane Repair Is Compromised in ALS
5. Therapeutic Potential of Exogenously Administered MG53 in ALS
6. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiernan, M.C.; Vucic, S.; Talbot, K.; McDermott, C.J.; Hardiman, O.; Shefner, J.M.; Al-Chalabi, A.; Huynh, W.; Cudkowicz, M.; Talman, P. Improving clinical trial outcomes in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2021, 17, 104–118. [Google Scholar] [CrossRef] [PubMed]
- Hardiman, O.; van den Berg, L.H.; Kiernan, M.C. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, J.P.; Picchiarelli, G.; Dupuis, L.; Gonzalez De Aguilar, J.L. The Role of Skeletal Muscle in Amyotrophic Lateral Sclerosis. Brain Pathol. 2016, 26, 227–236. [Google Scholar] [CrossRef]
- de Carvalho, M.; Swash, M.; Pinto, S. Diaphragmatic Neurophysiology and Respiratory Markers in ALS. Front. Neurol. 2019, 10, 143. [Google Scholar] [CrossRef] [Green Version]
- Niedermeyer, S.; Murn, M.; Choi, P.J. Respiratory Failure in Amyotrophic Lateral Sclerosis. Chest 2019, 155, 401–408. [Google Scholar] [CrossRef]
- Nguyen, Q.T.; Son, Y.J.; Sanes, J.R.; Lichtman, J.W. Nerve terminals form but fail to mature when postsynaptic differentiation is blocked: In vivo analysis using mammalian nerve-muscle chimeras. J. Neurosci. 2000, 20, 6077–6086. [Google Scholar] [CrossRef] [Green Version]
- Dobrowolny, G.; Aucello, M.; Rizzuto, E.; Beccafico, S.; Mammucari, C.; Boncompagni, S.; Belia, S.; Wannenes, F.; Nicoletti, C.; Del Prete, Z.; et al. Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab. 2008, 8, 425–436. [Google Scholar] [CrossRef]
- Wong, M.; Martin, L.J. Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum. Mol. Genet. 2010, 19, 2284–2302. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Ma, C.; Yi, J.; Wu, S.; Luo, G.; Xu, X.; Lin, P.H.; Sun, J.; Zhou, J. Suppressed autophagy flux in skeletal muscle of an amyotrophic lateral sclerosis mouse model during disease progression. Physiol. Rep. 2015, 3, e12271. [Google Scholar] [CrossRef]
- Luo, G.; Yi, J.; Ma, C.; Xiao, Y.; Yi, F.; Yu, T.; Zhou, J. Defective mitochondrial dynamics is an early event in skeletal muscle of an amyotrophic lateral sclerosis mouse model. PLoS ONE 2013, 8, e82112. [Google Scholar] [CrossRef]
- Wang, H.; Yi, J.; Li, X.; Xiao, Y.; Dhakal, K.; Zhou, J. ALS-associated mutation SOD1(G93A) leads to abnormal mitochondrial dynamics in osteocytes. Bone 2018, 106, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Ma, C.; Li, Y.; Weisleder, N.; Rios, E.; Ma, J.; Zhou, J. Mitochondrial calcium uptake regulates rapid calcium transients in skeletal muscle during excitation-contraction (E-C) coupling. J. Biol. Chem. 2011, 286, 32436–32443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Karam, C.; Yi, J.; Zhang, L.; Li, X.; Yoon, D.; Wang, H.; Dhakal, K.; Ramlow, P.; Yu, T.; et al. ROS-related mitochondrial dysfunction in skeletal muscle of an ALS mouse model during the disease progression. Pharm. Res. 2018, 138, 25–36. [Google Scholar] [CrossRef]
- Zhou, J.; Li, A.; Li, X.; Yi, J. Dysregulated mitochondrial Ca(2+) and ROS signaling in skeletal muscle of ALS mouse model. Arch. Biochem. Biophys. 2019, 663, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yi, J.; Fu, R.; Liu, E.; Siddique, T.; Rios, E.; Deng, H.X. Hyperactive intracellular calcium signaling associated with localized mitochondrial defects in skeletal muscle of an animal model of amyotrophic lateral sclerosis. J. Biol. Chem. 2010, 285, 705–712. [Google Scholar] [CrossRef] [Green Version]
- Zhu, K.; Yi, J.; Xiao, Y.; Lai, Y.; Song, P.; Zheng, W.; Jiao, H.; Fan, J.; Wu, C.; Chen, D.; et al. Impaired bone homeostasis in amyotrophic lateral sclerosis mice with muscle atrophy. J. Biol. Chem. 2015, 290, 8081–8094. [Google Scholar] [CrossRef] [Green Version]
- Dadon-Nachum, M.; Melamed, E.; Offen, D. The "dying-back" phenomenon of motor neurons in ALS. J. Mol. Neurosci. 2011, 43, 470–477. [Google Scholar] [CrossRef]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Frey, D.; Schneider, C.; Xu, L.; Borg, J.; Spooren, W.; Caroni, P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J. Neurosci. 2000, 20, 2534–2542. [Google Scholar] [CrossRef] [Green Version]
- Clark, J.A.; Southam, K.A.; Blizzard, C.A.; King, A.E.; Dickson, T.C. Axonal degeneration, distal collateral branching and neuromuscular junction architecture alterations occur prior to symptom onset in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. J. Chem. Neuroanat. 2016, 76, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Campanari, M.L.; Bourefis, A.R.; Kabashi, E. Diagnostic Challenge and Neuromuscular Junction Contribution to ALS Pathogenesis. Front. Neurol. 2019, 10, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martineau, E.; Di Polo, A.; Vande Velde, C.; Robitaille, R. Dynamic neuromuscular remodeling precedes motor-unit loss in a mouse model of ALS. Elife 2018, 7, e41973. [Google Scholar] [CrossRef] [PubMed]
- Cappello, V.; Francolini, M. Neuromuscular Junction Dismantling in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2017, 18, 2092. [Google Scholar] [CrossRef]
- Dupuis, L.; Gonzalez de Aguilar, J.L.; Echaniz-Laguna, A.; Eschbach, J.; Rene, F.; Oudart, H.; Halter, B.; Huze, C.; Schaeffer, L.; Bouillaud, F.; et al. Muscle mitochondrial uncoupling dismantles neuromuscular junction and triggers distal degeneration of motor neurons. PLoS ONE 2009, 4, e5390. [Google Scholar] [CrossRef] [Green Version]
- Karam, C.; Yi, J.; Xiao, Y.; Dhakal, K.; Zhang, L.; Li, X.; Manno, C.; Xu, J.; Li, K.; Cheng, H. Absence of physiological Ca 2+ transients is an initial trigger for mitochondrial dysfunction in skeletal muscle following denervation. Skelet. Muscle 2017, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Magrane, J.; Cortez, C.; Gan, W.B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef] [Green Version]
- Scaricamazza, S.; Salvatori, I.; Ferri, A.; Valle, C. Skeletal Muscle in ALS: An Unappreciated Therapeutic Opportunity? Cells 2021, 10, 525. [Google Scholar] [CrossRef]
- Cooper, S.T.; McNeil, P.L. Membrane Repair: Mechanisms and Pathophysiology. Physiol. Rev. 2015, 95, 1205–1240. [Google Scholar] [CrossRef] [Green Version]
- Han, R.; Campbell, K.P. Dysferlin and muscle membrane repair. Curr. Opin. Cell Biol. 2007, 19, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Demonbreun, A.R.; McNally, E.M. Plasma Membrane Repair in Health and Disease. Curr. Top. Membr. 2016, 77, 67–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gazzerro, E.; Bonetto, A.; Minetti, C. Caveolinopathies: Translational implications of caveolin-3 in skeletal and cardiac muscle disorders. Handb. Clin. Neurol. 2011, 101, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, L.; Yue, H.; Whitson, B.A.; Haggard, E.; Xu, X.; Ma, J. MG53, A Tissue Repair Protein with Broad Applications in Regenerative Medicine. Cells 2021, 10, 122. [Google Scholar] [CrossRef] [PubMed]
- Neel, B.A.; Lin, Y.; Pessin, J.E. Skeletal muscle autophagy: A new metabolic regulator. Trends Endocrinol. Metab. TEM 2013, 24, 635–643. [Google Scholar] [CrossRef] [Green Version]
- Larsen, S.; Nielsen, J.; Hansen, C.N.; Nielsen, L.B.; Wibrand, F.; Stride, N.; Schroder, H.D.; Boushel, R.; Helge, J.W.; Dela, F.; et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J. Physiol. 2012, 590, 3349–3360. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Carri, M.T.; D’Ambrosi, N.; Cozzolino, M. Pathways to mitochondrial dysfunction in ALS pathogenesis. Biochem. Biophys. Res. Commun 2017, 483, 1187–1193. [Google Scholar] [CrossRef]
- Echaniz-Laguna, A.; Zoll, J.; Ponsot, E.; N’Guessan, B.; Tranchant, C.; Loeffler, J.P.; Lampert, E. Muscular mitochondrial function in amyotrophic lateral sclerosis is progressively altered as the disease develops: A temporal study in man. Exp. Neurol. 2006, 198, 25–30. [Google Scholar] [CrossRef]
- Napoli, L.; Crugnola, V.; Lamperti, C.; Silani, V.; Di Mauro, S.; Bresolin, N.; Moggio, M. Ultrastructural mitochondrial abnormalities in patients with sporadic amyotrophic lateral sclerosis. Arch. Neurol. 2011, 68, 1612–1613. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, S.; Iwata, M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2007, 66, 10–16. [Google Scholar] [CrossRef]
- Siciliano, G.; Pastorini, E.; Pasquali, L.; Manca, M.L.; Iudice, A.; Murri, L. Impaired oxidative metabolism in exercising muscle from ALS patients. J. Neurol. Sci. 2001, 191, 61–65. [Google Scholar] [CrossRef]
- Soraru, G.; Vergani, L.; Fedrizzi, L.; D’Ascenzo, C.; Polo, A.; Bernazzi, B.; Angelini, C. Activities of mitochondrial complexes correlate with nNOS amount in muscle from ALS patients. Neuropathol. Appl. Neurobiol. 2007, 33, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, F.R.; Manfredi, G.; Mawrin, C.; Beal, M.F.; Schon, E.A. Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J. Neurochem. 2002, 80, 616–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carri, M.T.; Valle, C.; Bozzo, F.; Cozzolino, M. Oxidative stress and mitochondrial damage: Importance in non-SOD1 ALS. Front. Cell Neurosci. 2015, 9, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amico, E.; Factor-Litvak, P.; Santella, R.M.; Mitsumoto, H. Clinical perspective on oxidative stress in sporadic amyotrophic lateral sclerosis. Free Radic Biol. Med. 2013, 65, 509–527. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Yong, V.W. Oxidized phospholipids as novel mediators of neurodegeneration. Trends Neurosci. 2022, 45, 419–429. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Carri, M.T.; Cozzolino, M. SOD1 and mitochondria in ALS: A dangerous liaison. J. Bioenerg Biomembr. 2011, 43, 593–599. [Google Scholar] [CrossRef]
- Ivanova, M.I.; Sievers, S.A.; Guenther, E.L.; Johnson, L.M.; Winkler, D.D.; Galaleldeen, A.; Sawaya, M.R.; Hart, P.J.; Eisenberg, D.S. Aggregation-triggering segments of SOD1 fibril formation support a common pathway for familial and sporadic ALS. Proc. Natl. Acad. Sci. USA 2014, 111, 197–201. [Google Scholar] [CrossRef] [Green Version]
- Ludolph, A.C.; Bendotti, C.; Blaugrund, E.; Chio, A.; Greensmith, L.; Loeffler, J.P.; Mead, R.; Niessen, H.G.; Petri, S.; Pradat, P.F.; et al. Guidelines for preclinical animal research in ALS/MND: A consensus meeting. Amyotroph. Lateral Scler. 2010, 11, 38–45. [Google Scholar] [CrossRef]
- McGoldrick, P.; Joyce, P.I.; Fisher, E.M.; Greensmith, L. Rodent models of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2013, 1832, 1421–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, F.L.; Song, W.; Jang, Y.C.; Liu, Y.; Sabia, M.; Richardson, A.; Van Remmen, H. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am. J. Physiol. Regul Integr. Comp. Physiol. 2007, 293, R1159–R1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halter, B.; Gonzalez de Aguilar, J.L.; Rene, F.; Petri, S.; Fricker, B.; Echaniz-Laguna, A.; Dupuis, L.; Larmet, Y.; Loeffler, J.P. Oxidative stress in skeletal muscle stimulates early expression of Rad in a mouse model of amyotrophic lateral sclerosis. Free Radic Biol. Med. 2010, 48, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Han, S.M.; El Oussini, H.; Scekic-Zahirovic, J.; Vibbert, J.; Cottee, P.; Prasain, J.K.; Bellen, H.J.; Dupuis, L.; Miller, M.A. VAPB/ALS8 MSP ligands regulate striated muscle energy metabolism critical for adult survival in caenorhabditis elegans. PLoS Genet. 2013, 9, e1003738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stallings, N.R.; Puttaparthi, K.; Dowling, K.J.; Luther, C.M.; Burns, D.K.; Davis, K.; Elliott, J.L. TDP-43, an ALS linked protein, regulates fat deposition and glucose homeostasis. PLoS ONE 2013, 8, e71793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capitanio, D.; Vasso, M.; Ratti, A.; Grignaschi, G.; Volta, M.; Moriggi, M.; Daleno, C.; Bendotti, C.; Silani, V.; Gelfi, C. Molecular signatures of amyotrophic lateral sclerosis disease progression in hind and forelimb muscles of an SOD1(G93A) mouse model. Antioxid Redox Signal. 2012, 17, 1333–1350. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Fang, H.; Groom, L.; Cheng, A.; Zhang, W.; Liu, J.; Wang, X.; Li, K.; Han, P.; Zheng, M.; et al. Superoxide flashes in single mitochondria. Cell 2008, 134, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Fang, H.; Chen, M.; Ding, Y.; Shang, W.; Xu, J.; Zhang, X.; Zhang, W.; Li, K.; Xiao, Y.; Gao, F.; et al. Imaging superoxide flash and metabolism-coupled mitochondrial permeability transition in living animals. Cell Res. 2011, 21, 1295–1304. [Google Scholar] [CrossRef]
- Wei, L.; Salahura, G.; Boncompagni, S.; Kasischke, K.A.; Protasi, F.; Sheu, S.S.; Dirksen, R.T. Mitochondrial superoxide flashes: Metabolic biomarkers of skeletal muscle activity and disease. FASEB J. 2011, 25, 3068–3078. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Fang, H.; Shang, W.; Xiao, Y.; Sun, T.; Hou, N.; Pan, L.; Sun, X.; Ma, Q.; Zhou, J.; et al. Mitoflash altered by metabolic stress in insulin-resistant skeletal muscle. J. Mol. Med. (Berl) 2015, 93, 1119–1130. [Google Scholar] [CrossRef]
- Batandier, C.; Leverve, X.; Fontaine, E. Opening of the mitochondrial permeability transition pore induces reactive oxygen species production at the level of the respiratory chain complex I. J. Biol. Chem. 2004, 279, 17197–17204. [Google Scholar] [CrossRef] [Green Version]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Li, A.; Yi, J.; Li, X.; Zhou, J. Physiological Ca2+ Transients Versus Pathological Steady-State Ca2+ Elevation, Who Flips the ROS Coin in Skeletal Muscle Mitochondria. Front. Physiol. 2020, 11, 595800. [Google Scholar] [CrossRef]
- Ott, M.; Robertson, J.D.; Gogvadze, V.; Zhivotovsky, B.; Orrenius, S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc. Natl. Acad. Sci. USA 2002, 99, 1259–1263. [Google Scholar] [CrossRef] [Green Version]
- Turrens, J.F.; Alexandre, A.; Lehninger, A.L. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 1985, 237, 408–414. [Google Scholar] [CrossRef]
- Muller, F.; Crofts, A.R.; Kramer, D.M. Multiple Q-cycle bypass reactions at the Qo site of the cytochrome bc 1 complex. Biochemistry 2002, 41, 7866–7874. [Google Scholar] [CrossRef]
- Muller, F.L.; Roberts, A.G.; Bowman, M.K.; Kramer, D.M. Architecture of the Qo site of the cytochrome bc 1 complex probed by superoxide production. Biochemistry 2003, 42, 6493–6499. [Google Scholar] [CrossRef]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef]
- Basso, E.; Fante, L.; Fowlkes, J.; Petronilli, V.; Forte, M.A.; Bernardi, P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin, D.J. Biol. Chem. 2005, 280, 18558–18561. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T.; Shimizu, S.; Watanabe, T.; Yamaguchi, O.; Otsu, K.; Yamagata, H.; Inohara, H.; Kubo, T.; Tsujimoto, Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005, 434, 652–658. [Google Scholar] [CrossRef]
- Li, A.; Li, X.; Yi, J.; Ma, J.; Zhou, J. Butyrate Feeding Reverses CypD-Related Mitoflash Phenotypes in Mouse Myofibers. Int. J. Mol. Sci. 2021, 22, 7412. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Dirksen, R.T. Mitochondrial superoxide flashes: From discovery to new controversies. J. Gen. Physiol. 2012, 139, 425–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonora, M.; Giorgi, C.; Pinton, P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat. Rev. Mol. Cell Biol. 2022, 23, 266–285. [Google Scholar] [CrossRef] [PubMed]
- Hamer, P.W.; McGeachie, J.M.; Davies, M.J.; Grounds, M.D. Evans Blue Dye as an in vivo marker of myofibre damage: Optimising parameters for detecting initial myofibre membrane permeability. J. Anat. 2002, 200, 69–79. [Google Scholar] [CrossRef]
- Wooddell, C.I.; Radley-Crabb, H.G.; Griffin, J.B.; Zhang, G. Myofiber Damage Evaluation by Evans Blue Dye Injection. Curr. Protoc. Mouse Biol. 2011, 1, 463–488. [Google Scholar] [CrossRef]
- Matsuda, R.; Nishikawa, A.; Tanaka, H. Visualization of dystrophic muscle fibers in mdx mouse by vital staining with Evans blue: Evidence of apoptosis in dystrophin-deficient muscle. J. Biochem. 1995, 118, 959–964. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; Masumiya, H.; Weisleder, N.; Matsuda, N.; Nishi, M.; Hwang, M.; Ko, J.K.; Lin, P.; Thornton, A.; Zhao, X.; et al. MG53 nucleates assembly of cell membrane repair machinery. Nat. Cell Biol. 2009, 11, 56–64. [Google Scholar] [CrossRef] [Green Version]
- Yi, J.; Li, A.; Li, X.; Park, K.; Zhou, X.; Yi, F.; Xiao, Y.; Yoon, D.; Tan, T.; Ostrow, L.W.; et al. MG53 Preserves Neuromuscular Junction Integrity and Alleviates ALS Disease Progression. Antioxidants 2021, 10, 1522. [Google Scholar] [CrossRef]
- McNeil, P.L.; Miyake, K.; Vogel, S.S. The endomembrane requirement for cell surface repair. Proc. Natl. Acad. Sci. USA 2003, 100, 4592–4597. [Google Scholar] [CrossRef] [Green Version]
- Fucile, S. Ca2+ permeability of nicotinic acetylcholine receptors. Cell Calcium 2004, 35, 1–8. [Google Scholar] [CrossRef]
- Dobrowolny, G.; Giacinti, C.; Pelosi, L.; Nicoletti, C.; Winn, N.; Barberi, L.; Molinaro, M.; Rosenthal, N.; Musaro, A. Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. J. Cell Biol. 2005, 168, 193–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cejas, P.; Casado, E.; Belda-Iniesta, C.; De Castro, J.; Espinosa, E.; Redondo, A.; Sereno, M.; Garcia-Cabezas, M.A.; Vara, J.A.; Dominguez-Caceres, A.; et al. Implications of oxidative stress and cell membrane lipid peroxidation in human cancer (Spain). Cancer Causes Control 2004, 15, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Ozato, K.; Shin, D.M.; Chang, T.H.; Morse, H.C. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 2008, 8, 849–860. [Google Scholar] [CrossRef] [Green Version]
- Weisleder, N.; Takeshima, H.; Ma, J. Immuno-proteomic approach to excitation–contraction coupling in skeletal and cardiac muscle: Molecular insights revealed by the mitsugumins. Cell Calcium 2008, 43, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; Weisleder, N.; Ko, J.K.; Komazaki, S.; Sunada, Y.; Nishi, M.; Takeshima, H.; Ma, J. Membrane repair defects in muscular dystrophy are linked to altered interaction between MG53, caveolin-3, and dysferlin. J. Biol. Chem. 2009, 284, 15894–15902. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.; Zhu, H.; Cai, C.; Wang, X.; Cao, C.; Xiao, R.; Pan, Z.; Weisleder, N.; Takeshima, H.; Ma, J. Nonmuscle myosin IIA facilitates vesicle trafficking for MG53-mediated. cell membrane repair. FASEB J. 2012, 26, 1875–1883. [Google Scholar] [CrossRef]
- Zhu, H.; Lin, P.; De, G.; Choi, K.H.; Takeshima, H.; Weisleder, N.; Ma, J. Polymerase transcriptase release factor (PTRF) anchors MG53 protein to cell injury site for initiation of membrane repair. J. Biol. Chem. 2011, 286, 12820–12824. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; Masumiya, H.; Weisleder, N.; Pan, Z.; Nishi, M.; Komazaki, S.; Takeshima, H.; Ma, J. MG53 regulates membrane budding and exocytosis in muscle cells. J. Biol. Chem. 2009, 284, 3314–3322. [Google Scholar] [CrossRef] [Green Version]
- Cao, C.M.; Zhang, Y.; Weisleder, N.; Ferrante, C.; Wang, X.; Lv, F.; Zhang, Y.; Song, R.; Hwang, M.; Jin, L.; et al. MG53 constitutes a primary determinant of cardiac ischemic preconditioning. Circulation 2010, 121, 2565–2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, Z.; Wang, Q.; Zhou, X.; Tan, T.; Park, K.H.; Kramer, H.F.; McDougal, A.; Laping, N.J.; Kumar, S.; Adesanya, T.M.A.; et al. Sustained elevation of MG53 in the bloodstream increases tissue regenerative capacity without compromising metabolic function. Nat. Commun. 2019, 10, 4659. [Google Scholar] [CrossRef] [PubMed]
- Shefner, J.M. Effects of Strength Training in Amyotrophic Lateral Sclerosis: How Much Do We Know? Muscle Nerve 2019, 59, 6–7. [Google Scholar] [CrossRef] [Green Version]
- Tsitkanou, S.; Della Gatta, P.; Foletta, V.; Russell, A. The Role of Exercise as a Non-pharmacological Therapeutic Approach for Amyotrophic Lateral Sclerosis: Beneficial or Detrimental? Front. Neurol. 2019, 10, 783. [Google Scholar] [CrossRef] [Green Version]
- Rosenbohm, A.; Peter, R.; Dorst, J.; Kassubek, J.; Rothenbacher, D.; Nagel, G.; Ludolph, A.C.; Group, A.R.S.S. Life Course of Physical Activity and Risk and Prognosis of Amyotrophic Lateral Sclerosis in a German ALS Registry. Neurology 2021, 97, e1955–e1963. [Google Scholar] [CrossRef]
- Julian, T.H.; Glascow, N.; Barry, A.D.F.; Moll, T.; Harvey, C.; Klimentidis, Y.C.; Newell, M.; Zhang, S.; Snyder, M.P.; Cooper-Knock, J. Physical exercise is a risk factor for amyotrophic lateral sclerosis: Convergent evidence from Mendelian randomisation, transcriptomics and risk genotypes. EBioMedicine 2021, 68, 103397. [Google Scholar] [CrossRef]
- Raymond, J.; Mehta, P.; Larson, T.; Factor-Litvak, P.; Davis, B.; Horton, K. History of vigorous leisure-time physical activity and early onset amyotrophic lateral sclerosis (ALS), data from the national ALS registry: 2010–2018. Amyotroph. Lateral Scler. Front. Degener. 2021, 22, 535–544. [Google Scholar] [CrossRef]
- Harwood, C.A.; Westgate, K.; Gunstone, S.; Brage, S.; Wareham, N.J.; McDermott, C.J.; Shaw, P.J. Long-term physical activity: An exogenous risk factor for sporadic amyotrophic lateral sclerosis? Amyotroph. Lateral Scler. Front. Degener. 2016, 17, 377–384. [Google Scholar] [CrossRef] [Green Version]
- Di, P.W.C.; Di, P.S.G.C. Safety and efficacy of diaphragm pacing in patients with respiratory insufficiency due to amyotrophic lateral sclerosis (DiPALS): A multicentre, open-label, randomised controlled trial. Lancet Neurol. 2015, 14, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Bermejo, J.; Morelot-Panzini, C.; Tanguy, M.L.; Meininger, V.; Pradat, P.F.; Lenglet, T.; Bruneteau, G.; Forestier, N.L.; Couratier, P.; Guy, N.; et al. Early diaphragm pacing in patients with amyotrophic lateral sclerosis (RespiStimALS): A randomised controlled triple-blind trial. Lancet Neurol. 2016, 15, 1217–1227. [Google Scholar] [CrossRef]
- McDermott, C.J.; Bradburn, M.J.; Maguire, C.; Cooper, C.L.; Baird, W.O.; Baxter, S.K.; Cohen, J.; Cantrill, H.; Dixon, S.; Ackroyd, R.; et al. DiPALS: Diaphragm Pacing in patients with Amyotrophic Lateral Sclerosis - a randomised controlled trial. Health Technol. Assess 2016, 20, 1–186. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.G.; Lewis, R.A. Diaphragm pacing in patients with amyotrophic lateral sclerosis. Lancet Neurol. 2016, 15, 542. [Google Scholar] [CrossRef]
- Wood, H. Motor neuron disease: Diaphragm pacing is associated with reduced survival in ALS patients with respiratory insufficiency. Nat. Rev. Neurol. 2015, 11, 484. [Google Scholar] [CrossRef]
- Jablonka, S.; Holtmann, B.; Sendtner, M.; Metzger, F. Therapeutic effects of PEGylated insulin-like growth factor I in the pmn mouse model of motoneuron disease. Exp. Neurol. 2011, 232, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.; Didier, E.; Harris, P.; Siegel, N.; Stadler, J.; Tilbury, L.; Smith, D. PEGylated proteins: Evaluation of their safety in the absence of definitive metabolism studies. Drug Metab. Dispos 2007, 35, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisleder, N.; Takizawa, N.; Lin, P.; Wang, X.; Cao, C.; Zhang, Y.; Tan, T.; Ferrante, C.; Zhu, H.; Chen, P.J.; et al. Recombinant MG53 protein modulates therapeutic cell membrane repair in treatment of muscular dystrophy. Sci. Transl. Med. 2012, 4, 139ra185. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Hou, J.; Roe, J.L.; Park, K.H.; Tan, T.; Zheng, Y.; Li, L.; Zhang, C.; Liu, J.; Liu, Z.; et al. Amelioration of ischemia-reperfusion-induced muscle injury by the recombinant human MG53 protein. Muscle Nerve 2015, 52, 852–858. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Liu, J.; Bian, Z.; Cui, Y.; Zhou, X.; Zhou, X.; Zhang, B.; Adesanya, T.M.; Yi, F.; Park, K.H.; et al. Effect of metabolic syndrome on mitsugumin 53 expression and function. PLoS ONE 2015, 10, e0124128. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Bian, Z.; Jiang, Q.; Wang, X.; Zhou, X.; Park, K.H.; Hsueh, W.; Whitson, B.A.; Haggard, E.; Li, H.; et al. MG53 does not manifest the development of diabetes in db/db mice. Diabetes 2020, 69, 1052–1064. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, X.; Ong, H.; Tan, T.; Park, K.H.; Bian, Z.; Zou, X.; Haggard, E.; Janssen, P.M.; Merritt, R.E.; et al. MG53 suppresses NF-kappaB activation to mitigate age-related heart failure. JCI Insight 2021, 6, e148375. [Google Scholar] [CrossRef]
- Jia, Y.; Chen, K.; Lin, P.; Lieber, G.; Nishi, M.; Yan, R.; Wang, Z.; Yao, Y.; Li, Y.; Whitson, B.A.; et al. Treatment of acute lung injury by targeting MG53-mediated cell membrane repair. Nat. Commun. 2014, 5, 4387. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zhu, H.; Zheng, Y.; Xu, Z.; Li, L.; Tan, T.; Park, K.H.; Hou, J.; Zhang, C.; Li, D.; et al. Cardioprotection of recombinant human MG53 protein in a porcine model of ischemia and reperfusion injury. J. Mol. Cell Cardiol. 2015, 80, 10–19. [Google Scholar] [CrossRef]
- Duann, P.; Li, H.; Lin, P.; Tan, T.; Wang, Z.; Chen, K.; Zhou, X.; Gumpper, K.; Zhu, H.; Ludwig, T.; et al. MG53-mediated cell membrane repair protects against acute kidney injury. Sci. Transl. Med. 2015, 7, 279ra236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Zhang, B.; Zhu, H.; Li, H.; Han, Y.; Chen, K.; Wang, Z.; Zeng, J.; Liu, Y.; Wang, X.; et al. MG53 permeates through blood-brain barrier to protect ischemic brain injury. Oncotarget 2016, 7, 22474–22485. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, A.; Yi, J.; Li, X.; Dong, L.; Ostrow, L.W.; Ma, J.; Zhou, J. Deficient Sarcolemma Repair in ALS: A Novel Mechanism with Therapeutic Potential. Cells 2022, 11, 3263. https://doi.org/10.3390/cells11203263
Li A, Yi J, Li X, Dong L, Ostrow LW, Ma J, Zhou J. Deficient Sarcolemma Repair in ALS: A Novel Mechanism with Therapeutic Potential. Cells. 2022; 11(20):3263. https://doi.org/10.3390/cells11203263
Chicago/Turabian StyleLi, Ang, Jianxun Yi, Xuejun Li, Li Dong, Lyle W. Ostrow, Jianjie Ma, and Jingsong Zhou. 2022. "Deficient Sarcolemma Repair in ALS: A Novel Mechanism with Therapeutic Potential" Cells 11, no. 20: 3263. https://doi.org/10.3390/cells11203263
APA StyleLi, A., Yi, J., Li, X., Dong, L., Ostrow, L. W., Ma, J., & Zhou, J. (2022). Deficient Sarcolemma Repair in ALS: A Novel Mechanism with Therapeutic Potential. Cells, 11(20), 3263. https://doi.org/10.3390/cells11203263