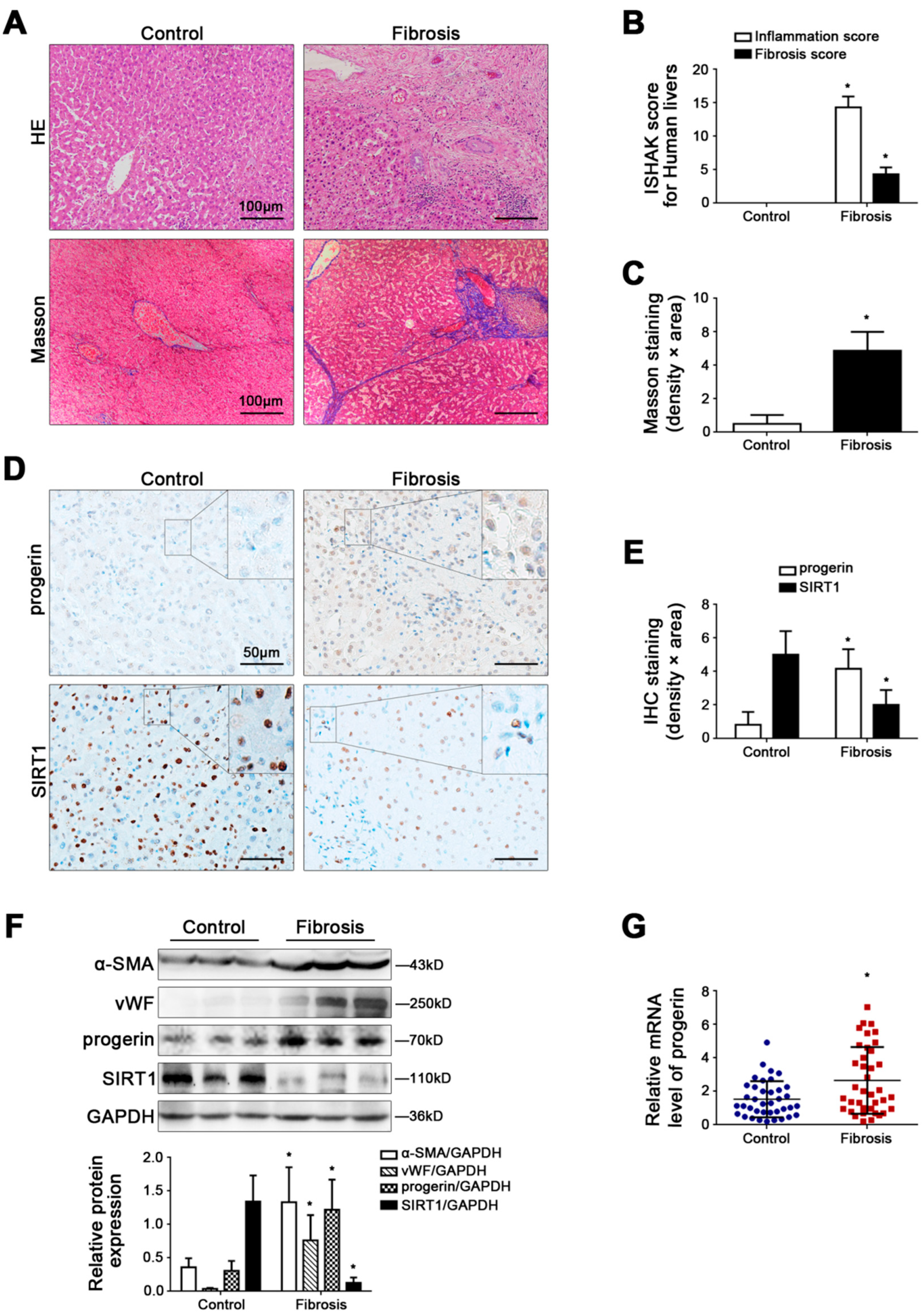
Figure 1.
Progerin is elevated in human liver fibrosis, along with depletion of SIRT1. (A) HE and Masson staining in human liver specimens (Scale bar: 100 μm). (B) The quantified analysis of liver inflammation and fibrosis with ISHAK score in the graph. * p < 0.05 versus the control group. (C) The semi-quantified analysis for the area density of Masson staining in the graph. * p < 0.05 versus the control group. (D) IHC staining for progerin and SIRT1 in human liver specimens (scale bar: 50 μm). (E) The semi-quantified analysis for the area density of IHC staining of progerin and SIRT1 in the graph. * p < 0.05 versus the control group. (F) Representative immunoblots of α-SMA, vWF, progerin, and SIRT1 in human liver tissue. The relative protein levels were quantified in the graph (below). * p < 0.05 versus the control group. (G) RT-qPCR analysis for progerin mRNA level in human peripheral blood granulocytes. * p < 0.05 versus the control group.
Figure 1.
Progerin is elevated in human liver fibrosis, along with depletion of SIRT1. (A) HE and Masson staining in human liver specimens (Scale bar: 100 μm). (B) The quantified analysis of liver inflammation and fibrosis with ISHAK score in the graph. * p < 0.05 versus the control group. (C) The semi-quantified analysis for the area density of Masson staining in the graph. * p < 0.05 versus the control group. (D) IHC staining for progerin and SIRT1 in human liver specimens (scale bar: 50 μm). (E) The semi-quantified analysis for the area density of IHC staining of progerin and SIRT1 in the graph. * p < 0.05 versus the control group. (F) Representative immunoblots of α-SMA, vWF, progerin, and SIRT1 in human liver tissue. The relative protein levels were quantified in the graph (below). * p < 0.05 versus the control group. (G) RT-qPCR analysis for progerin mRNA level in human peripheral blood granulocytes. * p < 0.05 versus the control group.
Figure 2.
Abnormal accumulation of progerin and Cav-1-related autophagy emerge in defenestrated and capillarized hepatic sinusoidal endothelium, along with loss of SIRT1. (A) IHC staining of progerin and SIRT1 in liver tissue of CCl4-induced rat models at different time points (day 0, day 3, day 6, and day 28) (scale bar: 50 μm). The black triangles indicate progerin- and SIRT1-positive LSECs. (B) The semi-quantified analysis for the area density of IHC staining of progerin and SIRT1 in the graph. * p < 0.05 versus the day 0 group. (C) Representative immunoblots of progerin, Lamin B1, SIRT1, Cav-1, and LC3 II/I in primary LSECs, which were isolated from CCl4-induced rat models at different time points (day 0, day 3, day 6, day 14, and day 28). The relative protein expression was quantified in the graph (right). * p < 0.05 versus the day 0 group. (D) The co-localization of LC3B (green) with vWF (red) in liver tissue of CCl4-induced rat models at different time points (day 0, day 3, day 6, and day 28) (scale bar: 10 μm). Nuclei are shown by DAPI (blue). N = 6 per group.
Figure 2.
Abnormal accumulation of progerin and Cav-1-related autophagy emerge in defenestrated and capillarized hepatic sinusoidal endothelium, along with loss of SIRT1. (A) IHC staining of progerin and SIRT1 in liver tissue of CCl4-induced rat models at different time points (day 0, day 3, day 6, and day 28) (scale bar: 50 μm). The black triangles indicate progerin- and SIRT1-positive LSECs. (B) The semi-quantified analysis for the area density of IHC staining of progerin and SIRT1 in the graph. * p < 0.05 versus the day 0 group. (C) Representative immunoblots of progerin, Lamin B1, SIRT1, Cav-1, and LC3 II/I in primary LSECs, which were isolated from CCl4-induced rat models at different time points (day 0, day 3, day 6, day 14, and day 28). The relative protein expression was quantified in the graph (right). * p < 0.05 versus the day 0 group. (D) The co-localization of LC3B (green) with vWF (red) in liver tissue of CCl4-induced rat models at different time points (day 0, day 3, day 6, and day 28) (scale bar: 10 μm). Nuclei are shown by DAPI (blue). N = 6 per group.
![Cells 11 03918 g002]()
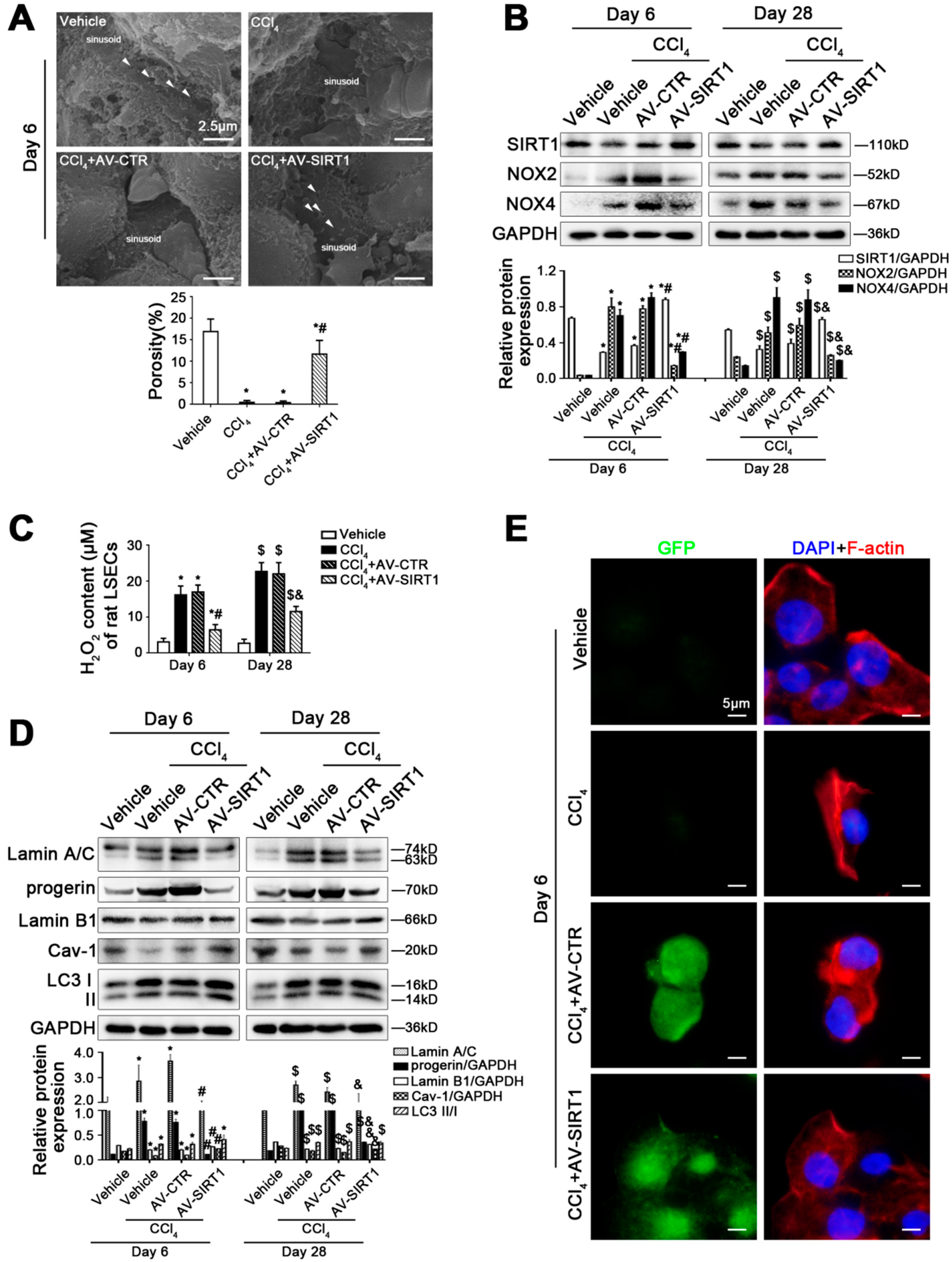
Figure 3.
SIRT1 gene transfer to CCl4-induced rat models alleviates LSEC defenestration and liver fibrosis through reducing progerin and reversing Cav-1 degradation. (A) The magnification of SEM for fenestrae in the liver sinusoidal endothelium of CCl4-induced rat models in the four groups (vehicle, CCl4, CCl4+AV-CTR, CCl4+AV-SIRT1) on day 6 (Scale bar: 2.5 μm). The white triangles indicate fenestrae in liver sinusoidal endothelium. The porosity is quantified in the graph (below). * p < 0.05 versus the vehicle group; # p < 0.05 versus the CCl4+AV-CTR group. (B) Representative immunoblots of SIRT1, NOX2, and NOX4 in primary LSECs, isolated from CCl4-induced rat models on day 6 and day 28. The relative protein expression was quantified in the graph (below). * p < 0.05 versus the vehicle group on day 6; # p < 0.05 versus the CCl4+AV-CTR group on day 6; $ p < 0.05 versus the vehicle group on day 28; & p < 0.05 versus the CCl4+AV-CTR group on day 28. (C) The H2O2 content of primary LSECs, isolated from CCl4-induced rat models on day 6 and day 28. * p < 0.05 versus the vehicle group on day 6; # p < 0.05 versus the CCl4+AV-CTR group on day 6; $ p < 0.05 versus the vehicle group on day 28; & p < 0.05 versus the CCl4+AV-CTR group on day 28. (D) Representative immunoblots of Lamin A/C, progerin, Lamin B1, Cav-1, and LC3 II/I in primary LSECs, isolated from CCl4-induced rat models on day 6 and day 28. The relative protein expression is quantified in the graph (below). * p < 0.05 versus the vehicle group on day 6; # p < 0.05 versus the CCl4+AV-CTR group on day 6; $ p < 0.05 versus the vehicle group on day 28; & p < 0.05 versus the CCl4+AV-CTR group on day 28. (E) ICC staining of F-actin (red) in primary LSECs, isolated from CCl4-induced rat models on day 6 (Scale bar: 5 μm). Adenovirus vectors are shown by GFP (green). Nuclei are shown by DAPI (blue). N = 12 per group.
Figure 3.
SIRT1 gene transfer to CCl4-induced rat models alleviates LSEC defenestration and liver fibrosis through reducing progerin and reversing Cav-1 degradation. (A) The magnification of SEM for fenestrae in the liver sinusoidal endothelium of CCl4-induced rat models in the four groups (vehicle, CCl4, CCl4+AV-CTR, CCl4+AV-SIRT1) on day 6 (Scale bar: 2.5 μm). The white triangles indicate fenestrae in liver sinusoidal endothelium. The porosity is quantified in the graph (below). * p < 0.05 versus the vehicle group; # p < 0.05 versus the CCl4+AV-CTR group. (B) Representative immunoblots of SIRT1, NOX2, and NOX4 in primary LSECs, isolated from CCl4-induced rat models on day 6 and day 28. The relative protein expression was quantified in the graph (below). * p < 0.05 versus the vehicle group on day 6; # p < 0.05 versus the CCl4+AV-CTR group on day 6; $ p < 0.05 versus the vehicle group on day 28; & p < 0.05 versus the CCl4+AV-CTR group on day 28. (C) The H2O2 content of primary LSECs, isolated from CCl4-induced rat models on day 6 and day 28. * p < 0.05 versus the vehicle group on day 6; # p < 0.05 versus the CCl4+AV-CTR group on day 6; $ p < 0.05 versus the vehicle group on day 28; & p < 0.05 versus the CCl4+AV-CTR group on day 28. (D) Representative immunoblots of Lamin A/C, progerin, Lamin B1, Cav-1, and LC3 II/I in primary LSECs, isolated from CCl4-induced rat models on day 6 and day 28. The relative protein expression is quantified in the graph (below). * p < 0.05 versus the vehicle group on day 6; # p < 0.05 versus the CCl4+AV-CTR group on day 6; $ p < 0.05 versus the vehicle group on day 28; & p < 0.05 versus the CCl4+AV-CTR group on day 28. (E) ICC staining of F-actin (red) in primary LSECs, isolated from CCl4-induced rat models on day 6 (Scale bar: 5 μm). Adenovirus vectors are shown by GFP (green). Nuclei are shown by DAPI (blue). N = 12 per group.
![Cells 11 03918 g003]()
Figure 4.
H2O2 induces excessive accumulation of progeirn, with depletion of Lamin B1 and Cav-1 to aggravate LSEC defenestration. Fresh rat primary LSECs, isolated from normal male SD rats, were treated with H2O2 (10 μM) in vitro for 48 h. (A) The magnification of SEM for fenestrae in primary LSECs in 48 h (scale bar: 2 μm). The black triangles indicate fenestrae in LSECs. The total fenestral diameter is quantified in the graph (right). * p < 0.05 versus the control group in 48 h. (B) RT-qPCR analysis for mRNA levels of LMNA, Lamin B1, and Cav-1 of primary LSECs in 24 h and 48 h. * p < 0.05 versus the control group in 24 h; # p < 0.05 versus the control group in 48 h. (C) Representative immunoblots of Lamin A/C, progerin, Lamin B1, Cav-1, and LC3 II/I in nucleus and cytoplasm of primary LSECs in 24 h and 48 h. The relative protein expression and LC3 II/I expression in nucleus and cytoplasm are quantified in the two graphs (right). * p < 0.05 versus the control group in 24 h; # p < 0.05 versus the control group in 48 h. (D) Renilla Luciferase Reporter Gene Assay for LMNA promoter activity of primary LSECs in 24 h and 48 h. * p < 0.05 versus the control group in 24 h. (E) The interaction of nuclear LC3B with acetyl Lysine, progerin, Lamin B1, and Cav-1 of primary LSECs in 24 h and 48 h was detected by the co-IP assay. Nuclear LC3B in primary LSECs was individually immunoprecipitated, and subsequently, acetyl Lysine, progerin, Lamin B1, Cav-1, and LC3 II/I in nucleus of primary LSECs were subjected to immunoblotting analysis. (F) The interaction of cytoplasmic LC3B with Cav-1 of primary LSECs in 24 h and 48 h was detected by the co-IP assay. Cytoplasmic LC3B in primary LSECs was individually immunoprecipitated, and subsequently, Cav-1 and LC3 II/I in cytoplasm of primary LSECs were subjected to immunoblotting analysis. (G) The immunocytochemical co-localization of progerin (red) with F-actin (purple) of primary LSECs in 48 h (Scale bar: 5 μm). Nuclei are shown by DAPI (blue).
Figure 4.
H2O2 induces excessive accumulation of progeirn, with depletion of Lamin B1 and Cav-1 to aggravate LSEC defenestration. Fresh rat primary LSECs, isolated from normal male SD rats, were treated with H2O2 (10 μM) in vitro for 48 h. (A) The magnification of SEM for fenestrae in primary LSECs in 48 h (scale bar: 2 μm). The black triangles indicate fenestrae in LSECs. The total fenestral diameter is quantified in the graph (right). * p < 0.05 versus the control group in 48 h. (B) RT-qPCR analysis for mRNA levels of LMNA, Lamin B1, and Cav-1 of primary LSECs in 24 h and 48 h. * p < 0.05 versus the control group in 24 h; # p < 0.05 versus the control group in 48 h. (C) Representative immunoblots of Lamin A/C, progerin, Lamin B1, Cav-1, and LC3 II/I in nucleus and cytoplasm of primary LSECs in 24 h and 48 h. The relative protein expression and LC3 II/I expression in nucleus and cytoplasm are quantified in the two graphs (right). * p < 0.05 versus the control group in 24 h; # p < 0.05 versus the control group in 48 h. (D) Renilla Luciferase Reporter Gene Assay for LMNA promoter activity of primary LSECs in 24 h and 48 h. * p < 0.05 versus the control group in 24 h. (E) The interaction of nuclear LC3B with acetyl Lysine, progerin, Lamin B1, and Cav-1 of primary LSECs in 24 h and 48 h was detected by the co-IP assay. Nuclear LC3B in primary LSECs was individually immunoprecipitated, and subsequently, acetyl Lysine, progerin, Lamin B1, Cav-1, and LC3 II/I in nucleus of primary LSECs were subjected to immunoblotting analysis. (F) The interaction of cytoplasmic LC3B with Cav-1 of primary LSECs in 24 h and 48 h was detected by the co-IP assay. Cytoplasmic LC3B in primary LSECs was individually immunoprecipitated, and subsequently, Cav-1 and LC3 II/I in cytoplasm of primary LSECs were subjected to immunoblotting analysis. (G) The immunocytochemical co-localization of progerin (red) with F-actin (purple) of primary LSECs in 48 h (Scale bar: 5 μm). Nuclei are shown by DAPI (blue).
![Cells 11 03918 g004]()
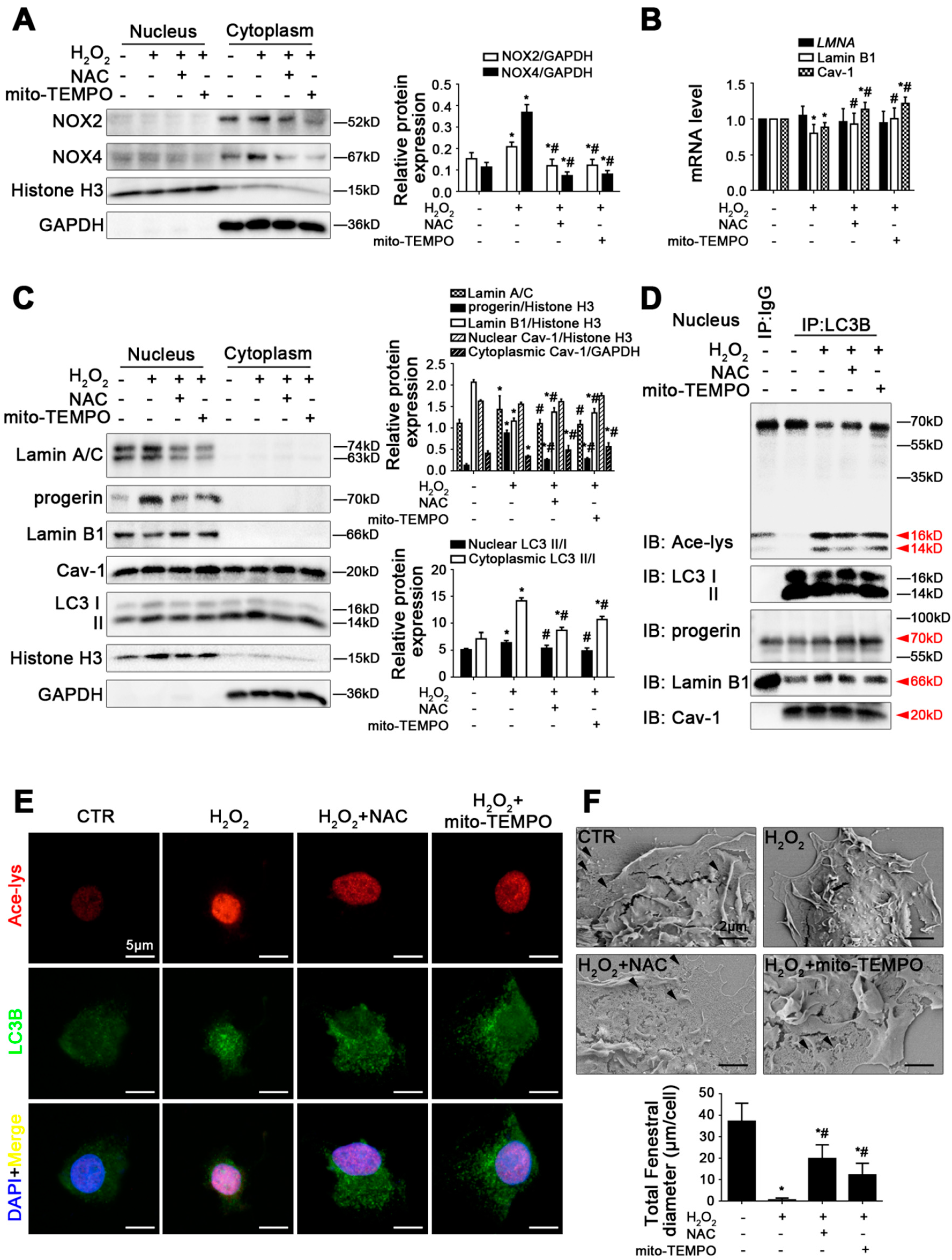
Figure 5.
Antioxidants promote progerin nucleophagic degradation and reverse depletion of Lamin B1 and Cav-1 to maintain LSEC fenestrae. Fresh rat primary LSECs, isolated from normal male SD rats, were pre-treated with NAC (1 mM) or mito-TEMPO (100 U/mL) and stimulated with H2O2 (10 μM) in vitro for 48 h. (A) Representative immunoblots of NOX2 and NOX4 in nucleus and cytoplasm of primary LSECs in the four groups (CTR, H2O2, H2O2+NAC, H2O2+mito-TEMPO). The relative protein expression was quantified in the graph (right). * p < 0.05 versus the control group; # p < 0.05 versus the H2O2 group. (B) RT-qPCR analysis for mRNA levels of LMNA, Lamin B1, and Cav-1 of primary LSECs in the four groups. * p < 0.05 versus the control group; # p < 0.05 versus the H2O2 group. (C) Representative immunoblots of Lamin A/C, progerin, Lamin B1, Cav-1, and LC3 II/I in nucleus and cytoplasm of primary LSECs in the four groups. The relative protein expression and LC3 II/I expression in nucleus and cytoplasm are quantified in the two graphs (right). * p < 0.05 versus the control group; # p < 0.05 versus the H2O2 group. (D) The interaction of nuclear LC3B with acetyl Lysine, progerin, Lamin B1, and Cav-1 of primary LSECs was detected by the co-IP assay. Nuclear LC3B in primary LSECs was individually immunoprecipitated, and subsequently, acetyl Lysine, progerin, Lamin B1, Cav-1, and LC3 II/I in nucleus of primary LSECs were subjected to immunoblotting analysis. (E) The immunocytochemical co-localization of acetyl Lysine (red) with LC3B (green) of primary LSECs in the four groups (scale bar: 5 μm). Nuclei are shown by DAPI (blue). (F) The magnification of SEM for fenestrae in primary LSECs in the four groups (scale bar: 2 μm). The black triangles indicate fenestrae in LSECs. The total fenestral diameter is quantified in the graph (below). * p < 0.05 versus the control group; # p < 0.05 versus the H2O2 group.
Figure 5.
Antioxidants promote progerin nucleophagic degradation and reverse depletion of Lamin B1 and Cav-1 to maintain LSEC fenestrae. Fresh rat primary LSECs, isolated from normal male SD rats, were pre-treated with NAC (1 mM) or mito-TEMPO (100 U/mL) and stimulated with H2O2 (10 μM) in vitro for 48 h. (A) Representative immunoblots of NOX2 and NOX4 in nucleus and cytoplasm of primary LSECs in the four groups (CTR, H2O2, H2O2+NAC, H2O2+mito-TEMPO). The relative protein expression was quantified in the graph (right). * p < 0.05 versus the control group; # p < 0.05 versus the H2O2 group. (B) RT-qPCR analysis for mRNA levels of LMNA, Lamin B1, and Cav-1 of primary LSECs in the four groups. * p < 0.05 versus the control group; # p < 0.05 versus the H2O2 group. (C) Representative immunoblots of Lamin A/C, progerin, Lamin B1, Cav-1, and LC3 II/I in nucleus and cytoplasm of primary LSECs in the four groups. The relative protein expression and LC3 II/I expression in nucleus and cytoplasm are quantified in the two graphs (right). * p < 0.05 versus the control group; # p < 0.05 versus the H2O2 group. (D) The interaction of nuclear LC3B with acetyl Lysine, progerin, Lamin B1, and Cav-1 of primary LSECs was detected by the co-IP assay. Nuclear LC3B in primary LSECs was individually immunoprecipitated, and subsequently, acetyl Lysine, progerin, Lamin B1, Cav-1, and LC3 II/I in nucleus of primary LSECs were subjected to immunoblotting analysis. (E) The immunocytochemical co-localization of acetyl Lysine (red) with LC3B (green) of primary LSECs in the four groups (scale bar: 5 μm). Nuclei are shown by DAPI (blue). (F) The magnification of SEM for fenestrae in primary LSECs in the four groups (scale bar: 2 μm). The black triangles indicate fenestrae in LSECs. The total fenestral diameter is quantified in the graph (below). * p < 0.05 versus the control group; # p < 0.05 versus the H2O2 group.
![Cells 11 03918 g005]()
Figure 6.
Excessive autophagy provokes depletion of Lamin B1 and Cav-1, as well as from progerin nucleophagic degradation. Fresh rat primary LSECs, isolated from normal male SD rats and cultured in vitro, were transfected with LC3B siRNA and nontarget siRNA (called NC) or pre-treated with rapamycin (10 nM) and then stimulated with H2O2 (10 μM) for 48 h. (A) Representative immunoblots of LC3 II/I, Cav-1, Lamin A/C, progerin, and Lamin B1 in nucleus and cytoplasm of primary LSECs in the four groups (CTR, H2O2, H2O2+rapamycin, rapamycin). The relative protein expression and LC3 II/I expression in nucleus and cytoplasm are quantified in the two graphs (right). * p < 0.05 versus the control group; # p < 0.05 versus the H2O2 group. (B) The interaction of cytoplasmic LC3B with Cav-1 was detected by the co-IP assay. Cytoplasmic LC3B in primary LSECs was individually immunoprecipitated, and subsequently, Cav-1 and LC3B in cytoplasm of primary LSECs in the four groups were subjected to immunoblotting analysis as indicated. (C) The interaction of nuclear LC3B with acetyl Lysine, progerin, Lamin B1, and Cav-1 was detected by the co-IP assay. Nuclear LC3B in primary LSECs was individually immunoprecipitated, and subsequently, acetyl Lysine, progerin, Lamin B1, Cav-1, and LC3 II/I in nucleus of primary LSECs in the four groups were subjected to immunoblotting analysis. (D) Representative immunoblots of LC3 II/I, Lamin A/C, progerin, Lamin B1, and Cav-1 in nucleus and cytoplasm of primary LSECs in the four groups (NC, NC+H2O2, si LC3+H2O2, si LC3). The relative protein expression and LC3 II/I expression in nucleus and cytoplasm are quantified in the two graphs (right). * p < 0.05 versus the NC group; # p < 0.05 versus the NC+H2O2 group.
Figure 6.
Excessive autophagy provokes depletion of Lamin B1 and Cav-1, as well as from progerin nucleophagic degradation. Fresh rat primary LSECs, isolated from normal male SD rats and cultured in vitro, were transfected with LC3B siRNA and nontarget siRNA (called NC) or pre-treated with rapamycin (10 nM) and then stimulated with H2O2 (10 μM) for 48 h. (A) Representative immunoblots of LC3 II/I, Cav-1, Lamin A/C, progerin, and Lamin B1 in nucleus and cytoplasm of primary LSECs in the four groups (CTR, H2O2, H2O2+rapamycin, rapamycin). The relative protein expression and LC3 II/I expression in nucleus and cytoplasm are quantified in the two graphs (right). * p < 0.05 versus the control group; # p < 0.05 versus the H2O2 group. (B) The interaction of cytoplasmic LC3B with Cav-1 was detected by the co-IP assay. Cytoplasmic LC3B in primary LSECs was individually immunoprecipitated, and subsequently, Cav-1 and LC3B in cytoplasm of primary LSECs in the four groups were subjected to immunoblotting analysis as indicated. (C) The interaction of nuclear LC3B with acetyl Lysine, progerin, Lamin B1, and Cav-1 was detected by the co-IP assay. Nuclear LC3B in primary LSECs was individually immunoprecipitated, and subsequently, acetyl Lysine, progerin, Lamin B1, Cav-1, and LC3 II/I in nucleus of primary LSECs in the four groups were subjected to immunoblotting analysis. (D) Representative immunoblots of LC3 II/I, Lamin A/C, progerin, Lamin B1, and Cav-1 in nucleus and cytoplasm of primary LSECs in the four groups (NC, NC+H2O2, si LC3+H2O2, si LC3). The relative protein expression and LC3 II/I expression in nucleus and cytoplasm are quantified in the two graphs (right). * p < 0.05 versus the NC group; # p < 0.05 versus the NC+H2O2 group.
![Cells 11 03918 g006]()
Figure 7.
Overexpression of SIRT1 promotes progerin nucleophagic degradation via deacetylation of nuclear LC3 and inhibits loss of Lamin B1 and Cav-1, contributing to maintaining LSEC fenestrae. Fresh rat primary LSECs were transfected with SIRT1 adenovirus vector (called AV-SIRT1) to overexpress SIRT1 or nontarget adenovirus vector (called AV-CTR) and then stimulated with H2O2 (10 μM) for 48 h. (A) RT-qPCR analysis for mRNA levels of LMNA, Lamin B1, and Cav-1 of primary LSECs in the four groups (AV-CTR, AV-CTR+H2O2, AV-SIRT1+H2O2, AV-SIRT1). * p < 0.05 versus the AV-CTR group; # p < 0.05 versus the AV-CTR+H2O2 group. (B) Renilla Luciferase Reporter Gene Assay for LMNA promoter activity of primary LSECs in the four groups. (C) Representative immunoblots of LC3 II/I, Lamin A/C, progerin, Lamin B1, and Cav-1 in nucleus and cytoplasm of primary LSECs in the four groups. The relative protein expression and LC3 II/I expression in nucleus and cytoplasm are quantified in the two graphs (right). * p < 0.05 versus the AV-CTR group; # p < 0.05 versus the AV-CTR+H2O2 group. (D) The immunocytochemical co-localization of progerin (red) with LC3B (green) and F-actin (purple) of primary LSECs in the four groups (scale bar: 5 μm). Nucleus is shown by DAPI (blue). (E) The interaction of nuclear LC3B with acetyl Lysine, progerin, Lamin B1, and Cav-1 was detected by the co-IP assay. Nuclear LC3B in primary LSECs was individually immunoprecipitated, and subsequently, acetyl Lysine, progerin, Lamin B1, Cav-1 and LC3 II/I in nucleus of LSECs in the four groups were subjected to immunoblotting analysis. (F) The magnification of SEM for fenestrae in primary LSECs in the four groups (scale bar: 2 μm). The black triangles indicate fenestrae in LSECs.
Figure 7.
Overexpression of SIRT1 promotes progerin nucleophagic degradation via deacetylation of nuclear LC3 and inhibits loss of Lamin B1 and Cav-1, contributing to maintaining LSEC fenestrae. Fresh rat primary LSECs were transfected with SIRT1 adenovirus vector (called AV-SIRT1) to overexpress SIRT1 or nontarget adenovirus vector (called AV-CTR) and then stimulated with H2O2 (10 μM) for 48 h. (A) RT-qPCR analysis for mRNA levels of LMNA, Lamin B1, and Cav-1 of primary LSECs in the four groups (AV-CTR, AV-CTR+H2O2, AV-SIRT1+H2O2, AV-SIRT1). * p < 0.05 versus the AV-CTR group; # p < 0.05 versus the AV-CTR+H2O2 group. (B) Renilla Luciferase Reporter Gene Assay for LMNA promoter activity of primary LSECs in the four groups. (C) Representative immunoblots of LC3 II/I, Lamin A/C, progerin, Lamin B1, and Cav-1 in nucleus and cytoplasm of primary LSECs in the four groups. The relative protein expression and LC3 II/I expression in nucleus and cytoplasm are quantified in the two graphs (right). * p < 0.05 versus the AV-CTR group; # p < 0.05 versus the AV-CTR+H2O2 group. (D) The immunocytochemical co-localization of progerin (red) with LC3B (green) and F-actin (purple) of primary LSECs in the four groups (scale bar: 5 μm). Nucleus is shown by DAPI (blue). (E) The interaction of nuclear LC3B with acetyl Lysine, progerin, Lamin B1, and Cav-1 was detected by the co-IP assay. Nuclear LC3B in primary LSECs was individually immunoprecipitated, and subsequently, acetyl Lysine, progerin, Lamin B1, Cav-1 and LC3 II/I in nucleus of LSECs in the four groups were subjected to immunoblotting analysis. (F) The magnification of SEM for fenestrae in primary LSECs in the four groups (scale bar: 2 μm). The black triangles indicate fenestrae in LSECs.
![Cells 11 03918 g007]()
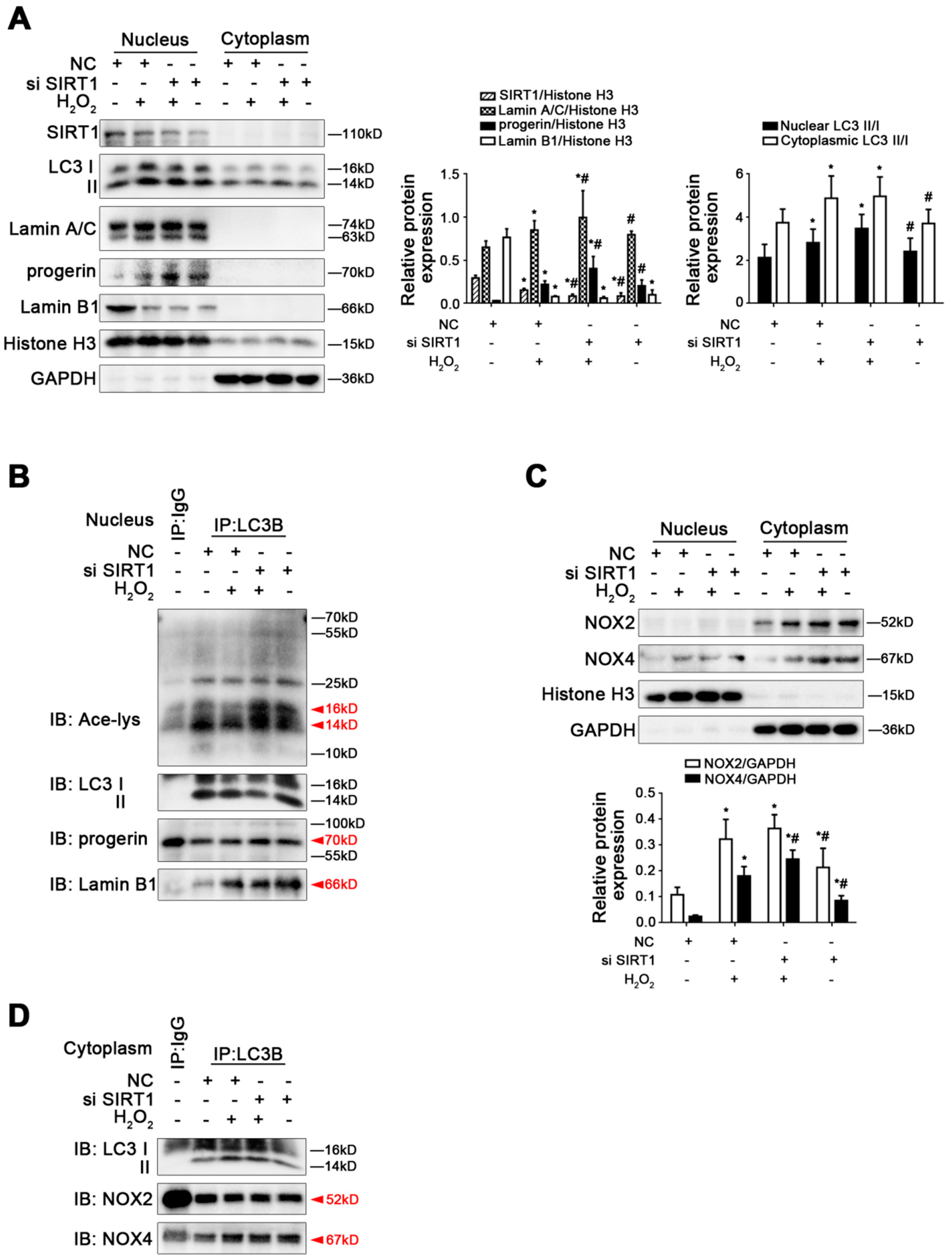
Figure 8.
Knockdown of SIRT1 with siRNA activated Lamin B1 nucleophagic degradation but did not change H2O2-induced elevated progerin. Rat primary LSECs were transfected with SIRT1 siRNA or nontarget siRNA (called NC) and then treated with H2O2 (10 μM) for 48 h. The transfection efficiency was about 75%. (A) Representative immunoblots of SIRT1, LC3 II/I, Lamin A/C, progerin, and Lamin B1 in nucleus and cytoplasm of primary LSECs in the four groups (NC, NC+H2O2, si SIRT1+H2O2, si SIRT1). The relative protein expression and LC3 II/I expression in nucleus and cytoplasm are quantified in the two graphs (right). * p < 0.05 versus the NC group; # p < 0.05 versus the NC+H2O2 group. (B) The interaction of nuclear LC3B with acetyl Lysine, progerin, and Lamin B1 was detected by the co-IP assay. Nuclear LC3B in primary LSECs was individually immunoprecipitated, and subsequently, acetyl Lysine, progerin, Lamin B1, and LC3 II/I in nucleus of LSECs in the four groups were subjected to immunoblotting analysis. (C) Representative immunoblots of NOX2 and NOX4 in nucleus and cytoplasm of primary LSECs in the four groups. The relative protein expression is quantified in the graph (below). * p < 0.05 versus the NC group; # p < 0.05 versus the NC+H2O2 group. (D) The interaction of cytoplasmic LC3B with NOX2 and NOX4 was detected by the co-IP assay. Cytoplasmic LC3B in primary LSECs was individually immunoprecipitated, and subsequently, NOX2, NOX4, and LC3 II/I in cytoplasm of LSECs in the four groups were subjected to immunoblotting analysis.
Figure 8.
Knockdown of SIRT1 with siRNA activated Lamin B1 nucleophagic degradation but did not change H2O2-induced elevated progerin. Rat primary LSECs were transfected with SIRT1 siRNA or nontarget siRNA (called NC) and then treated with H2O2 (10 μM) for 48 h. The transfection efficiency was about 75%. (A) Representative immunoblots of SIRT1, LC3 II/I, Lamin A/C, progerin, and Lamin B1 in nucleus and cytoplasm of primary LSECs in the four groups (NC, NC+H2O2, si SIRT1+H2O2, si SIRT1). The relative protein expression and LC3 II/I expression in nucleus and cytoplasm are quantified in the two graphs (right). * p < 0.05 versus the NC group; # p < 0.05 versus the NC+H2O2 group. (B) The interaction of nuclear LC3B with acetyl Lysine, progerin, and Lamin B1 was detected by the co-IP assay. Nuclear LC3B in primary LSECs was individually immunoprecipitated, and subsequently, acetyl Lysine, progerin, Lamin B1, and LC3 II/I in nucleus of LSECs in the four groups were subjected to immunoblotting analysis. (C) Representative immunoblots of NOX2 and NOX4 in nucleus and cytoplasm of primary LSECs in the four groups. The relative protein expression is quantified in the graph (below). * p < 0.05 versus the NC group; # p < 0.05 versus the NC+H2O2 group. (D) The interaction of cytoplasmic LC3B with NOX2 and NOX4 was detected by the co-IP assay. Cytoplasmic LC3B in primary LSECs was individually immunoprecipitated, and subsequently, NOX2, NOX4, and LC3 II/I in cytoplasm of LSECs in the four groups were subjected to immunoblotting analysis.
![Cells 11 03918 g008]()
Figure 9.
A schematic view of major signal pathways involved in the conclusion. Oxidative damage accelerates LSEC defenestration due to excessive accumulation of progerin and depletion of Cav-1. Overexpressing SIRT1 provokes progerin nucleophagic degradation via deacetylation of nuclear LC3 and inhibits depletion of Lamin B1 and Cav-1, contributing to reversing LSEC defenestration.
Figure 9.
A schematic view of major signal pathways involved in the conclusion. Oxidative damage accelerates LSEC defenestration due to excessive accumulation of progerin and depletion of Cav-1. Overexpressing SIRT1 provokes progerin nucleophagic degradation via deacetylation of nuclear LC3 and inhibits depletion of Lamin B1 and Cav-1, contributing to reversing LSEC defenestration.



 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








