
mRNA-Based Approaches to Treating Liver Diseases




Abstract
:1. Introduction
2. Therapeutic, mRNA Constructs
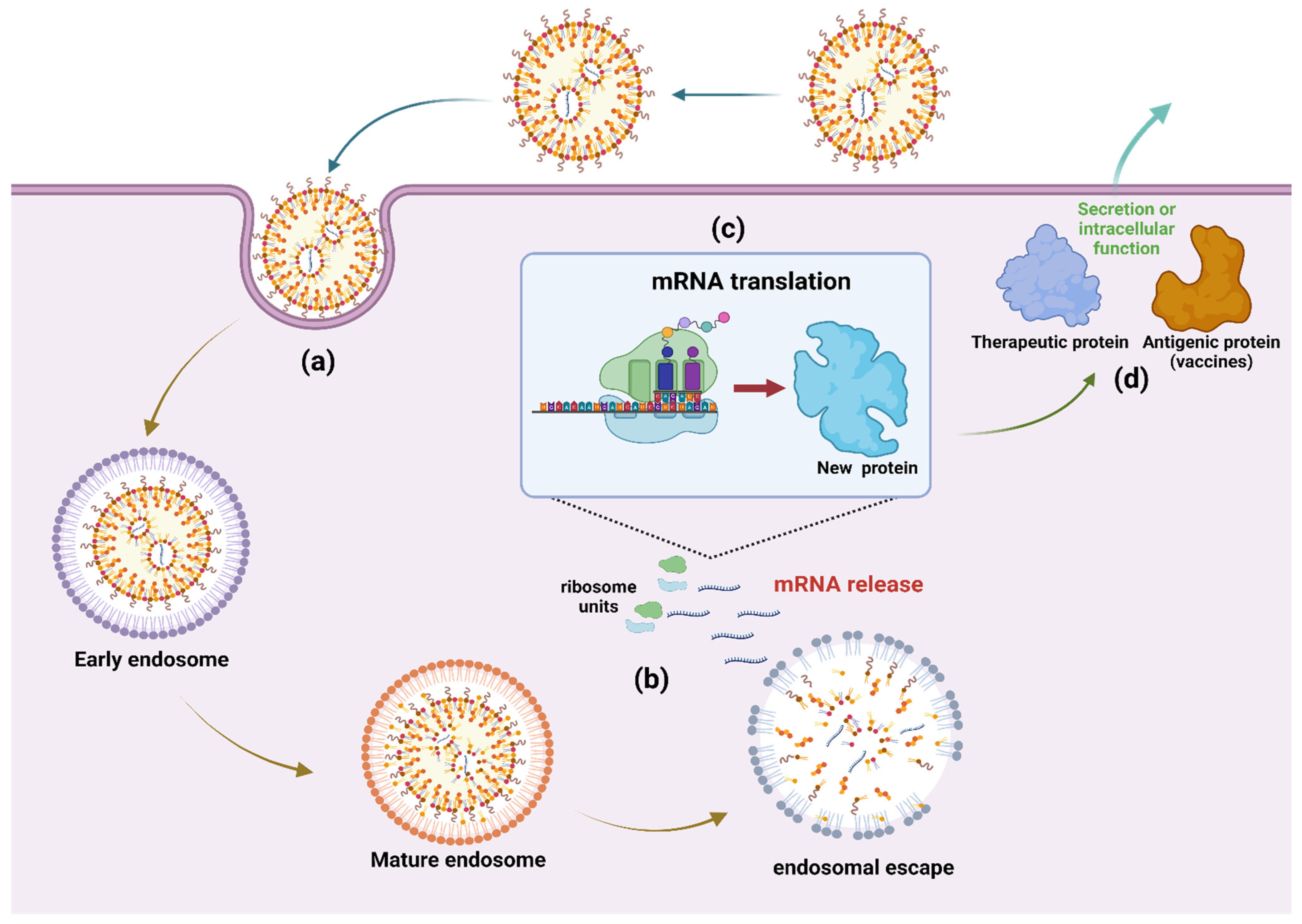
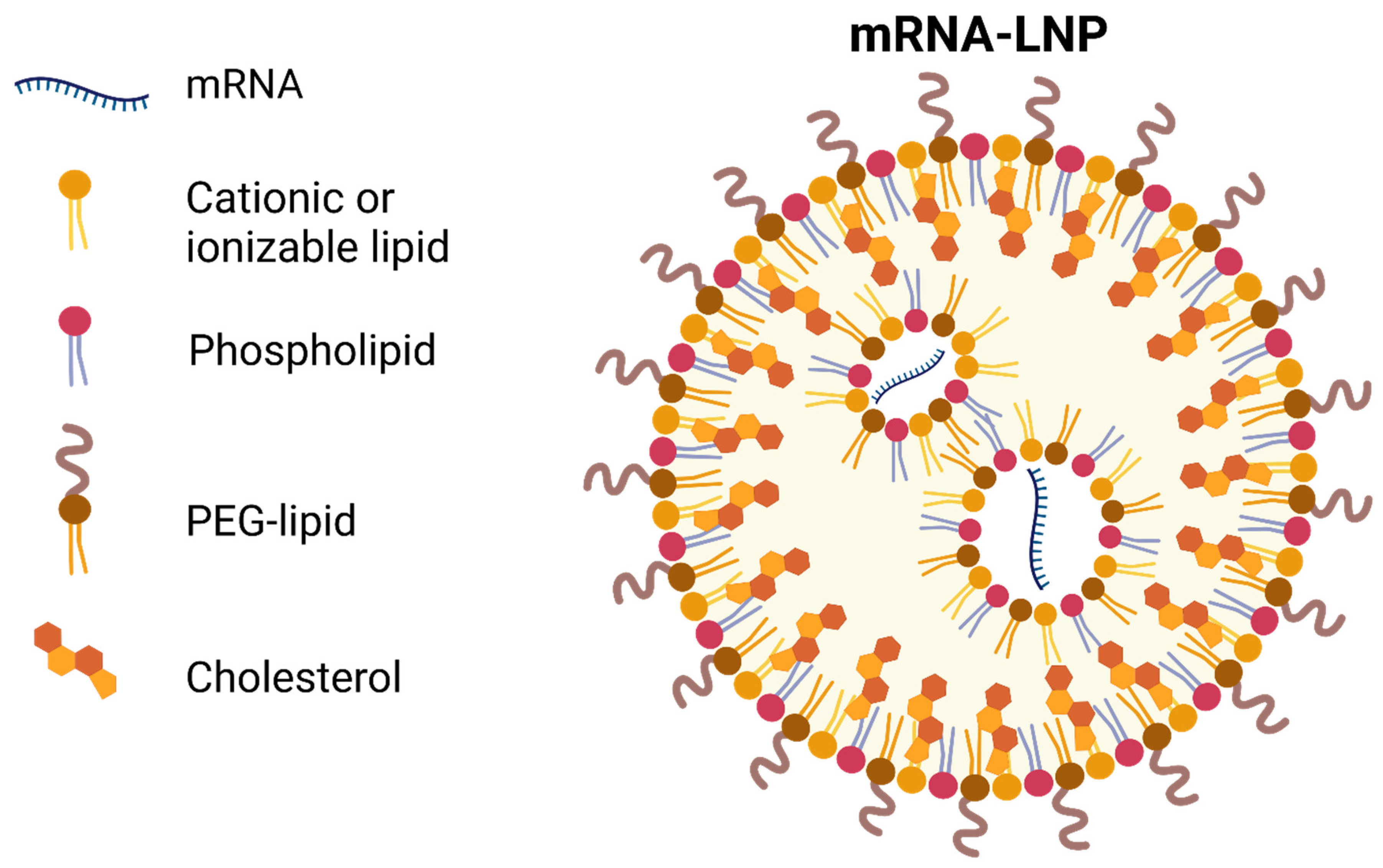
3. Delivery Vehicle
4. Clinical and Preclinical Applications
4.1. Inherited Metabolic Diseases
4.1.1. Hereditary Tyrosinemia Type 1 (HT1)
4.1.2. Phenylketonuria (PKU)
4.1.3. Methylmalonic Acidemia (MMA)
4.1.4. Propionic Acidemia (PA)
4.1.5. Glycogen Storage Disease Type 1a (GSD1a)
4.1.6. Ornithine Transcarbamylase (OTC) Deficiency
4.2. Acquired Liver Injury
4.3. Primary Liver Cancer
4.3.1. Checkpoint Inhibitors
4.3.2. Vaccines
4.3.3. Personalized RNA Mutanome Vaccines
4.3.4. Therapeutic Proteins
4.3.5. Adjuvants
4.4. Infectious Diseases
4.4.1. Hepatitis B Virus (HBV)
4.4.2. Cytomegalovirus (CMV)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.T.; Wang, J.J.; Luu, M.; Noureddin, M.; Kosari, K.; Agopian, V.G.; Rich, N.E.; Lu, S.C.; Tseng, H.R.; Nissen, N.N.; et al. The mortality and overall survival trends of primary liver cancer in the United States. J. Natl. Cancer Inst. 2021, 113, 1531–1541. [Google Scholar] [CrossRef] [PubMed]
- Valery, P.C.; Laversanne, M.; Clark, P.J.; Petrick, J.L.; McGlynn, K.A.; Bray, F. Projections of primary liver cancer to 2030 in 30 countries worldwide. Hepatology 2018, 67, 600–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witzigmann, D.; Kulkarni, J.A.; Leung, J.; Chen, S.; Cullis, P.R.; van der Meel, R. Lipid nanoparticle technology for therapeutic gene regulation in the liver. Adv. Drug Deliv. Rev. 2020, 159, 344–363. [Google Scholar] [CrossRef]
- Menon, J.; Vij, M.; Sachan, D.; Rammohan, A.; Shanmugam, N.; Kaliamoorthy, I.; Rela, M. Pediatric metabolic liver diseases: Evolving role of liver transplantation. World J. Transplant. 2021, 11, 161–179. [Google Scholar] [CrossRef]
- Ferreira, C.R.; Cassiman, D.; Blau, N. Clinical and biochemical footprints of inherited metabolic diseases. II. Metabolic liver diseases. Mol. Genet. Metab. 2019, 127, 117–121. [Google Scholar] [CrossRef]
- Maestro, S.; Weber, N.D.; Zabaleta, N.; Aldabe, R.; Gonzalez-Aseguinolaza, G. Novel vectors and approaches for gene therapy in liver diseases. JHEP Rep. 2021, 3, 100300. [Google Scholar] [CrossRef]
- Baruteau, J.; Waddington, S.N.; Alexander, I.E.; Gissen, P. Gene therapy for monogenic liver diseases: Clinical successes, current challenges and future prospects. J. Inherit. Metab. Dis. 2017, 40, 497–517. [Google Scholar] [CrossRef] [Green Version]
- Mak, K.Y.; Rajapaksha, I.G.; Angus, P.W.; Herath, C.B. The adeno-associated virus—A safe and promising vehicle for liverspecific gene therapy of inherited and non-inherited disorders. Curr. Gene Ther. 2017, 17, 4–16. [Google Scholar] [CrossRef]
- Naso, M.F.; Tomkowicz, B.; Perry, W.L., III; Strohl, W.R. Adeno-associated virus (AAV) as a vector for gene therapy. BioDrugs 2017, 31, 317–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trepotec, Z.; Lichtenegger, E.; Plank, C.; Aneja, M.K.; Rudolph, C. Delivery of mRNA therapeutics for the treatment of hepatic diseases. Mol. Ther. 2019, 27, 794–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jauze, L.; Monteillet, L.; Mithieux, G.; Rajas, F.; Ronzitti, G. Challenges of gene therapy for the treatment of glycogen storage diseases type I and type III. Hum. Gene Ther. 2019, 30, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Kariko, K.; Tureci, O. mRNA-based therapeutics—developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—a new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [Green Version]
- Kowalzik, F.; Schreiner, D.; Jensen, C.; Teschner, D.; Gehring, S.; Zepp, F. mRNA-based vaccines. Vaccines 2021, 9, 390. [Google Scholar] [CrossRef]
- Zabaleta, N.; Torella, L.; Weber, N.D.; Gonzalez-Aseguinolaza, G. mRNA and gene editing: Late breaking therapies in liver diseases. Hepatology 2022, 76, 869–887. [Google Scholar] [CrossRef]
- Wong, C.H. Protein glycosylation: New challenges and opportunities. J. Org. Chem. 2005, 70, 4219–4225. [Google Scholar] [CrossRef]
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef]
- Devarkar, S.C.; Wang, C.; Miller, M.T.; Ramanathan, A.; Jiang, F.; Khan, A.G.; Patel, S.S.; Marcotrigiano, J. Structural basis for m7G recognition and 2′-O-methyl discrimination in capped RNAs by the innate immune receptor RIG-I. Proc. Natl. Acad. Sci. USA 2016, 113, 596–601. [Google Scholar] [CrossRef]
- Leppek, K.; Das, R.; Barna, M. Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol. 2018, 19, 158–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanguay, R.L.; Gallie, D.R. Translational efficiency is regulated by the length of the 3′ untranslated region. Mol. Cell Biol. 1996, 16, 146–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstrohm, A.C.; Wickens, M. Multifunctional deadenylase complexes diversify mRNA control. Nat. Rev. Mol. Cell Biol. 2008, 9, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Lima, S.A.; Chipman, B.L.; Nicholson, L.A.; Chen, Y.H.; Yee, B.A.; Yeo, G.W.; Coller, J.; Pasquinelli, A.E. Short poly(A) tails are a conserved feature of highly expressed genes. Nat. Struct. Mol. Biol. 2017, 24, 1057–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kariko, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, C.; Govindarajan, S.; Minshull, J. Codon bias and heterologous protein expression. Trends Biotechnol. 2004, 22, 346–353. [Google Scholar] [CrossRef]
- Milligan, J.F.; Groebe, D.R.; Witherell, G.W.; Uhlenbeck, O.C. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 1987, 15, 8783–8798. [Google Scholar] [CrossRef] [Green Version]
- Triana-Alonso, F.J.; Dabrowski, M.; Wadzack, J.; Nierhaus, K.H. Self-coded 3′-extension of run-off transcripts produces aberrant products during in vitro transcription with T7 RNA polymerase. J. Biol. Chem. 1995, 270, 6298–6307. [Google Scholar] [CrossRef] [Green Version]
- Kariko, K.; Muramatsu, H.; Ludwig, J.; Weissman, D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011, 39, e142. [Google Scholar] [CrossRef] [Green Version]
- Houseley, J.; Tollervey, D. The many pathways of RNA degradation. Cell 2009, 136, 763–776. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Weissman, D. Recent advances in mRNA vaccine technology. Curr. Opin. Immunol. 2020, 65, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Stanton, M.G. Current status of messenger RNA delivery systems. Nucleic Acid Ther. 2018, 28, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Alfagih, I.M.; Aldosari, B.; AlQuadeib, B.; Almurshedi, A.; Alfagih, M.M. Nanoparticles as adjuvants and nanodelivery systems for mRNA-based vaccines. Pharmaceutics 2020, 13, 45. [Google Scholar] [CrossRef] [PubMed]
- Durymanov, M.; Reineke, J. Non-viral delivery of nucleic acids: Insight Into mechanisms of overcoming intracellular barriers. Front. Pharmacol. 2018, 9, 971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalski, P.S.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the messenger: Advances in technologies for therapeutic mRNA delivery. Mol. Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef] [Green Version]
- Harayama, T.; Riezman, H. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. [Google Scholar] [CrossRef]
- Jackson, N.A.C.; Kester, K.E.; Casimiro, D.; Gurunathan, S.; DeRosa, F. The promise of mRNA vaccines: A biotech and industrial perspective. NPJ Vaccines 2020, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Semple, S.C.; Akinc, A.; Chen, J.; Sandhu, A.P.; Mui, B.L.; Cho, C.K.; Sah, D.W.; Stebbing, D.; Crosley, E.J.; Yaworski, E.; et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010, 28, 172–176. [Google Scholar] [CrossRef]
- Buschmann, M.D.; Carrasco, M.J.; Alishetty, S.; Paige, M.; Alameh, M.G.; Weissman, D. Nanomaterial delivery systems for mRNA vaccines. Vaccines 2021, 9, 65. [Google Scholar] [CrossRef]
- Thess, A.; Grund, S.; Mui, B.L.; Hope, M.J.; Baumhof, P.; Fotin-Mleczek, M.; Schlake, T. Sequence-engineered mRNA without chemical nucleoside modifications enables an effective protein therapy in large animals. Mol. Ther. 2015, 23, 1456–1464. [Google Scholar] [CrossRef]
- Hassett, K.J.; Benenato, K.E.; Jacquinet, E.; Lee, A.; Woods, A.; Yuzhakov, O.; Himansu, S.; Deterling, J.; Geilich, B.M.; Ketova, T.; et al. Optimization of lipid nanoparticles for intramuscular administration of mRNA vaccines. Mol. Ther. Nucleic Acids 2019, 15, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabhan, J.F.; Wood, K.M.; Rao, V.P.; Morin, J.; Bhamidipaty, S.; LaBranche, T.P.; Gooch, R.L.; Bozal, F.; Bulawa, C.E.; Guild, B.C. Intrathecal delivery of frataxin mRNA encapsulated in lipid nanoparticles to dorsal root ganglia as a potential therapeutic for Friedreich’s ataxia. Sci. Rep. 2016, 6, 20019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabnis, S.; Kumarasinghe, E.S.; Salerno, T.; Mihai, C.; Ketova, T.; Senn, J.J.; Lynn, A.; Bulychev, A.; McFadyen, I.; Chan, J.; et al. A novel amino lipid series for mRNA delivery: Improved endosomal escape and sustained pharmacology and safety in non-human primates. Mol. Ther. 2018, 26, 1509–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacicedo, M.L.; Weinl-Tenbruck, C.; Frank, D.; Wirsching, S.; Straub, B.K.; Hauke, J.; Okun, J.G.; Horscroft, N.; Hennermann, J.B.; Zepp, F.; et al. mRNA-based therapy proves superior to the standard of care for treating hereditary tyrosinemia 1 in a mouse model. Mol. Ther. Methods Clin. Dev. 2022, 26, 294–308. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Maruggi, G.; Shan, H.; Li, J. Advances in mRNA vaccines for infectious diseases. Front. Immunol. 2019, 10, 594. [Google Scholar] [CrossRef] [Green Version]
- Pardi, N.; Tuyishime, S.; Muramatsu, H.; Kariko, K.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; Hope, M.J.; Weissman, D. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Control. Release 2015, 217, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S.; Kim, K.M.; Oh, S.H.; Kim, H.J.; Cho, J.M.; Yoo, H.W.; Namgoong, J.M.; Kim, D.Y.; Kim, K.H.; Hwang, S.; et al. Liver transplantation for metabolic liver disease: Experience at a living donor dominant liver transplantation center. Pediatr. Gastroenterol. Hepatol. Nutr. 2015, 18, 48–54. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Kaczmarek, J.C.; Kowalski, P.S.; Anderson, D.G. Advances in the delivery of RNA therapeutics: From concept to clinical reality. Genome Med. 2017, 9, 60. [Google Scholar] [CrossRef] [Green Version]
- Wetterstrand, K.A. DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP). 2022. Available online: www.genome.gov/sequencingcostsdata (accessed on 19 August 2022).
- Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Cantley, W.L.; Dorkin, J.R.; et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther. 2010, 18, 1357–1364. [Google Scholar] [CrossRef]
- Angileri, F.; Roy, V.; Morrow, G.; Scoazec, J.Y.; Gadot, N.; Orejuela, D.; Tanguay, R.M. Molecular changes associated with chronic liver damage and neoplastic lesions in a murine model of hereditary tyrosinemia type 1. Biochim. Biophys. Acta 2015, 1852, 2603–2617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.M. Clinical utility of nitisinone for the treatment of hereditary tyrosinemia type-1 (HT-1). Appl. Clin. Genet. 2017, 10, 43–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, W.S.; Mondal, G.; Vanlith, C.J.; Kaiser, R.A.; Lillegard, J.B. The future of gene-targeted therapy for hereditary tyrosinemia type 1 as a lead indication among the inborn errors of metabolism. Expert. Opin. Orphan. Drugs 2020, 8, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Macias, I.; Lain, A.; Bernardo-Seisdedos, G.; Gil, D.; Gonzalez, E.; Falcon-Perez, J.M.; Millet, O. Hereditary tyrosinemia type I-associated mutations in fumarylacetoacetate hydrolase reduce the enzyme stability and increase its aggregation rate. J. Biol. Chem. 2019, 294, 13051–13060. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Q.; Wei, T.; Jia, Y.; Farbiak, L.; Zhou, K.; Zhang, S.; Wei, Y.; Zhu, H.; Siegwart, D.J. Dendrimer-based lipid nanoparticles deliver therapeutic FAH mRNA to normalize liver function and extend survival in a mouse model of hepatorenal tyrosinemia type I. Adv. Mater. 2018, 30, e1805308. [Google Scholar] [CrossRef]
- Lichter-Konecki, U.; Vockley, J. Phenylketonuria: Current treatments and future developments. Drugs 2019, 79, 495–500. [Google Scholar] [CrossRef]
- van Spronsen, F.J.; Blau, N.; Harding, C.; Burlina, A.; Longo, N.; Bosch, A.M. Phenylketonuria. Nat. Rev. Dis. Primers 2021, 7, 36. [Google Scholar] [CrossRef]
- Cacicedo, M.L.; Weinl-Tenbruck, C.; Frank, D.; Limeres, M.J.; Wirsching, S.; Hilbert, K.; Famian, M.A.P.; Hennermann, J.B.; Zepp, F.; Chevessier-Tunnesen, F.; et al. Repeated intravenous injections of formulated phenylalanine hydroxylase mRNA rescues the phenylketonuria phenotype in mice. Front. Bioeng. Biotechnol. 2022, 10, 993298. [Google Scholar] [CrossRef]
- Perez-Garcia, C.G.; Diaz-Trelles, R.; Vega, J.B.; Bao, Y.; Sablad, M.; Limphong, P.; Chikamatsu, S.; Yu, H.; Taylor, W.; Karmali, P.P.; et al. Development of an mRNA replacement therapy for phenylketonuria. Mol. Ther. Nucleic Acids 2022, 28, 87–98. [Google Scholar] [CrossRef]
- Almasi, T.; Guey, L.T.; Lukacs, C.; Csetneki, K.; Voko, Z.; Zelei, T. Systematic literature review and meta-analysis on the epidemiology of methylmalonic acidemia (MMA) with a focus on MMA caused by methylmalonyl-CoA mutase (mut) deficiency. Orphanet. J. Rare. Dis. 2019, 14, 84. [Google Scholar] [CrossRef]
- Fraser, J.L.; Venditti, C.P. Methylmalonic and propionic acidemias: Clinical management update. Curr. Opin. Pediatr. 2016, 28, 682–693. [Google Scholar] [CrossRef] [PubMed]
- An, D.; Schneller, J.L.; Frassetto, A.; Liang, S.; Zhu, X.; Park, J.S.; Theisen, M.; Hong, S.J.; Zhou, J.; Rajendran, R.; et al. Systemic messenger RNA therapy as a treatment for methylmalonic acidemia. Cell Rep. 2017, 21, 3548–3558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, D.; Frassetto, A.; Jacquinet, E.; Eybye, M.; Milano, J.; DeAntonis, C.; Nguyen, V.; Laureano, R.; Milton, J.; Sabnis, S.; et al. Long-term efficacy and safety of mRNA therapy in two murine models of methylmalonic acidemia. EBioMedicine 2019, 45, 519–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wongkittichote, P.; Ah, M.N.; Chapman, K.A. Propionyl-CoA carboxylase—A review. Mol. Genet. Metab. 2017, 122, 145–152. [Google Scholar] [CrossRef]
- Jiang, L.; Park, J.S.; Yin, L.; Laureano, R.; Jacquinet, E.; Yang, J.; Liang, S.; Frassetto, A.; Zhuo, J.; Yan, X.; et al. Dual mRNA therapy restores metabolic function in long-term studies in mice with propionic acidemia. Nat. Commun. 2020, 11, 5339. [Google Scholar] [CrossRef] [PubMed]
- Sever, S.; Weinstein, D.A.; Wolfsdorf, J.I.; Gedik, R.; Schaefer, E.J. Glycogen storage disease type Ia: Linkage of glucose, glycogen, lactic acid, triglyceride, and uric acid metabolism. J. Clin. Lipidol. 2012, 6, 596–600. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Choi, M.; Guadagnin, E.; Soty, M.; Silva, M.; Verzieux, V.; Weisser, E.; Markel, A.; Zhuo, J.; Liang, S.; et al. mRNA therapy restores euglycemia and prevents liver tumors in murine model of glycogen storage disease. Nat. Commun. 2021, 12, 3090. [Google Scholar] [CrossRef]
- Lichter-Konecki, U.; Caldovic, L.; Morizono, H.; Simpson, K.; Ah, M.N.; MacLeod, E. Ornithine Transcarbamylase Deficiency. In GeneReviews®; Adams, M., Ardinger, H., Pagon, R., Wallace, S., Bean, L., Stephens, K., Eds.; University of Washington: Seattle, DC, USA, 1993. [Google Scholar]
- Prieve, M.G.; Harvie, P.; Monahan, S.D.; Roy, D.; Li, A.G.; Blevins, T.L.; Paschal, A.E.; Waldheim, M.; Bell, E.C.; Galperin, A.; et al. Targeted mRNA therapy for ornithine transcarbamylase deficiency. Mol. Ther. 2018, 26, 801–813. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Yin, L.; Theisen, M.; Zhuo, J.; Siddiqui, S.; Levy, B.; Presnyak, V.; Frassetto, A.; Milton, J.; Salerno, T.; et al. Systemic mRNA therapy for the treatment of Fabry disease: Preclinical studies in wild-type mice, Fabry mouse model, and wild-type non-human primates. Am. J. Hum. Genet. 2019, 104, 625–637. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Berraondo, P.; Jerico, D.; Guey, L.T.; Sampedro, A.; Frassetto, A.; Benenato, K.E.; Burke, K.; Santamaria, E.; Alegre, M.; et al. Systemic messenger RNA as an etiological treatment for acute intermittent porphyria. Nat. Med. 2018, 24, 1899–1909. [Google Scholar] [CrossRef]
- Cao, J.; An, D.; Galduroz, M.; Zhuo, J.; Liang, S.; Eybye, M.; Frassetto, A.; Kuroda, E.; Funahashi, A.; Santana, J.; et al. mRNA therapy improves metabolic and behavioral abnormalities in a murine model of citrin deficiency. Mol. Ther. 2019, 27, 1242–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balakrishnan, B.; An, D.; Nguyen, V.; DeAntonis, C.; Martini, P.G.V.; Lai, K. Novel mRNA-based therapy reduces toxic galactose metabolites and overcomes galactose sensitivity in a mouse model of classic galactosemia. Mol. Ther. 2020, 28, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Roseman, D.S.; Khan, T.; Rajas, F.; Jun, L.S.; Asrani, K.H.; Isaacs, C.; Farelli, J.D.; Subramanian, R.R. G6PC mRNA therapy positively regulates fasting blood glucose and decreases liver abnormalities in a mouse model of glycogen storage disease 1a. Mol. Ther. 2018, 26, 814–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, G.; Cao, J.; Huang, P.; An, P.; Badlani, D.; Vaid, K.A.; Zhao, S.; Wang, D.Q.; Zhuo, J.; Yin, L.; et al. Synthetic human ABCB4 mRNA therapy rescues severe liver disease phenotype in a BALB/c.Abcb4−/− mouse model of PFIC3. J. Hepatol. 2021, 74, 1416–1428. [Google Scholar] [CrossRef]
- Truong, B.; Allegri, G.; Liu, X.B.; Burke, K.E.; Zhu, X.; Cederbaum, S.D.; Haberle, J.; Martini, P.G.V.; Lipshutz, G.S. Lipid nanoparticle-targeted mRNA therapy as a treatment for the inherited metabolic liver disorder arginase deficiency. Proc. Natl. Acad. Sci. USA 2019, 116, 21150–21159. [Google Scholar] [CrossRef]
- Karadagi, A.; Cavedon, A.G.; Zemack, H.; Nowak, G.; Eybye, M.E.; Zhu, X.; Guadagnin, E.; White, R.A.; Rice, L.M.; Frassetto, A.L.; et al. Systemic modified messenger RNA for replacement therapy in a1-antitrypsin deficiency. Sci. Rep. 2020, 10, 7052. [Google Scholar] [CrossRef]
- Apgar, J.F.; Tang, J.P.; Singh, P.; Balasubramanian, N.; Burke, J.; Hodges, M.R.; Lasaro, M.A.; Lin, L.; Millard, B.L.; Moore, K.; et al. Quantitative systems pharmacology model of hUGT1A1-modRNA encoding for the UGT1A1 enzyme to treat Crigler-Najjar syndrome type 1. CPT Pharmacomet. Syst. Pharmacol. 2018, 7, 404–412. [Google Scholar] [CrossRef] [Green Version]
- Gill, R.Q.; Sterling, R.K. Acute liver failure. J. Clin. Gastroenterol. 2001, 33, 191–198. [Google Scholar] [CrossRef]
- Rangnekar, A.S.; Fontana, R.J. An update on drug induced liver injury. Minerva Gastroenterol. Dietol. 2011, 57, 213–229. [Google Scholar]
- Rizvi, F.; Everton, E.; Smith, A.R.; Liu, H.; Osota, E.; Beattie, M.; Tam, Y.; Pardi, N.; Weissman, D.; Gouon-Evans, V. Murine liver repair via transient activation of regenerative pathways in hepatocytes using lipid nanoparticle-complexed nucleoside-modified mRNA. Nat. Commun. 2021, 12, 613. [Google Scholar] [CrossRef]
- Yang, T.; Poenisch, M.; Khanal, R.; Hu, Q.; Dai, Z.; Li, R.; Song, G.; Yuan, Q.; Yao, Q.; Shen, X.; et al. Therapeutic HNF4A mRNA attenuates liver fibrosis in a preclinical model. J. Hepatol. 2021, 75, 1420–1433. [Google Scholar] [CrossRef] [PubMed]
- Dougan, M.; Dranoff, G.; Dougan, S.K. Cancer immunotherapy: Beyond checkpoint blockade. Annu. Rev. Cancer Biol. 2019, 3, 55–75. [Google Scholar] [CrossRef]
- Yang, J.D.; Luu, M.; Singal, A.G.; Noureddin, M.; Kuo, A.; Ayoub, W.S.; Sundaram, V.; Kotler, H.; Kim, I.K.; Todo, T.; et al. Factors associated with detection and survival of T1 hepatocellular carcinoma in the United States: National cancer database analysis. J. Natl. Compr. Cancer Netw. 2020, 18, 1210–1220. [Google Scholar] [CrossRef]
- Mathias-Machado, C.M.; da Fonseca, L.G. Neoadjuvant and adjuvant systemic treatment for hepatocellular carcinoma. Hepatol. Res. 2021, 7, 67–79. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Guo, X.; He, Y.; Zhou, J.; Lv, Q.; Huang, X.; Li, X. Comparative effectiveness of adjuvant treatment for resected hepatocellular carcinoma: A systematic review and network meta-analysis. Front. Oncol. 2021, 11, 709278. [Google Scholar] [CrossRef]
- Sangro, B.; Sarobe, P.; Hervas-Stubbs, S.; Melero, I. Advances in immunotherapy for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 525–543. [Google Scholar] [CrossRef]
- Dougan, M.; Pietropaolo, M. Time to dissect the autoimmune etiology of cancer antibody immunotherapy. J. Clin. Investig. 2020, 130, 51–61. [Google Scholar] [CrossRef]
- Mellman, I.; Hubbard-Lucey, V.M.; Tontonoz, M.J.; Kalos, M.D.; Chen, D.S.; Allison, J.P.; Drake, C.G.; Levitsky, H.; Lonberg, N.; van der Burg, S.H.; et al. De-risking immunotherapy: Report of a consensus workshop of the Cancer Immunotherapy Consortium of the Cancer Research Institute. Cancer Immunol. Res. 2016, 4, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Conry, R.M.; LoBuglio, A.F.; Wright, M.; Sumerel, L.; Pike, M.J.; Johanning, F.; Benjamin, R.; Lu, D.; Curiel, D.T. Characterization of a messenger RNA polynucleotide vaccine vector. Cancer Res. 1995, 55, 1397–1400. [Google Scholar]
- Melief, C.J.; van, H.T.; Arens, R.; Ossendorp, F.; van der Burg, S.H. Therapeutic cancer vaccines. J. Clin. Investig. 2015, 125, 3401–3412. [Google Scholar] [CrossRef]
- Yang, H.; Song, K.; Xue, T.; Xue, X.P.; Huyan, T.; Wang, W.; Wang, H. The distribution and expression profiles of human Aspartyl/Asparaginyl beta-hydroxylase in tumor cell lines and human tissues. Oncol. Rep. 2010, 24, 1257–1264. [Google Scholar] [PubMed] [Green Version]
- Wirsching, S.; Fichter, M.; Cacicedo, M.L.; Landfester, K.; Gehring, S. Modification of regulatory T cell epitopes promotes effector T cell responses to aspartyl/asparaginyl ß-hydroxylase. Int. J. Mol. Sci. 2022, 23, 12444. [Google Scholar] [CrossRef]
- Sahin, U.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Vormehr, M.; Gold, M.; Maurus, D.; Schwarck-Kokarakis, D.; Kuhn, A.N.; Omokoko, T.; et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Tureci, O. Personalized vaccines for cancer immunotherapy. Science 2018, 359, 1355–1360. [Google Scholar] [CrossRef] [Green Version]
- Fiedler, K.; Lazzaro, S.; Lutz, J.; Rauch, S.; Heidenreich, R. mRNA cancer vaccines. Recent Results Cancer Res. 2016, 209, 61–85. [Google Scholar]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Lower, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrors, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Vormehr, M.; Diken, M.; Boegel, S.; Kreiter, S.; Tureci, O.; Sahin, U. Mutanome directed cancer immunotherapy. Curr. Opin. Immunol. 2016, 39, 14–22. [Google Scholar] [CrossRef]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef] [Green Version]
- Alexander, W. The checkpoint immunotherapy revolution: What started as a trickle has become a flood, despite some daunting adverse effects; new drugs, indications, and combinations continue to emerge. Pharm. Ther. 2016, 41, 185–191. [Google Scholar]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [Green Version]
- Richman, L.P.; Vonderheide, R.H.; Rech, A.J. Neoantigen dissimilarity to the self-proteome predicts immunogenicity and response to immune checkpoint blockade. Cell Syst. 2019, 9, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Friedman, C.F.; Proverbs-Singh, T.A.; Postow, M.A. Treatment of the immune-related adverse effects of immune checkpoint inhibitors: A review. JAMA Oncol. 2016, 2, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.K.; Wang, J.L.; Li, W.X.; Wu, T.Q.; Chen, M.S.; Zhou, Z.G. Anti-programmed cell death protein-1 therapy in intrahepatic cholangiocarcinoma induced type 1 diabetes: A case report and literature review. Front. Public Health 2022, 10, 917679. [Google Scholar] [CrossRef] [PubMed]
- Richard, G.; Princiotta, M.F.; Bridon, D.; Martin, W.D.; Steinberg, G.D.; de Groot, A.S. Neoantigen-based personalized cancer vaccines: The emergence of precision cancer immunotherapy. Expert Rev. Vaccines 2022, 21, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Reparaz, D.; Ruiz, M.; Llopiz, D.; Silva, L.; Vercher, E.; Aparicio, B.; Egea, J.; Tamayo-Uria, I.; Hervas-Stubbs, S.; Garcia-Balduz, J.; et al. Neoantigens as potential vaccines in hepatocellular carcinoma. J. Immunother. Cancer 2022, 10, e003978. [Google Scholar] [CrossRef] [PubMed]
- Van, H.L.; Verbeke, R.; Dewitte, H.; Lentacker, I.; Vermaelen, K.; Breckpot, K.; Van, L.S. mRNA in cancer immunotherapy: Beyond a source of antigen. Mol. Cancer 2021, 20, 48. [Google Scholar]
- Deal, C.E.; Carfi, A.; Plante, O.J. Advancements in mRNA encoded antibodies for passive immunotherapy. Vaccines 2021, 9, 108. [Google Scholar] [CrossRef]
- Milling, L.; Zhang, Y.; Irvine, D.J. Delivering safer immunotherapies for cancer. Adv. Drug Deliv. Rev. 2017, 114, 79–101. [Google Scholar] [CrossRef]
- Zidek, Z.; Anzenbacher, P.; Kmonickova, E. Current status and challenges of cytokine pharmacology. Br. J. Pharmacol. 2009, 157, 342–361. [Google Scholar] [CrossRef] [Green Version]
- Tugues, S.; Burkhard, S.H.; Ohs, I.; Vrohlings, M.; Nussbaum, K.; Vom, B.J.; Kulig, P.; Becher, B. New insights into IL-12-mediated tumor suppression. Cell Death Differ. 2015, 22, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Lai, I.; Swaminathan, S.; Baylot, V.; Mosley, A.; Dhanasekaran, R.; Gabay, M.; Felsher, D.W. Lipid nanoparticles that deliver IL-12 messenger RNA suppress tumorigenesis in MYC oncogene-driven hepatocellular carcinoma. J. Immunother. Cancer 2018, 6, 125. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.A.; Rice, J.; Reesor, E.; Zope, H.; Tao, W.; Lim, M.; Ding, J.; Chen, Y.; Aduluso, D.; Zetter, B.R.; et al. Adjuvant-pulsed mRNA vaccine nanoparticle for immunoprophylactic and therapeutic tumor suppression in mice. Biomaterials 2021, 266, 120431. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Kim, S.Y.; Seo, Y.; Kim, M.H.; Chang, J.; Lee, H. Adjuvant incorporated lipid nanoparticles for enhanced mRNA-mediated cancer immunotherapy. Biomater. Sci. 2020, 8, 1101–1105. [Google Scholar] [CrossRef] [PubMed]
- Tse, S.W.; McKinney, K.; Walker, W.; Nguyen, M.; Iacovelli, J.; Small, C.; Hopson, K.; Zaks, T.; Huang, E. mRNA-encoded, constitutively active STING (V155M) is a potent genetic adjuvant of antigen-specific CD8+ T cell response. Mol. Ther. 2021, 29, 2227–2238. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Pelc, R.S.; Muramatsu, H.; Andersen, H.; DeMaso, C.R.; Dowd, K.A.; Sutherland, L.L.; Scearce, R.M.; Parks, R.; et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 2017, 543, 248–251. [Google Scholar] [CrossRef]
- Richner, J.M.; Himansu, S.; Dowd, K.A.; Butler, S.L.; Salazar, V.; Fox, J.M.; Julander, J.G.; Tang, W.W.; Shresta, S.; Pierson, T.C.; et al. Modified mRNA vaccines protect against Zika virus infection. Cell 2017, 168, 1114–1125. [Google Scholar] [CrossRef] [Green Version]
- Lutz, J.; Lazzaro, S.; Habbeddine, M.; Schmidt, K.E.; Baumhof, P.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; Hope, M.J.; Heidenreich, R.; et al. Unmodified mRNA in LNPs constitutes a competitive technology for prophylactic vaccines. NPJ Vaccines 2017, 2, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanning, G.C.; Zoulim, F.; Hou, J.; Bertoletti, A. Therapeutic strategies for hepatitis B virus infection: Towards a cure. Nat. Rev. Drug Discov. 2019, 18, 827–844. [Google Scholar] [CrossRef] [PubMed]
- Bertoletti, A.; Ferrari, C. Innate and adaptive immune responses in chronic hepatitis B virus infections: Towards restoration of immune control of viral infection. Gut 2012, 61, 1754–1764. [Google Scholar] [CrossRef]
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015, 64, 1972–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ely, A.; Singh, P.; Smith, T.S.; Arbuthnot, P. In vitro transcribed mRNA for expression of designer nucleases: Advantages as a novel therapeutic for the management of chronic HBV infection. Adv. Drug Deliv. Rev. 2021, 168, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Gorsuch, C.L.; Nemec, P.; Yu, M.; Xu, S.; Han, D.; Smith, J.; Lape, J.; van, B.N.; Ramirez, R.; Muench, R.C.; et al. Targeting the hepatitis B cccDNA with a sequence-specific ARCUS nuclease to eliminate hepatitis B virus in vivo. Mol. Ther. 2022, 30, 2909–2922. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Chen, Y.; Li, J.; Wang, C.; Song, W.; Wen, Y.; Lin, J.; Wu, Y.; Ying, T. A single dose of anti-HBsAg antibody-encoding mRNA-LNPs suppressed HBsAg expression: A potential cure of chronic hepatitis B virus infection. MBio 2022, 13, e0161222. [Google Scholar] [CrossRef] [PubMed]
- Lugade, A.A.; Kalathil, S.; Miller, A.; Iyer, R.; Thanavala, Y. High immunosuppressive burden in advanced hepatocellular carcinoma patients: Can effector functions be restored? Oncoimmunology 2013, 2, e24679. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disorder | Protein Affected | Manifestation | mRNA Construct 1 | Animal Model | Reference |
|---|---|---|---|---|---|
| Fabry disease | Alpha-galactosidase A | Accumulation glyco- sphingolipids | h-a-Gal A | Gla-deficient, B6;129-Glatm1Kul/J mouse; wild-type NHP | [71] |
| Type II citrullinemia | Liver-specific mitochondrial aspartate/glutamate transporter (citrin) | Elevated: hepatic citrulline, blood ammonia | hCitrin | Ctrn/mGPD-double KO mouse | [73] |
| Classic galactosemia (CG) | Galactose-1 phosphate uridylyltransferase | Elevated: galactose-1 phosphate and plasma galactose | hGALT or mGalT | GalT−/− mouse | [74] |
| Glycogen storage disease type 1a (GSD1a) | Glucose-6- phosphatase | Hypoglycemia | hG6PC | L-G6PC−/− mouse | [75] |
| Acute intermittent porphyria (AIP) | Porphobilinogen deaminase | Accumulation porphyrin precursors | hPBGD | (Pbgdtm1(neo)UAM) X (Pbgdtm2(neo)UAM) mouse; porphyric rabbit; wild-type NHP | [72] |
| Progressive familial intra-hepatic cholestasis type 3 (PFIC3) | Liver-specific phosphatidylcholine transporter (ABCB4/MDR3) | Cholestasis; progressive biliary fibrosis | hABCB4 | BALB/c Abcb4−/− mouse | [76] |
| Arginase deficiency | Arginase 1 | Hyperargininemia; guanidino compounds | hARG1 | Conditional arginase deficient Arg1flox/flox mouse | [77] |
| Alpha-1 antitrypsin (AAT) deficiency | SERPINA1 2 | Uncontrolled elastolytic activity | hAAT | NSG-PiZ mouse 3 | [78] |
| Crigler–Najjar syndrome type 1 (CN1) | Uridine-diphosphate- glucuronosyltransferase (UGT1A1) | Unconjugated hyperbilirubinemia | hUGT1A1 | Gunn-UGT1a1j/BluHsdRrrc rat | [79] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cacicedo, M.L.; Limeres, M.J.; Gehring, S. mRNA-Based Approaches to Treating Liver Diseases. Cells 2022, 11, 3328. https://doi.org/10.3390/cells11203328
Cacicedo ML, Limeres MJ, Gehring S. mRNA-Based Approaches to Treating Liver Diseases. Cells. 2022; 11(20):3328. https://doi.org/10.3390/cells11203328
Chicago/Turabian StyleCacicedo, Maximiliano L., María José Limeres, and Stephan Gehring. 2022. "mRNA-Based Approaches to Treating Liver Diseases" Cells 11, no. 20: 3328. https://doi.org/10.3390/cells11203328
APA StyleCacicedo, M. L., Limeres, M. J., & Gehring, S. (2022). mRNA-Based Approaches to Treating Liver Diseases. Cells, 11(20), 3328. https://doi.org/10.3390/cells11203328