
Musashi-1 and miR-147 Precursor Interaction Mediates Synergistic Oncogenicity Induced by Co-Infection of Two Avian Retroviruses



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Viruses, and Plasmids
2.2. Establishment of a Tumor Model and Sample Collection in Specific Pathogen-Free Chickens
2.3. Hematoxylin and Eosin Staining
2.4. Illumina Small-RNA Deep Sequencing
2.5. TMT-Labeled LC−MS/MS
2.6. Luciferase Reporter Assays
2.7. Western Blotting
2.8. Quantitative Real-Time Polymerase Chain Reaction
2.9. Determination of NF-κB p65 Nuclear Translocation
2.10. Senescence-Associated β-Gal Staining
2.11. ELISA for NF-κB p65 and P-IκBα Assays
2.12. RNA ChIP Assay
2.13. Statistical Analysis
3. Results
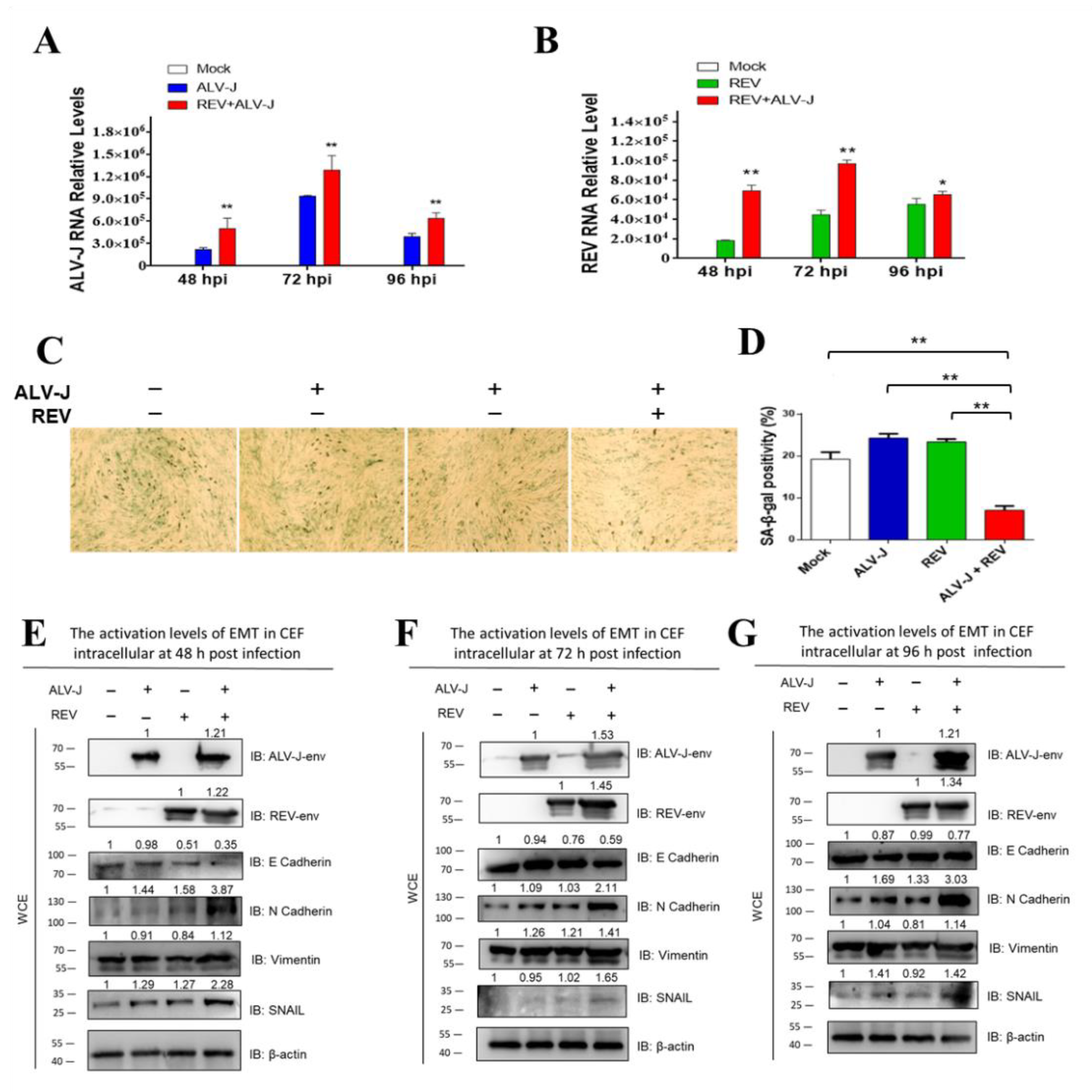
3.1. ALV-J and REV Synergistically Suppress Cellular Senescence and Activate Epithelial–Mesenchymal Transition (EMT) In Vitro
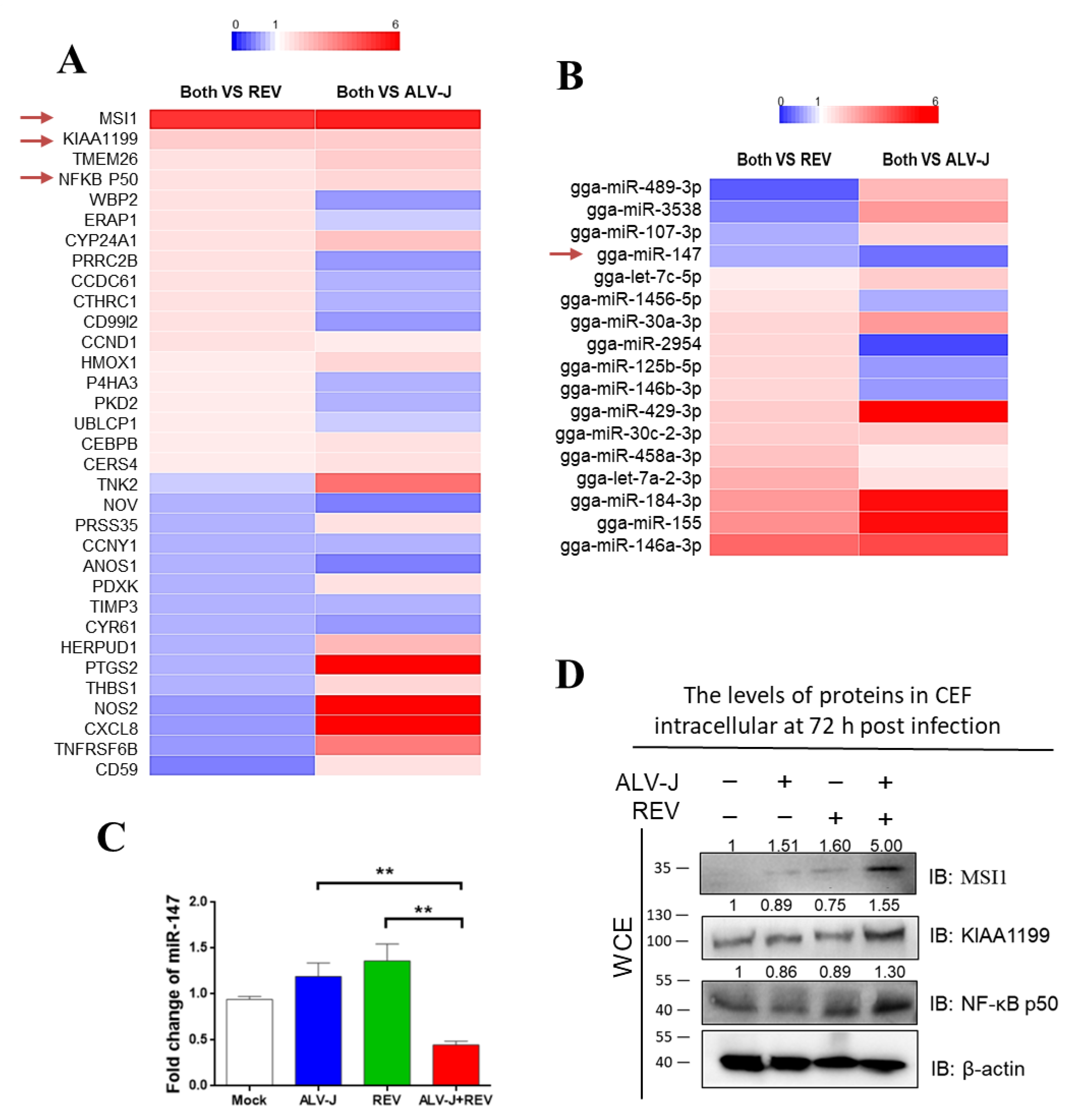
3.2. Identification of Key Host Molecules Responsible for Synergistic Oncogenicity Induced by ALV-J and REV
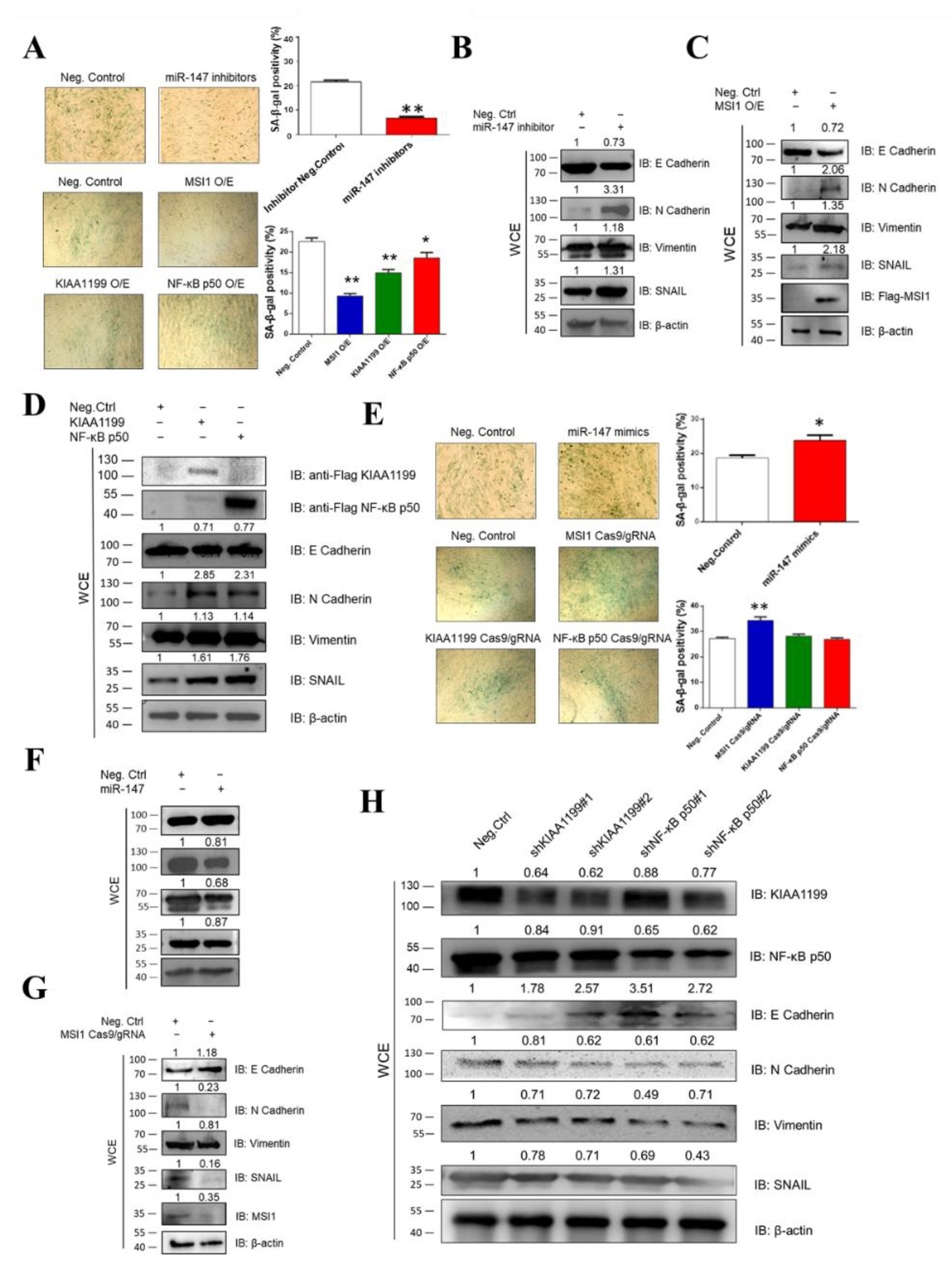
3.3. Ectopic Expression of miR-147, MSI1, KIAA1199, and NF-κB p50 Is Associated with Oncogenicity
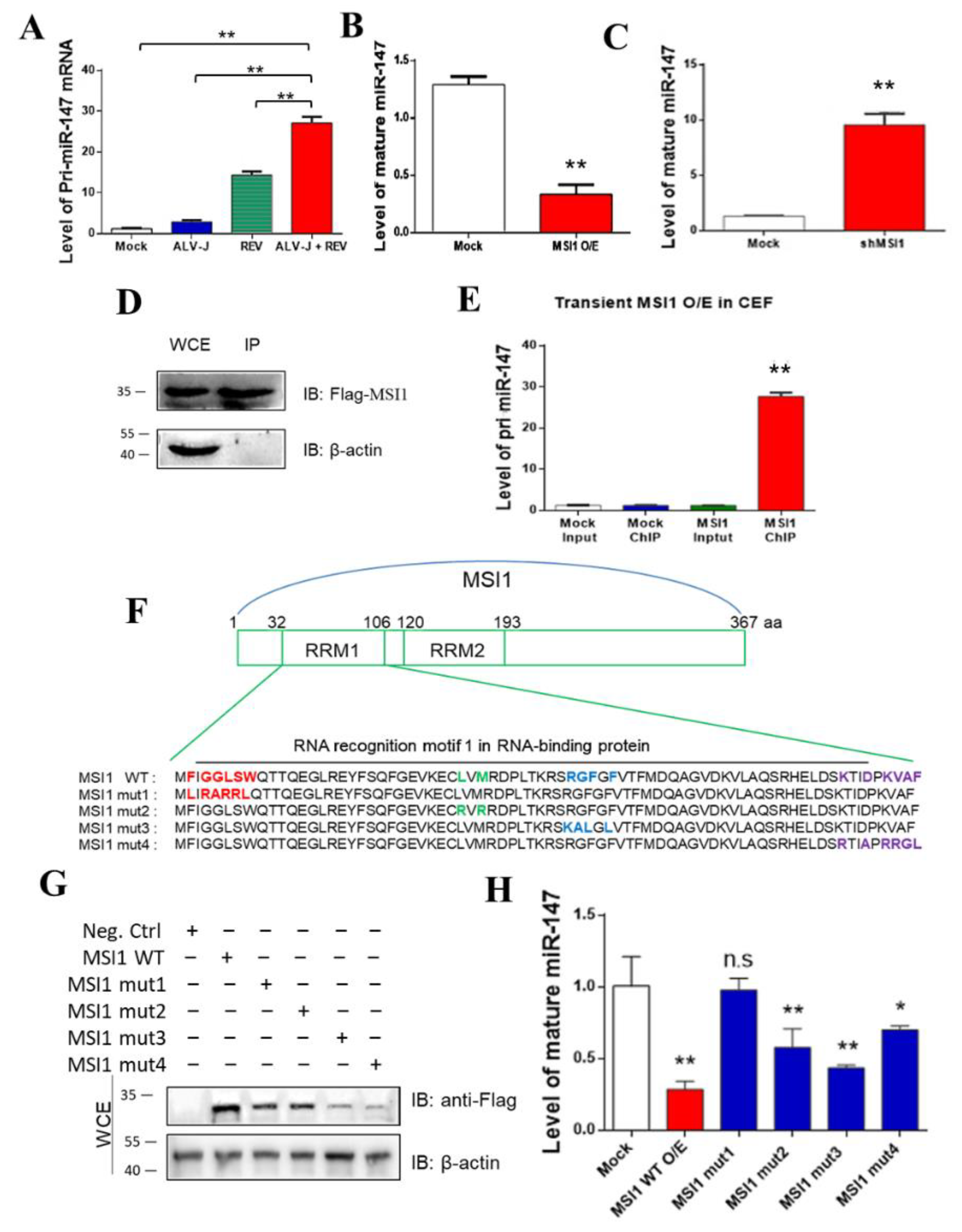
3.4. MSI1 Directly Targeted pri-miR-147 to Inhibit miR-147 Maturation
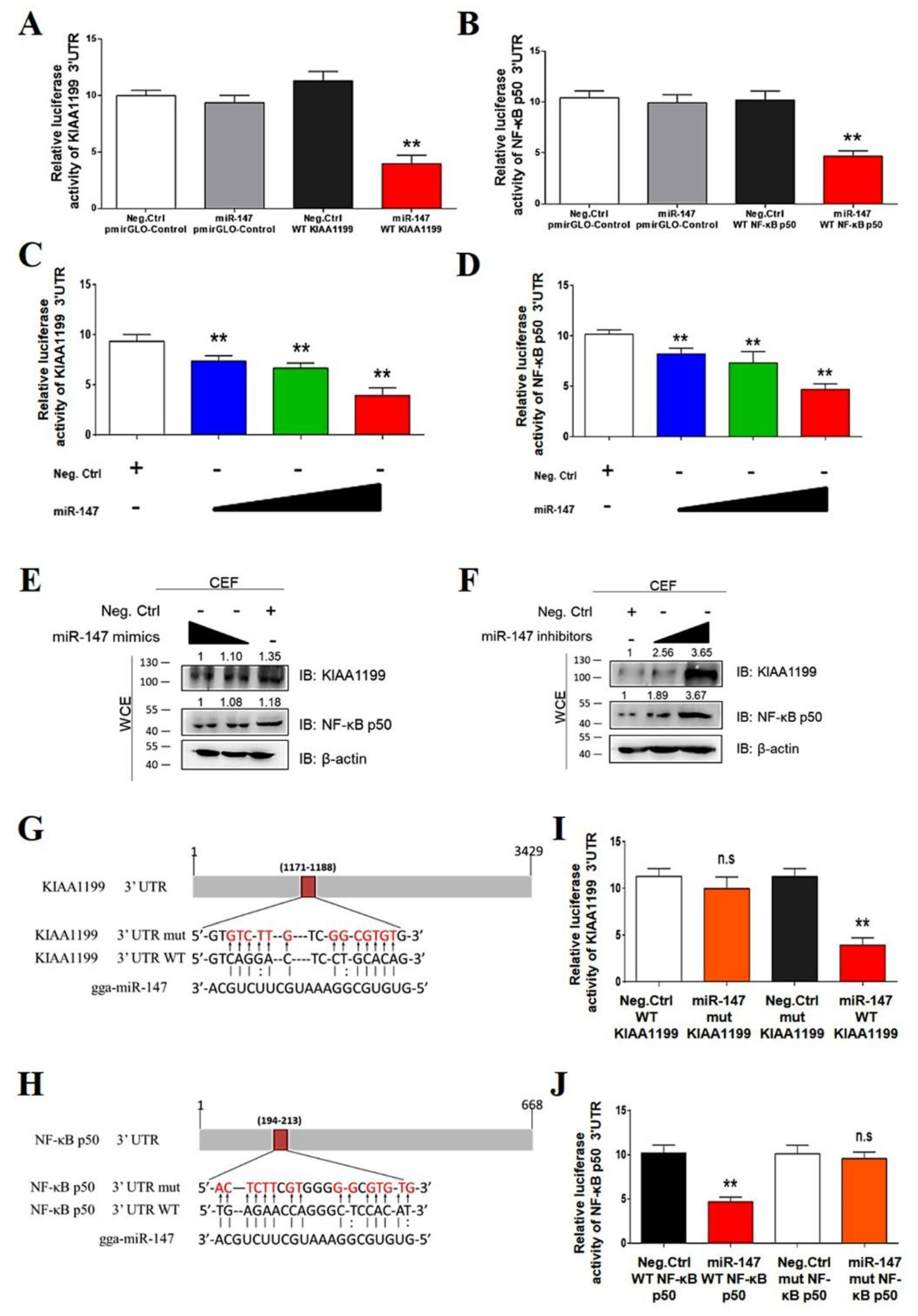
3.5. miR-147 Targets NF-κB p50 and KIAA1199
3.6. ALV-J and REV Synergistically Activated the NF-κB/KIAA1199/EGFR Signaling Pathway
3.7. Structural Proteins, Especially Gags from ALV-J and REV, Synergistically Activate MSI1
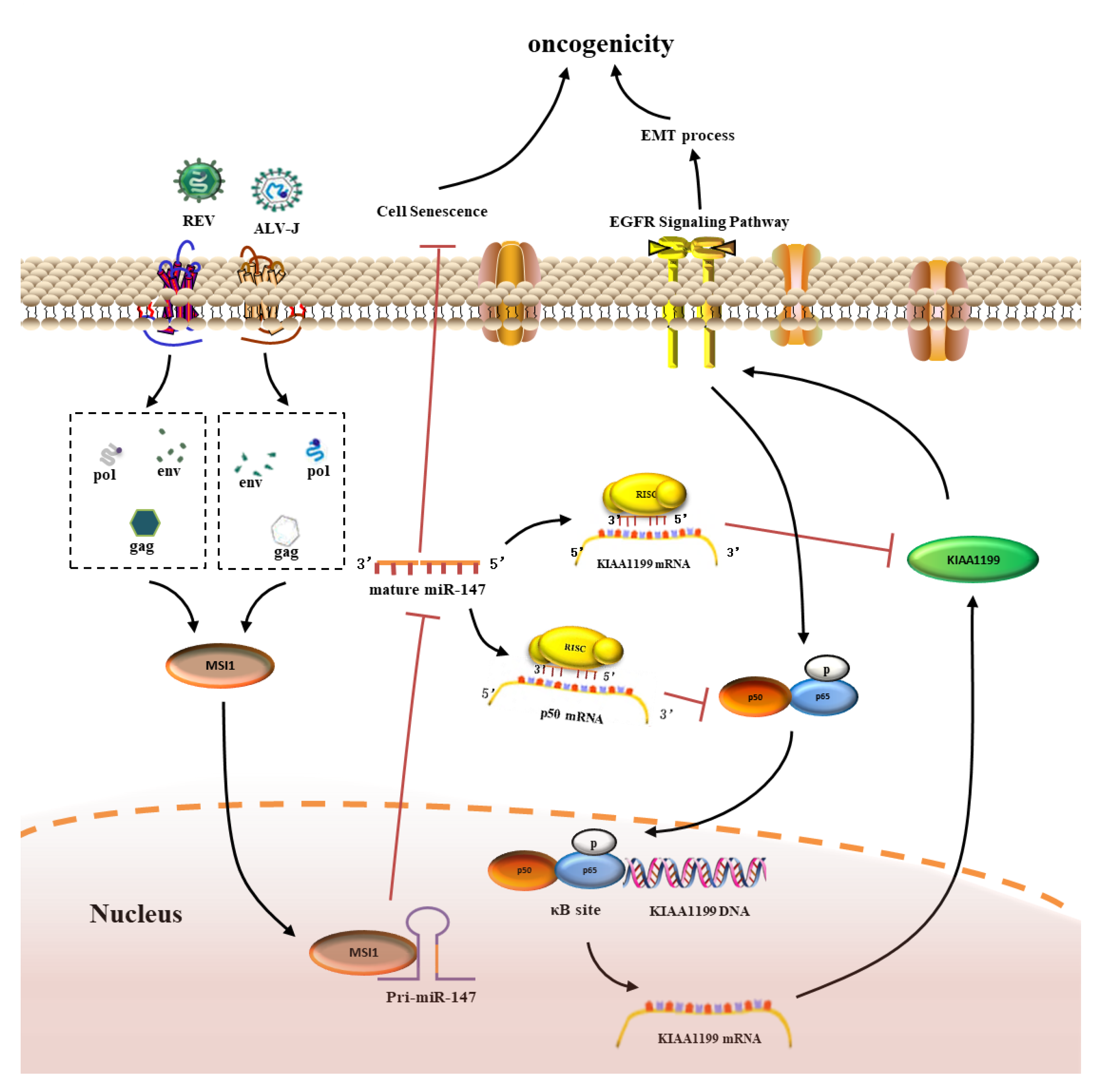
3.8. MSI1-miR-147 Regulated NF-κB/KIAA1199/EGFR Pathways Present in Tumors Induced by ALV-J and REV
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Payne, L.N.; Brown, S.R.; Bumstead, N.; Howes, K.; Frazier, J.A.; Thouless, M.E. A novel subgroup of exogenous avian leukosis virus in chickens. J. Gen. Virol. 1991, 72, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, K. Avian leukosis virus subgroup J: A rapidly evolving group of oncogenic retroviruses. Res. Vet. Sci. 1999, 67, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Bose, H.R., Jr.; Levine, A.S. Replication of the reticuloendotheliosis virus (strain T) in chicken embryo cell culture. J. Virol. 1967, 1, 1117–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niewiadomska, A.M.; Gifford, R.J. The extraordinary evolutionary history of the reticuloendotheliosis viruses. PLoS Biol. 2013, 11, e1001642. [Google Scholar] [CrossRef] [Green Version]
- Fadly, A.M. Avian retroviruses. Vet. Clin. North Am. Food Anim. Pract. 1997, 13, 71–85. [Google Scholar] [CrossRef]
- Payne, L.N. Retrovirus-induced disease in poultry. Poult. Sci. 1998, 77, 1204–1212. [Google Scholar] [CrossRef]
- Reece, R.L. Some observations on naturally occurring neoplasms of domestic fowls in the State of Victoria, Australia (1977-87). Avian Pathol. 1996, 25, 407–447. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Hang, F.; Qian, K.; Shao, H.; Ye, J.; Qin, A. Chicken hepatomegaly and splenomegaly associated with novel subgroup J avian leukosis virus infection. BMC Vet. Res. 2022, 18, 32. [Google Scholar] [CrossRef]
- Cui, N.; Cui, X.; Huang, Q.; Yang, S.; Su, S.; Xu, C.; Li, J.; Li, W.; Li, C. Isolation and Identification of Subgroup J Avian Leukosis Virus Inducing Multiple Systemic Tumors in Parental Meat-Type Chickens. Front. Vet. Sci. 2020, 7, 614854. [Google Scholar] [CrossRef]
- Xu, A.; Huo, C.; Zhong, Q.; Xu, M.; Yang, Y.; Tian, H.; Zhang, G.; Hu, Y. Isolation and pathogenicity testing of avian reticuloendotheliosis virus from layer chickens in China. J. Vet. Diagn. Invest. 2020, 32, 389–393. [Google Scholar] [CrossRef]
- Cooper, G.M.; Temin, H.M. Infectious rous sarcoma virus and reticuloendotheliosis virus DNAs. J. Virol. 1974, 14, 1132–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, I.; Borenstein, R. Multiple infection of chickens and turkeys with avian oncogenic viruses: Prevalence and molecular analysis. Acta Virol. 1999, 43, 136–142. [Google Scholar] [PubMed]
- Cui, Z.; Sun, S.; Zhang, Z.; Meng, S. Simultaneous endemic infections with subgroup J avian leukosis virus and reticuloendotheliosis virus in commercial and local breeds of chickens. Avian Pathol. 2009, 38, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Gopal, S.; Manoharan, P.; Kathaperumal, K.; Chidambaram, B.; Divya, K.C. Differential detection of avian oncogenic viruses in poultry layer farms and Turkeys by use of multiplex PCR. J. Clin. Microbiol. 2012, 50, 2668–2673. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Ma, K.; Liu, M.; Yang, C.; Huang, X.; Zhao, Y.; Qi, K. Histologic findings and viral antigen distribution in natural coinfection of layer hens with subgroup J avian leukosis virus, Marek’s disease virus, and reticuloendotheliosis virus. J. Vet. Diagn. Invest. 2019, 31, 761–765. [Google Scholar] [CrossRef]
- Kannaki, T.R.; Edigi, P.; Yalagandula, N.; Haunshi, S. Simultaneous detection and differentiation of three oncogenic viral diseases of chicken by use of multiplex PCR. Anim. Biotechnol. 2021, 30, 1–6. [Google Scholar] [CrossRef]
- Dong, X.; Ju, S.; Zhao, P.; Li, Y.; Meng, F.; Sun, P.; Cui, Z. Synergetic effects of subgroup J avian leukosis virus and reticuloendotheliosis virus co-infection on growth retardation and immunosuppression in SPF chickens. Vet. Microbiol. 2014, 172, 425–431. [Google Scholar] [CrossRef]
- Dong, X.; Zhao, P.; Chang, S.; Ju, S.; Li, Y.; Meng, F.; Sun, P.; Cui, Z. Synergistic pathogenic effects of co-infection of subgroup J avian leukosis virus and reticuloendotheliosis virus in broiler chickens. Avian Pathol. 2015, 44, 43–49. [Google Scholar] [CrossRef]
- Zhou, D.; Xue, J.; He, S.; Du, X.; Zhou, J.; Li, C.; Huang, L.; Nair, V.; Yao, Y.; Cheng, Z. Reticuloendotheliosis virus and avian leukosis virus subgroup J synergistically increase the accumulation of exosomal miRNAs. Retrovirology 2018, 15, 45. [Google Scholar] [CrossRef] [Green Version]
- Alais, S.; Pasquier, A.; Jegado, B.; Journo, C.; Rua, R.; Gessain, A.; Tobaly-Tapiero, J.; Lacoste, R.; Turpin, J.; Mahieux, R. STLV-1 co-infection is correlated with an increased SFV proviral load in the peripheral blood of SFV/STLV-1 naturally infected non-human primates. PLoS Negl. Trop. Dis. 2018, 12, e0006812. [Google Scholar] [CrossRef]
- Calattini, S.; Betsem, E.; Bassot, S.; Chevalier, S.A.; Tortevoye, P.; Njouom, R.; Mahieux, R.; Froment, A.; Gessain, A. Multiple retroviral infection by HTLV type 1, 2, 3 and simian foamy virus in a family of Pygmies from Cameroon. Virology 2011, 410, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Traina-Dorge, V.L.; Martin, L.N.; Lorino, R.; Winsor, E.L.; Beilke, M.A. Human T cell leukemia virus type 1 up-regulation after simian immunodeficiency virus-1 coinfection in the nonhuman primate. J. Infect Dis. 2007, 195, 562–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leendertz, S.A.J.; Junglen, S.; Hedemann, C.; Goffe, A.; Calvignac, S.; Boesch, C.; Leendertz, F.H. High prevalence, coinfection rate, and genetic diversity of retroviruses in wild red colobus monkeys (Piliocolobus badius badius) in Tai National Park, Cote d’Ivoire. J. Virol. 2010, 84, 7427–7436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, L.N.; Venugopal, K. Neoplastic diseases: Marek’s disease, avian leukosis and reticuloendotheliosis. Rev. Sci. Tech. 2000, 19, 544–564. [Google Scholar] [CrossRef]
- Loo, T.M.; Miyata, K.; Tanaka, Y.; Takahashi, A. Cellular senescence and senescence-associated secretory phenotype via the cGAS-STING signaling pathway in cancer. Cancer Sci. 2020, 111, 304–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; You, Y.; Jiang, H.; Wang, Z.Z. Epithelial-mesenchymal transition (EMT): A biological process in the development, stem cell differentiation, and tumorigenesis. J. Cell Physiol. 2017, 232, 3261–3272. [Google Scholar] [CrossRef]
- Calcinotto, A.; Kohli, J.; Zagato, E.; Pellegrini, L.; Demaria, M.; Alimonti, A. Cellular Senescence: Aging, Cancer, and Injury. Physiol. Rev. 2019, 99, 1047–1078. [Google Scholar] [CrossRef]
- De Lope, C.; Martín-Alonso, S.; Auzmendi-Iriarte, J.; Escudero, C.; Mulet, I.; Larrasa-Alonso, J.; Palmero, I. SIX1 represses senescence and promotes SOX2-mediated cellular plasticity during tumorigenesis. Sci. Rep. 2019, 9, 1412. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.; Yu, T.; Han, Y.; Jiang, H.; Wang, C.; You, T.; Zhao, X.; Shan, H.; Yang, R.; Yang, L.; et al. LncRNA PTAR promotes EMT and invasion-metastasis in serous ovarian cancer by competitively binding miR-101-3p to regulate ZEB1 expression. Mol. Cancer 2018, 17, 119. [Google Scholar] [CrossRef]
- Zhou, J.; Zhou, D.; Du, X.; Xue, J.; Yang, J.; Wang, G.; Cheng, Z. Interaction between Avian Leukosis Virus Subgroup J Surface Protein and Doublecortin-Like Kinase 1 Accelerates Cell Proliferation and Epithelial-Mesenchymal Transition. J. Virol. 2022, 96, e0165721. [Google Scholar] [CrossRef]
- Laher, A.E.; Ebrahim, O. HTLV-1, ATLL, severe hypercalcaemia and HIV-1 co-infection: An overview. Pan. Afr. Med. J. 2018, 30, 61. [Google Scholar] [CrossRef] [PubMed]
- Yasunaga, J.I.; Matsuoka, M. Oncogenic spiral by infectious pathogens: Cooperation of multiple factors in cancer development. Cancer Sci. 2018, 109, 24–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burmeister, T. Oncogenic retroviruses in animals and humans. Rev. Med. Virol. 2001, 11, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Robinson, H.L.; Coffin, J.M.; Tsichlis, P.N.; Shank, P.R.; Schatz, P.; Jensen, L. Cancer induction by insertional mutagenesis: The role of viral genes in avian leukosis virus induced cancers. Prog. Clin. Biol. Res. 1983, 119, 37–42. [Google Scholar]
- Pizer, E.; Humphries, E.H. RAV-1 insertional mutagenesis: Disruption of the c-myb locus and development of avian B-cell lymphomas. J. Virol. 1989, 63, 1630–1640. [Google Scholar] [CrossRef] [Green Version]
- Kung, H.J.; Pelley, R.J.; Shu, H.K.; Maihle, N.; Raines, M.; Carter, T.; Boerkoel, C.; Moscovici, G.; Moscovici, C. Retroviral insertional mutagenesis: The making of a receptor-oncogene. Dev. Biol. Stand. 1990, 72, 139–144. [Google Scholar]
- Noori-Daloii, M.R.; Swift, R.A.; Kung, H.-J.; Crittenden, L.B.; Witter, R. Specific integration of REV proviruses in avian bursal lymphomas. Nature 1981, 294, 574–576. [Google Scholar] [CrossRef]
- Salter, D.W.; Smith, E.J.; Hughes, S.H.; Wright, S.E.; Fadly, A.M.; Witter, R.L.; Crittenden, L.B. Gene insertion into the chicken germ line by retroviruses. Poult. Sci. 1986, 65, 1445–1458. [Google Scholar] [CrossRef]
- Lax, I.; Kris, R.; Sasson, I.; Ullrich, A.; Hayman, M.J.; Beug, H.; Schlessinger, J. Activation of c-erbB in avian leukosis virus-induced erythroblastosis leads to the expression of a truncated EGF receptor kinase. Embo. J. 1985, 4, 3179–3182. [Google Scholar] [CrossRef]
- Yao, Y.; Zhang, Y.; Tang, N.; Pedrera, M.; Shen, Z.; Nair, V. Inhibition of v-rel-Induced Oncogenesis through microRNA Targeting. Viruses 2018, 10, 242. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, H.; Imai, T.; Imataka, H.; Tsujimoto, M.; Matsumoto, K.; Okano, H. Neural RNA-binding protein Musashi1 inhibits translation initiation by competing with eIF4G for PABP. J. Cell Biol. 2008, 181, 639–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohyama, T.; Nagata, T.; Tsuda, K.; Kobayashi, N.; Imai, T.; Okano, H.; Yamazaki, T.; Katahira, M. Structure of Musashi1 in a complex with target RNA: The role of aromatic stacking interactions. Nucleic Acids Res. 2012, 40, 3218–3231. [Google Scholar] [CrossRef] [PubMed]
- Shostak, K.; Chariot, A. EGFR and NF-kappaB: Partners in cancer. Trends Mol. Med. 2015, 21, 385–393. [Google Scholar] [CrossRef]
- Shostak, K.; Zhang, X.; Hubert, P.; Göktuna, S.I.; Jiang, Z.; Klevernic, I.; Chariot, A. NF-κB-induced KIAA1199 promotes survival through EGFR signalling. Nat. Commun. 2014, 5, 5232. [Google Scholar] [CrossRef] [Green Version]
- Chesters, P.M.; Howes, K.; McKay, J.C.; Payne, L.N.; Venugopal, K. Acutely transforming avian leukosis virus subgroup J strain 966: Defective genome encodes a 72-kilodalton Gag-Myc fusion protein. J. Virol. 2001, 75, 4219–4225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Hu, K. MiR-147: Functions and Implications in Inflammation and Diseases. Microrna 2021, 10, 91–96. [Google Scholar] [CrossRef]
- Ning, X.; Wang, C.; Zhang, M.; Wang, K. Ectopic Expression of miR-147 Inhibits Stem Cell Marker and Epithelial-Mesenchymal Transition (EMT)-Related Protein Expression in Colon Cancer Cells. Oncol. Res. 2019, 27, 399–406. [Google Scholar] [CrossRef]
- Li, F.; Wang, X.; Yang, L. MicroRNA-147 targets BDNF to inhibit cell proliferation, migration and invasion in non-small cell lung cancer. Oncol. Lett. 2020, 20, 1931–1937. [Google Scholar] [CrossRef]
- Lee, C.G.; McCarthy, S.; Gruidl, M.; Timme, C.; Yeatman, T.J. MicroRNA-147 induces a mesenchymal-to-epithelial transition (MET) and reverses EGFR inhibitor resistance. PLoS ONE 2014, 9, e84597. [Google Scholar] [CrossRef]
- Kudinov, A.E.; Karanicolas, J.; Golemis, E.A.; Boumber, Y. Musashi RNA-Binding Proteins as Cancer Drivers and Novel Therapeutic Targets. Clin. Cancer Res. 2017, 23, 2143–2153. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Yan, W.; Han, P.; Tian, D. The emerging role of KIAA1199 in cancer development and therapy. Biomed. Pharm. 2021, 138, 111507. [Google Scholar] [CrossRef] [PubMed]
- Tilborghs, S.; Corthouts, J.; Verhoeven, Y.; Arias, D.; Rolfo, C.; Trinh, X.B.; van Dam, P.A. The role of Nuclear Factor-kappa B signaling in human cervical cancer. Crit. Rev. Oncol. Hematol. 2017, 120, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Swanson, I.; Jude, B.A.; Zhang, A.R.; Pucker, A.; Smith, Z.E.; Golovkina, T.V. Sequences within the gag gene of mouse mammary tumor virus needed for mammary gland cell transformation. J. Virol. 2006, 80, 3215–3224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, C.R.; Rosenberg, N. Gag influences transformation by Abelson murine leukemia virus and suppresses nuclear localization of the v-Abl protein. J. Virol. 2007, 81, 9461–9468. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, D.; Ding, L.; Xu, M.; Liu, X.; Xue, J.; Zhang, X.; Du, X.; Zhou, J.; Cui, X.; Cheng, Z. Musashi-1 and miR-147 Precursor Interaction Mediates Synergistic Oncogenicity Induced by Co-Infection of Two Avian Retroviruses. Cells 2022, 11, 3312. https://doi.org/10.3390/cells11203312
Zhou D, Ding L, Xu M, Liu X, Xue J, Zhang X, Du X, Zhou J, Cui X, Cheng Z. Musashi-1 and miR-147 Precursor Interaction Mediates Synergistic Oncogenicity Induced by Co-Infection of Two Avian Retroviruses. Cells. 2022; 11(20):3312. https://doi.org/10.3390/cells11203312
Chicago/Turabian StyleZhou, Defang, Longying Ding, Menglu Xu, Xiaoyao Liu, Jingwen Xue, Xinyue Zhang, Xusheng Du, Jing Zhou, Xiyao Cui, and Ziqiang Cheng. 2022. "Musashi-1 and miR-147 Precursor Interaction Mediates Synergistic Oncogenicity Induced by Co-Infection of Two Avian Retroviruses" Cells 11, no. 20: 3312. https://doi.org/10.3390/cells11203312
APA StyleZhou, D., Ding, L., Xu, M., Liu, X., Xue, J., Zhang, X., Du, X., Zhou, J., Cui, X., & Cheng, Z. (2022). Musashi-1 and miR-147 Precursor Interaction Mediates Synergistic Oncogenicity Induced by Co-Infection of Two Avian Retroviruses. Cells, 11(20), 3312. https://doi.org/10.3390/cells11203312