
FGFR Inhibitors in Cholangiocarcinoma—A Novel Yet Primary Approach: Where Do We Stand Now and Where to Head Next in Targeting This Axis?

 , ,
, ,  and
and
Abstract
:1. Introduction
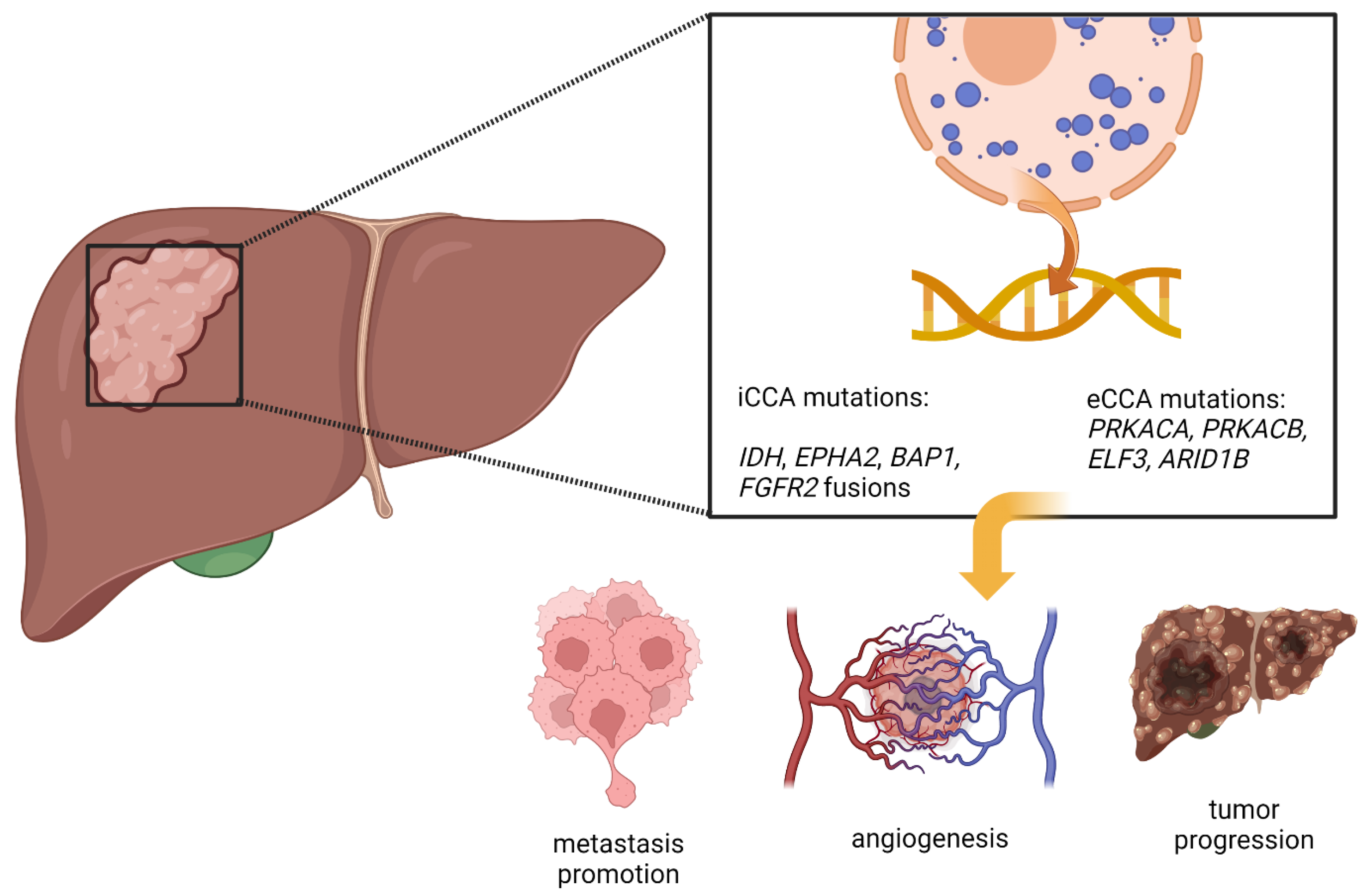
2. Cholangiocarcinoma—In a Summary
3. Genetic Aberrations in Cholangiocarcinoma
4. FGF/FGFR Interplay in Cholangiocarcinoma
5. Targeted Therapies
5.1. Ponatinib
5.2. Debio 1347
5.3. Derazantanib
5.4. Erdafitinib
5.5. Infigratinib
5.6. Futibatinib
5.7. Pemigatinib

{kind=link}
{kind=link}
{kind=link}
| The Current Stage of Development | Inhibitor Generation/Potency | Efficacy Results | Adverse Events and Disadvantages of the Therapy | NCT/Reference | |
|---|---|---|---|---|---|
| Ponatinib | The first study was conducted, based on the results of 12 patients with CCA. | Third-generation TKI; FGFR1-4; VEGFR2; RET; c-KIT; BCR-ABL1 | mPFS 2.4 months; mOS 15.7 months | Rash, fatigue, lymphopenia | [107] |
| Debio 1347 | Two main clinical trials with mixed results; phase II study showed great results in patients with CCA, however, the FUZE study has been terminated due to low antitumor activity. | Third generation TKI; highly selective for FGFR1-3 | mPFS 18.3 weeks | fatigue, hyperphosphatemia, anaemia, alopecia, nausea, vomiting, constipation, and palmar-plantar erythrodysesthesia syndrome | NCT01948297 [108,109] |
| Derazantanib | Trial with hopeful results followed by ongoing FIDES-01 trial with tumours harbouring FGFR2 alterations. | FGFR1-3, RET, VEGFR1, DDR, KIT | mPFS 5.7 months | hyperphosphatemia, dry mouth and nausea, asthenia, fatigue, dysgeusia, vomiting, dry eye, conjunctivitis, blurred vision, photophobia | NCT01752920 NCT03230318 [111,112,113] |
| Erdafitinib | Trial for patients harbouring FGFR2 mutations in the Asian population. | First-generation TKI inhibitor; FGFR1-4 and to lesser extent VEGFR | mPFS 2.35 months | Hyperphosphatemia, stomatitis, dry mouth, elevated AST, elevated ALT | NCT02699606 [115] |
| Infigratinib | Approved by FDA for unresectable, locally advanced, or metastatic CCA with FGFR2 fusion or another rearrangement. Ongoing phase III trial versus chemotherapy in patients with CCA. | FGFR1-3 selective inhibitor | mPFS 7.3 months | hyperphosphatemia, eye disorders, stomatitis, and fatigue | NCT01004224 NCT03773302 [117,118,119] |
| Futibatinib | Approved by FDA for locally advanced or metastatic CCA harbouring an FGFR2 rearrangement or fusion. Phase III FOENIX-CCA3 trial recruiting. | FGFR1-4 selective inhibitor | mPFS 9 months | Hyperphosphatemia, diarrhoea, dry mouth | NCT02052778 NCT04093362 [122,124,125,126] |
| Pemigatinib | Approved by FDA for previously treated, unresectable, advanced/ metastatic CCA with FGFR2 alterations. Phase III trial (FIGHT-302) versus chemotherapy as first-line treatment in CCA is ongoing. | FGFR1-3 and weaker activity against FGFR4 | mOS 21.1 months mPFS 6.9 months | Hyperphosphatemia, alopecia, diarrhoea, fatigue, dysgeusia | NCT02393248 NCT02924376 NCT03656536 [8,128,129,130] |
6. Key Questions and How to Address Them
6.1. Primary and Acquired Resistance Mechanisms
6.2. Crucial Disadvantages of FGFR-Targeted Therapy
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CCA | cholangiocarcinoma |
| iCCA | intrahepatic cholangiocarcinoma |
| pCCA | perihilar cholangiocarcinoma |
| dCCA | distal cholangiocarcinoma |
| eCCA | extrahepatic cholangiocarcinoma |
| FGFR | fibroblast growth factor receptor |
| FGF | fibroblast growth factor |
| TK | tyrosine kinase |
| TKI | tyrosine kinase inhibitor |
| FDA | Food and Drug Administration |
| IDH1 | Isocitrate dehydrogenase 1 |
| ARID1A/ARID1B | AT-Rich Interaction Domain 1A/1B |
| BAP1 BRCA1 | Associated Protein 1 |
| TP53 | tumour protein p53 |
| RAS | rat sarcoma viral proto-oncogene |
| PTEN | Phosphatase And Tensin Homolog |
| APC | Regulator Of WNT Signalling Pathway |
| EPHA2 | Epithelial Cell Receptor Protein Tyrosine Kinase A2 |
| PRKACA/PRKACB | Protein Kinase CAMP-Activated Catalytic Subunit Alpha/Beta |
| ELF3 E74 | Like ETS Transcription Factor 3 |
| PD-1/PD-L1 | Programmed cell death protein 1/Programmed death-ligand 1 |
| ERBB2 | Erb-B2 Receptor Tyrosine Kinase 2 |
| PI3K | phosphoinositide 3-kinase |
| AKT | kinases protein kinase B family |
| mTOR | Mammalian target of rapamycin |
| PLCγ | phospholipase C gamma |
| DAG | dystroglycan |
| PKC | protein kinase C |
| RAF | rapidly accelerated fibrosarcoma kinase |
| MEK | Mitogen-activated protein kinase kinase |
| MAPK | Mitogen activated protein kinase |
| JAK | kinase Janus kinase |
| STAT | signal transducer and activator of transcription |
| IP3 | inositol trisphosphate |
| BICC1 | Protein Bicaudal C Homolog 1 |
| PPHLN1 | Periphilin 1 |
| TACC3 | Transforming Acidic Coiled-Coil Containing Protein 3 |
| MGEA5 | meningioma expressed antigen 5 |
| RET | Ret Proto-Oncogene |
| VEGFR1 | Vascular endothelial growth factor receptor 1 |
| DDR DNA | damage response and repair gene |
| CDKN2A/B | cyclin-dependent kinase inhibitor 2A/B |
| PBRM1 | Polybromo 1 gene |
| cfDNA | cell-free circulating tumour DNA |
| siRNA | small interfering RNA |
| OS | overall survival |
| DFS | disease-free survival |
| ORR | overall response rate |
| PFS | progression-free survival |
| SD | stable disease |
| PR | partial response |
| PD | progressed disease |
| DCR | disease control rate |
| TEAE | treatment-emergent adverse events |
| AE | adverse events |
References
- De Luca, A.; Abate, R.E.; Rachiglio, A.M.; Maiello, M.R.; Esposito, C.; Schettino, C.; Izzo, F.; Nasti, G.; Normanno, N. FGFR Fusions in Cancer: From Diagnostic Approaches to Therapeutic Intervention. Int. J. Mol. Sci. 2020, 21, 6856. [Google Scholar] [CrossRef] [PubMed]
- Bale, T.A. FGFR- gene family alterations in low-grade neuroepithelial tumors. Acta Neuropathol. Commun. 2020, 8, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Gores, G.J. Pathogenesis, Diagnosis, and Management of Cholangiocarcinoma. Gastroenterology 2013, 145, 1215–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Ghedini, G.C.; Ronca, R.; Presta, M.; Giacomini, A. Future applications of FGF/FGFR inhibitors in cancer. Expert Rev. Anticancer. Ther. 2018, 18, 861–872. [Google Scholar] [CrossRef]
- Kang, C. Infigratinib: First Approval. Drugs 2021, 81, 1355–1360. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: A multicentre, open-label, phase 2 study. Lancet Oncol. 2020, 21, 671–684. [Google Scholar] [CrossRef]
- Zhou, Y.; Wu, C.; Lu, G.; Hu, Z.; Chen, Q.; Du, X. FGF/FGFR signaling pathway involved resistance in various cancer types. J. Cancer 2020, 11, 2000–2007. [Google Scholar] [CrossRef]
- Razumilava, N.; Gores, G.J. Classification, Diagnosis, and Management of Cholangiocarcinoma. Clin. Gastroenterol. Hepatol. 2013, 11, 13–21.e1. [Google Scholar] [CrossRef]
- Blechacz, B.; Komuta, M.; Roskams, T.; Gores, G.J. Clinical diagnosis and staging of cholangiocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 512–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Garrido, P.; Rodrigues, P.M. The jigsaw of dual hepatocellular–intrahepatic cholangiocarcinoma tumours. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 653–655. [Google Scholar] [CrossRef] [PubMed]
- Brindley, P.J.; Bachini, M.; Ilyas, S.I.; Khan, S.A.; Loukas, A.; Sirica, A.E.; Teh, B.T.; Wongkham, S.; Gores, G.J. Cholangiocarcinoma. Nat. Rev. Dis. Prim. 2021, 7, 56. [Google Scholar] [CrossRef]
- Rizvi, S.; Khan, S.A.; Hallemeier, C.L.; Kelley, R.K.; Gores, G.J. Cholangiocarcinoma—Evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 2018, 15, 95–111. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Yang, T.; Wu, M.; Shen, F. Intrahepatic cholangiocarcinoma: Epidemiology, risk factors, diagnosis and surgical management. Cancer Lett. 2016, 379, 198–205. [Google Scholar] [CrossRef]
- Gad, M.M.; Saad, A.M.; Faisaluddin, M.; Găman, M.-A.; Ruhban, I.A.; Jazieh, K.A.; Al-Husseini, M.J.; Simons-Linares, C.R.; Sonbol, M.B.; Estfan, B.N. Epidemiology of Cholangiocarcinoma; United States Incidence and Mortality Trends. Clin. Res. Hepatol. Gastroenterol. 2020, 44, 885–893. [Google Scholar] [CrossRef]
- DeOliveira, M.L.; Cunningham, S.C.; Cameron, J.L.; Kamangar, F.; Winter, J.M.; Lillemoe, K.D.; Choti, M.A.; Yeo, C.J.; Schulick, R.D. Cholangiocarcinoma. Ann. Surg. 2007, 245, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Patel, T. Worldwide trends in mortality from biliary tract malignancies. BMC Cancer 2002, 2, 10. [Google Scholar] [CrossRef] [Green Version]
- Taylor-Robinson, S.D.; Toledano, M.B.; Arora, S.; Keegan, T.J.; Hargreaves, S.; Beck, A.; A Khan, S.; Elliott, P.; Thomas, H.C. Increase in mortality rates from intrahepatic cholangiocarcinoma in England and Wales 1968–1998. Gut 2001, 48, 816–820. [Google Scholar] [CrossRef]
- Saha, S.K.; Zhu, A.X.; Fuchs, C.S.; Brooks, G.A. Forty-Year Trends in Cholangiocarcinoma Incidence in the U.S.: Intrahepatic Disease on the Rise. Oncologist 2016, 21, 594–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, K.J.; Jabbour, S.; Parekh, N.; Lin, Y.; Moss, R.A. Increasing mortality in the United States from cholangiocarcinoma: An analysis of the National Center for Health Statistics Database. BMC Gastroenterol. 2016, 16, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alabraba, E.; Joshi, H.; Bird, N.; Griffin, R.; Sturgess, R.; Stern, N.; Sieberhagen, C.; Cross, T.; Camenzuli, A.; Davis, R.; et al. Increased multimodality treatment options has improved survival for Hepatocellular carcinoma but poor survival for biliary tract cancers remains unchanged. Eur. J. Surg. Oncol. 2019, 45, 1660–1667. [Google Scholar] [CrossRef] [PubMed]
- Lindnér, P.; Rizell, M.; Hafström, L. The Impact of Changed Strategies for Patients with Cholangiocarcinoma in This Millenium. HPB Surg. 2015, 2015, 736049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komaya, K.; Ebata, T.; Yokoyama, Y.; Igami, T.; Sugawara, G.; Mizuno, T.; Yamaguchi, J.; Nagino, M. Recurrence after curative-intent resection of perihilar cholangiocarcinoma: Analysis of a large cohort with a close postoperative follow-up approach. Surgery 2018, 163, 732–738. [Google Scholar] [CrossRef]
- Krasinskas, A.M. Cholangiocarcinoma. Surg. Pathol. Clin. 2018, 11, 403–429. [Google Scholar] [CrossRef]
- Plentz, R.R.; Malek, N.P. Clinical presentation, risk factors and staging systems of cholangiocarcinoma. Best Pract. Res. Clin. Gastroenterol. 2015, 29, 245–252. [Google Scholar] [CrossRef]
- Gupta, A.; Dixon, E. Epidemiology and risk factors: Intrahepatic cholangiocarcinoma. HepatoBiliary Surg. Nutr. 2017, 6, 101–104. [Google Scholar] [CrossRef] [Green Version]
- Tyson, G.L.; El-Serag, H.B. Risk factors for cholangiocarcinoma. Hepatology 2011, 54, 173–184. [Google Scholar] [CrossRef] [Green Version]
- Petrick, J.L.; Thistle, J.E.; Zeleniuch-Jacquotte, A.; Zhang, X.; Wactawski-Wende, J.; Van Dyke, A.L.; Stampfer, M.J.; Sinha, R.; Sesso, H.D.; Schairer, C.; et al. Body Mass Index, Diabetes and Intrahepatic Cholangiocarcinoma Risk: The Liver Cancer Pooling Project and Meta-analysis. Am. J. Gastroenterol. 2018, 113, 1494–1505. [Google Scholar] [CrossRef]
- Welzel, T.M.; Mellemkjaer, L.; Gloria, G.; Sakoda, L.C.; Hsing, A.W.; El Ghormli, L.; Olsen, J.H.; McGlynn, K.A. Risk factors for intrahepatic cholangiocarcinoma in a low-risk population: A nationwide case-control study. Int. J. Cancer 2007, 120, 638–641. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Tavolari, S.; Brandi, G. Cholangiocarcinoma: Epidemiology and risk factors. Liver Int. 2019, 39, 19–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Diwany, R.; Pawlik, T.M.; Ejaz, A. Intrahepatic Cholangiocarcinoma. Surg. Oncol. Clin. N. Am. 2019, 28, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Sarcognato, S.; Sacchi, D.; Fassan, M.; Fabris, L.; Cadamuro, M.; Zanus, G.; Cataldo, I.; Capelli, P.; Baciorri, F.; Cacciatore, M.; et al. Cholangiocarcinoma. Pathologica 2021, 113, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Joo, I.; Lee, J.M.; Yoon, J.H. Imaging Diagnosis of Intrahepatic and Perihilar Cholangiocarcinoma: Recent Advances and Challenges. Radiology 2018, 288, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Wildner, D.; Bernatik, T.; Greis, C.; Seitz, K.; Neurath, M.F.; Strobel, D. CEUS in Hepatocellular Carcinoma and Intrahepatic Cholangiocellular Carcinoma in 320 Patients—Early or Late Washout Matters: A Subanalysis of the DEGUM Multicenter Trial. Ultraschall Der Med.-Eur. J. Ultrasound 2015, 36, 132–139. [Google Scholar] [CrossRef]
- Jhaveri, K.S.; Hosseini-Nik, H. MRI of cholangiocarcinoma. J. Magn. Reson. Imaging 2015, 42, 1165–1179. [Google Scholar] [CrossRef]
- Rizvi, S.; Eaton, J.; Yang, J.D.; Chandrasekhara, V.; Gores, G.J. Emerging Technologies for the Diagnosis of Perihilar Cholangiocarcinoma. Semin. Liver Dis. 2018, 38, 160–169. [Google Scholar] [CrossRef]
- Meyer, C.G.; Penn, I.; James, L. Liver Transplantation for Cholangiocarcinoma: Results in 207 Patients1. Transplantation 2000, 69, 1633–1637. [Google Scholar] [CrossRef]
- Rizzo, A. Targeted Therapies in Advanced Cholangiocarcinoma: A Focus on FGFR Inhibitors. Medicina 2021, 57, 458. [Google Scholar] [CrossRef]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus Gemcitabine versus Gemcitabine for Biliary Tract Cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigt, J.; Malfertheiner, P. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. Expert Rev. Gastroenterol. Hepatol. 2010, 4, 395–397. [Google Scholar] [CrossRef]
- Greten, T.F. Erstmals ein Chemotherapiestandard in der Behandlung von Patienten mit malignen Gallenwegserkrankungen. Z. Gastroenterol. 2010, 48, 850–851. [Google Scholar] [CrossRef]
- Razumilava, N.; Gores, G.J. Combination of gemcitabine and cisplatin for biliary tract cancer: A platform to build on. J. Hepatol. 2011, 54, 577–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murad, S.D.; Kim, W.R.; Harnois, D.M.; Douglas, D.D.; Burton, J.; Kulik, L.M.; Botha, J.F.; Mezrich, J.D.; Chapman, W.C.; Schwartz, J.J.; et al. Efficacy of Neoadjuvant Chemoradiation, Followed by Liver Transplantation, for Perihilar Cholangiocarcinoma at 12 US Centers. Gastroenterology 2012, 143, 88–98.e3. [Google Scholar] [CrossRef] [Green Version]
- Seehofer, D.; Thelen, A.; Neumann, U.P.; Veltzke-Schlieker, W.; Denecke, T.; Kamphues, C.; Pratschke, J.; Jonas, S.; Neuhaus, P. Extended bile duct resection liver and transplantation in patients with hilar cholangiocarcinoma: Long-term results. Liver Transplant. 2009, 15, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Kelley, R.K.; Furuse, J.; Edeline, J.; Ren, Z.; Su, S.-C.; Malhotra, U.; Siegel, A.B.; Valle, J.W. Abstract CT283: KEYNOTE-966: A randomized, double-blind, placebo-controlled, phase 3 study of pembrolizumab in combination with gemcitabine and cisplatin for the treatment of advanced biliary tract carcinoma. Cancer Res. 2020, 80, CT283. [Google Scholar] [CrossRef]
- Klein, O.; Kee, D.; Nagrial, A.; Markman, B.; Underhill, C.; Michael, M.; Jackett, L.; Lum, C.; Behren, A.; Palmer, J.; et al. Evaluation of Combination Nivolumab and Ipilimumab Immunotherapy in Patients with Advanced Biliary Tract Cancers. JAMA Oncol. 2020, 6, 1405–1409. [Google Scholar] [CrossRef]
- Massironi, S.; Pilla, L.; Elvevi, A.; Longarini, R.; Rossi, R.E.; Bidoli, P.; Invernizzi, P. New and Emerging Systemic Therapeutic Options for Advanced Cholangiocarcinoma. Cells 2020, 9, 688. [Google Scholar] [CrossRef] [Green Version]
- Chong, D.Q.; Zhu, A.X. The landscape of targeted therapies for cholangiocarcinoma: Current status and emerging targets. Oncotarget 2016, 7, 46750–46767. [Google Scholar] [CrossRef]
- Rizvi, S.; Gores, G.J. Emerging molecular therapeutic targets for cholangiocarcinoma. J. Hepatol. 2017, 67, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Tella, S.H.; Kommalapati, A.; Borad, M.J.; Mahipal, A. Second-line therapies in advanced biliary tract cancers. Lancet Oncol. 2020, 21, e29–e41. [Google Scholar] [CrossRef]
- Goyal, L.; Kongpetch, S.; Crolley, V.E.; Bridgewater, J. Targeting FGFR inhibition in cholangiocarcinoma. Cancer Treat. Rev. 2021, 95, 102170. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; ElZawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Lowery, M.A.; Ptashkin, R.; Jordan, E.; Berger, M.F.; Zehir, A.; Capanu, M.; Kemeny, N.E.; O’Reilly, E.M.; El-Dika, I.; Jarnagin, W.R.; et al. Comprehensive Molecular Profiling of Intrahepatic and Extrahepatic Cholangiocarcinomas: Potential Targets for Intervention. Clin. Cancer Res. 2018, 24, 4154–4161. [Google Scholar] [CrossRef] [Green Version]
- Zou, S.; Li, J.; Zhou, H.; Frech, C.; Jiang, X.; Chu, J.S.C.; Zhao, X.; Li, Y.; Li, Q.; Wang, H.; et al. Mutational landscape of intrahepatic cholangiocarcinoma. Nat. Commun. 2014, 5, 5696. [Google Scholar] [CrossRef] [Green Version]
- Ong, C.K.; Subimerb, C.; Pairojkul, C.; Wongkham, S.; Cutcutache, I.; Yu, W.; McPherson, J.R.; E Allen, G.; Ng, C.C.Y.; Wong, B.H.; et al. Exome sequencing of liver fluke–associated cholangiocarcinoma. Nat. Genet. 2012, 44, 690–693. [Google Scholar] [CrossRef]
- Jusakul, A.; Cutcutache, I.; Yong, C.H.; Lim, J.Q.; Ni Huang, M.; Padmanabhan, N.; Nellore, V.; Kongpetch, S.; Ng, A.W.T.; Ng, L.M.; et al. Whole-Genome and Epigenomic Landscapes of Etiologically Distinct Subtypes of Cholangiocarcinoma. Cancer Discov. 2017, 7, 1116–1135. [Google Scholar] [CrossRef] [Green Version]
- Nepal, C.; O'Rourke, C.J.; Oliveira, D.N.P.; Taranta, A.; Shema, S.; Gautam, P.; Calderaro, J.; Barbour, A.; Raggi, C.; Wennerberg, K.; et al. Genomic perturbations reveal distinct regulatory networks in intrahepatic cholangiocarcinoma. Hepatology 2018, 68, 949–963. [Google Scholar] [CrossRef] [Green Version]
- Farshidfar, F.; Zheng, S.; Gingras, M.-C.; Newton, Y.; Shih, J.; Robertson, A.G.; Hinoue, T.; Hoadley, K.A.; Gibb, E.A.; Roszik, J.; et al. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell Rep. 2017, 18, 2780–2794. [Google Scholar] [CrossRef]
- Chaisaingmongkol, J.; Budhu, A.; Dang, H.; Rabibhadana, S.; Pupacdi, B.; Kwon, S.M.; Forgues, M.; Pomyen, Y.; Bhudhisawasdi, V.; Lertprasertsuke, N.; et al. Common Molecular Subtypes among Asian Hepatocellular Carcinoma and Cholangiocarcinoma. Cancer Cell 2017, 32, 57–70.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merino-Azpitarte, M.; Lozano, E.; Perugorria, M.J.; Esparza-Baquer, A.; Erice, O.; Santos-Laso, Á.; O’Rourke, C.J.; Andersen, J.B.; Jiménez-Agüero, R.; Lacasta, A.; et al. SOX17 regulates cholangiocyte differentiation and acts as a tumor suppressor in cholangiocarcinoma. J. Hepatol. 2017, 67, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Goeppert, B.; Konermann, C.; Schmidt, C.R.; Bogatyrova, O.; Geiselhart, L.; Ernst, C.; Gu, L.; Becker, N.; Zucknick, M.; Mehrabi, A.; et al. Global alterations of DNA methylation in cholangiocarcinoma target the Wnt signaling pathway. Hepatology 2014, 59, 544–554. [Google Scholar] [CrossRef] [PubMed]
- Tischoff, I.; Wittekind, C.; Tannapfel, A. Role of epigenetic alterations in cholangiocarcinoma. J. Hepato-Biliary-Pancreat. Surg. 2006, 13, 274–279. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, C.J.; Lafuente-Barquero, J.; Andersen, J.B. Epigenome Remodeling in Cholangiocarcinoma. Trends Cancer 2019, 5, 335–350. [Google Scholar] [CrossRef]
- Javle, M.; Lowery, M.; Shroff, R.T.; Weiss, K.H.; Springfeld, C.; Borad, M.J.; Ramanathan, R.K.; Goyal, L.; Sadeghi, S.; Macarulla, T.; et al. Phase II Study of BGJ398 in Patients with FGFR-Altered Advanced Cholangiocarcinoma. J. Clin. Oncol. 2018, 36, 276–282. [Google Scholar] [CrossRef]
- Benson, A.B.; D’Angelica, M.I.; Abbott, D.E.; Anaya, D.A.; Anders, R.; Are, C.; Bachini, M.; Borad, M.; Brown, D.; Burgoyne, A.; et al. Hepatobiliary Cancers, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 541–565. [Google Scholar] [CrossRef]
- Itoh, N.; Ornitz, D.M. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004, 20, 563–569. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Legeai-Mallet, L. Achondroplasia: Development, pathogenesis, and therapy. Dev. Dyn. 2017, 246, 291–309. [Google Scholar] [CrossRef] [Green Version]
- Thisse, B.; Thisse, C. Functions and regulations of fibroblast growth factor signaling during embryonic development. Dev. Biol. 2005, 287, 390–402. [Google Scholar] [CrossRef]
- Zhou, M.; Luo, J.; Chen, M.; Yang, H.; Learned, R.M.; DePaoli, A.M.; Tian, H.; Ling, L. Mouse species-specific control of hepatocarcinogenesis and metabolism by FGF19/FGF15. J. Hepatol. 2017, 66, 1182–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inagaki, T.; Choi, M.; Moschetta, A.; Peng, L.; Cummins, C.L.; McDonald, J.G.; Luo, G.; Jones, S.A.; Goodwin, B.; Richardson, J.A.; et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005, 2, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Quarles, L.D. Skeletal secretion of FGF-23 regulates phosphate and vitamin D metabolism. Nat. Rev. Endocrinol. 2012, 8, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Cross, M.J.; Claesson-Welsh, L. FGF and VEGF function in angiogenesis: Signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol. Sci. 2001, 22, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef] [Green Version]
- Kalinina, J.; Dutta, K.; Ilghari, D.; Beenken, A.; Goetz, R.; Eliseenkova, A.V.; Cowburn, D.; Mohammadi, M. The Alternatively Spliced Acid Box Region Plays a Key Role in FGF Receptor Autoinhibition. Structure 2012, 20, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Jin, C.; Wang, F.; Wu, X.; Yu, C.; Luo, Y.; McKeehan, W.L. Directionally specific paracrine communication mediated by epithelial FGF9 to stromal FGFR3 in two-compartment premalignant prostate tumors. Cancer Res. 2004, 64, 4555–4562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sleeman, M.; Fraser, J.; McDonald, M.; Yuan, S.; White, D.; Grandison, P.; Kumble, K.; Watson, J.D.; Murison, J.G. Identification of a new fibroblast growth factor receptor, FGFR5. Gene 2001, 271, 171–182. [Google Scholar] [CrossRef]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Tomlinson, D.C.; Hurst, C.D.; A Knowles, M. Knockdown by shRNA identifies S249C mutant FGFR3 as a potential therapeutic target in bladder cancer. Oncogene 2007, 26, 5889–5899. [Google Scholar] [CrossRef]
- Teven, C.M.; Farina, E.M.; Rivas, J.; Reid, R.R. Fibroblast growth factor (FGF) signaling in development and skeletal diseases. Genes Dis. 2014, 1, 199–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetz, R.; Mohammadi, M. Exploring mechanisms of FGF signalling through the lens of structural biology. Nat. Rev. Mol. Cell Biol. 2013, 14, 166–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kommalapati, A.; Tella, S.; Borad, M.; Javle, M.; Mahipal, A. FGFR Inhibitors in Oncology: Insight on the Management of Toxicities in Clinical Practice. Cancers 2021, 13, 2968. [Google Scholar] [CrossRef] [PubMed]
- Leelawat, K. Basic fibroblast growth factor induces cholangiocarcinoma cell migration via activation of the MEK1/2 pathway. Oncol. Lett. 2011, 2, 821–825. [Google Scholar] [CrossRef] [PubMed]
- Sinha, J.; Chen, F.; Miloh, T.; Burns, R.C.; Yu, Z.; Shneider, B.L. β-Klotho and FGF-15/19 inhibit the apical sodium-dependent bile acid transporter in enterocytes and cholangiocytes. Am. J. Physiol. Liver Physiol. 2008, 295, G996–G1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Torrecuadrada, J.; Cifuentes, G.; López-Serra, P.; Saenz, P.; Martínez, A.; Casal, J.I. Targeting the Extracellular Domain of Fibroblast Growth Factor Receptor 3 with Human Single-Chain Fv Antibodies Inhibits Bladder Carcinoma Cell Line Proliferation. Clin. Cancer Res. 2005, 11, 6280–6290. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Dubrovska, A.; Salamone, R.J.; Walker, J.R.; Grandinetti, K.B.; Bonamy, G.M.C.; Orth, A.P.; Elliott, J.; Porta, D.G.; Garcia-Echeverria, C.; et al. FGFR2 Promotes Breast Tumorigenicity through Maintenance of Breast Tumor-Initiating Cells. PLoS ONE 2013, 8, e51671. [Google Scholar] [CrossRef] [Green Version]
- Coleman, S.J.; Chioni, A.; Ghallab, M.; Anderson, R.K.; Lemoine, N.R.; Kocher, H.M.; Grose, R.P. Nuclear translocation ofFGFR1 andFGF2 in pancreatic stellate cells facilitates pancreatic cancer cell invasion. EMBO Mol. Med. 2014, 6, 467–481. [Google Scholar] [CrossRef]
- Harding, T.C.; Long, L.; Palencia, S.; Zhang, H.; Sadra, A.; Hestir, K.; Patil, N.; Levin, A.; Hsu, A.W.; Charych, D.; et al. Blockade of Nonhormonal Fibroblast Growth Factors by FP-1039 Inhibits Growth of Multiple Types of Cancer. Sci. Transl. Med. 2013, 5, 178ra39. [Google Scholar] [CrossRef]
- Wang, Y.; Becker, D. Antisense targeting of basic fibroblast growth factor and dibroblast growth factor receptor-1 in human melanomas blocks intratumoral angiogenesis and tumor growth. Nat. Med. 1997, 3, 887–893. [Google Scholar] [CrossRef]
- Wu, Y.-M.; Su, F.; Kalyana-Sundaram, S.; Khazanov, N.; Ateeq, B.; Cao, X.; Lonigro, R.J.; Vats, P.; Wang, R.; Lin, S.-F.; et al. Identification of Targetable FGFR Gene Fusions in Diverse Cancers. Cancer Discov. 2013, 3, 636–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.-F.; Yang, X.-Q.; Lu, X.-F.; Guo, S.; Liu, Y.; Iqbal, M.; Ning, S.-L.; Yang, H.; Suo, N.; Chen, Y.-X. Fibroblast growth factor receptor 4 promotes progression and correlates to poor prognosis in cholangiocarcinoma. Biochem. Biophys. Res. Commun. 2014, 446, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Totoki, Y.; Hosoda, F.; Shirota, T.; Hama, N.; Nakamura, H.; Ojima, H.; Furuta, K.; Shimada, K.; Okusaka, T.; et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014, 59, 1427–1434. [Google Scholar] [CrossRef]
- Ross, J.S.; Wang, K.; Gay, L.; Al-Rohil, R.; Rand, J.V.; Jones, D.M.; Lee, H.J.; Sheehan, C.E.; Otto, G.A.; Palmer, G.; et al. New Routes to Targeted Therapy of Intrahepatic Cholangiocarcinomas Revealed by Next-Generation Sequencing. Oncologist 2014, 19, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borad, M.J.; Champion, M.D.; Egan, J.B.; Liang, W.S.; Fonseca, R.; Bryce, A.H.; McCullough, A.E.; Barrett, M.T.; Hunt, K.; Patel, M.; et al. Integrated Genomic Characterization Reveals Novel, Therapeutically Relevant Drug Targets in FGFR and EGFR Pathways in Sporadic Intrahepatic Cholangiocarcinoma. PLoS Genet. 2014, 10, e1004135. [Google Scholar] [CrossRef]
- Sia, D.; Losic, B.; Moeini, A.; Cabellos, L.; Hao, K.; Revill, K.; Bonal, D.M.; Miltiadous, O.; Zhang, Z.; Hoshida, Y.; et al. Massive parallel sequencing uncovers actionable FGFR2–PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nat. Commun. 2015, 6, 6087. [Google Scholar] [CrossRef] [Green Version]
- Pu, X.-H.; Ye, Q.; Yang, J.; Wu, H.-Y.; Ding, X.-W.; Shi, J.; Mao, L.; Fan, X.-S.; Chen, J.; Qiu, Y.-D.; et al. Low-level clonal FGFR2 amplification defines a unique molecular subtype of intrahepatic cholangiocarcinoma in a Chinese population. Hum. Pathol. 2018, 76, 100–109. [Google Scholar] [CrossRef]
- Pu, X.; Ye, Q.; Cai, J.; Yang, X.; Fu, Y.; Fan, X.; Wu, H.; Chen, J.; Qiu, Y.; Yue, S. Typing FGFR2 translocation determines the response to targeted therapy of intrahepatic cholangiocarcinomas. Cell Death Dis. 2021, 12, 256. [Google Scholar] [CrossRef]
- Yoo, C.; Kang, J.; Kim, D.; Kim, K.-P.; Ryoo, B.-Y.; Hong, S.-M.; Hwang, J.J.; Jeong, S.-Y.; Hwang, S.; Kim, K.-H.; et al. Multiplexed gene expression profiling identifies the FGFR4 pathway as a novel biomarker in intrahepatic cholangiocarcinoma. Oncotarget 2017, 8, 38592–38601. [Google Scholar] [CrossRef] [Green Version]
- Saborowski, A.; Lehmann, U.; Vogel, A. FGFR inhibitors in cholangiocarcinoma: What’s now and what’s next? Ther. Adv. Med. Oncol. 2020, 12, 175883592095329. [Google Scholar] [CrossRef]
- Fostea, R.M.; Fontana, E.; Torga, G.; Arkenau, H.-T. Recent Progress in the Systemic Treatment of Advanced/Metastatic Cholangiocarcinoma. Cancers 2020, 12, 2599. [Google Scholar] [CrossRef] [PubMed]
- King, G.; Javle, M. FGFR Inhibitors: Clinical Activity and Development in the Treatment of Cholangiocarcinoma. Curr. Oncol. Rep. 2021, 23, 108. [Google Scholar] [CrossRef] [PubMed]
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef]
- Touat, M.; Ileana, E.; Postel-Vinay, S.; André, F.; Soria, J.-C. Targeting FGFR Signaling in Cancer. Clin. Cancer Res. 2015, 21, 2684–2694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massaro, F.; Molica, M.; Breccia, M.; Massaro, M.M.A.M.B.F. Ponatinib: A Review of Efficacy and Safety. Curr. Cancer Drug Targets 2018, 18, 847–856. [Google Scholar] [CrossRef]
- Cortes, J.E.; Kim, D.-W.; Pinilla-Ibarz, J.; Le Coutre, P.D.; Paquette, R.; Chuah, C.; Nicolini, F.E.; Apperley, J.F.; Khoury, H.J.; Talpaz, M.; et al. Ponatinib efficacy and safety in Philadelphia chromosome–positive leukemia: Final 5-year results of the phase 2 PACE trial. Blood 2018, 132, 393–404. [Google Scholar] [CrossRef]
- Ahn, D.H.; Junior, P.L.S.U.; Masci, P.; Kosiorek, H.; Halfdanarson, T.R.; Mody, K.; Babiker, H.; DeLeon, T.; Sonbol, M.B.; Gores, G.; et al. A pilot study of Pan-FGFR inhibitor ponatinib in patients with FGFR-altered advanced cholangiocarcinoma. Investig. New Drugs 2022, 40, 134–141. [Google Scholar] [CrossRef]
- Voss, M.H.; Hierro, C.; Heist, R.S.; Cleary, J.M.; Meric-Bernstam, F.; Tabernero, J.; Janku, F.; Gandhi, L.; Iafrate, A.J.; Borger, D.R.; et al. A Phase I, Open-Label, Multicenter, Dose-escalation Study of the Oral Selective FGFR Inhibitor Debio 1347 in Patients with Advanced Solid Tumors Harboring FGFR Gene Alterations. Clin. Cancer Res. 2019, 25, 2699–2707. [Google Scholar] [CrossRef] [Green Version]
- Cleary, J.M.; Iyer, G.; Oh, D.-Y.; Mellinghoff, I.K.; Goyal, L.; Ng, M.C.; Meric-Bernstam, F.; Matos, I.; Chao, T.-Y.; Sarkouh, R.A.; et al. Final results from the phase I study expansion cohort of the selective FGFR inhibitor Debio 1347 in patients with solid tumors harboring an FGFR gene fusion. J. Clin. Oncol. 2020, 38, 3603. [Google Scholar] [CrossRef]
- Hyman, D.M.; Goyal, L.; Grivas, P.; Meric-Bernstam, F.; Tabernero, J.; Hu, Y.; Kirpicheva, Y.; Nicolas-Metral, V.; Pokorska-Bocci, A.; Vaslin, A.; et al. FUZE clinical trial: A phase 2 study of Debio 1347 in FGFR fusion-positive advanced solid tumors irrespectively of tumor histology. J. Clin. Oncol. 2019, 37, TPS3157. [Google Scholar] [CrossRef]
- Hall, T.G.; Yu, Y.; Eathiraj, S.; Wang, Y.; Savage, R.E.; Lapierre, J.-M.; Schwartz, B.; Abbadessa, G. Preclinical Activity of ARQ 087, a Novel Inhibitor Targeting FGFR Dysregulation. PLoS ONE 2016, 11, e0162594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzaferro, V.; El-Rayes, B.F.; Droz Dit Busset, M.; Cotsoglou, C.; Harris, W.P.; Damjanov, N.; Masi, G.; Rimassa, L.; Personeni, N.; Braiteh, F.; et al. Derazantinib (ARQ 087) in advanced or inoperable FGFR2 gene fusion-positive intrahepatic cholangiocarcinoma. Br. J. Cancer 2019, 120, 165–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javle, M.M.; Shaib, W.L.; Braun, S.; Engelhardt, M.; Borad, M.J.; Abou-Alfa, G.K.; Boncompagni, A.; Friedmann, S.; Gahlemann, C.G. FIDES-01, a phase II study of derazantinib in patients with unresectable intrahepatic cholangiocarcinoma (iCCA) and FGFR2 fusions and mutations or amplifications (M/A). J. Clin. Oncol. 2020, 38, TPS597. [Google Scholar] [CrossRef]
- Perera, T.P.; Jovcheva, E.; Mevellec, L.; Vialard, J.; De Lange, D.; Verhulst, T.; Paulussen, C.; Van De Ven, K.; King, P.; Freyne, E.; et al. Discovery and Pharmacological Characterization of JNJ-42756493 (Erdafitinib), a Functionally Selective Small-Molecule FGFR Family Inhibitor. Mol. Cancer Ther. 2017, 16, 1010–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.O.; Feng, Y.-H.; Chen, Y.-Y.; Su, W.-C.; Oh, D.-Y.; Shen, L.; Kim, K.-P.; Liu, X.; Bai, Y.; Liao, H.; et al. Updated results of a phase IIa study to evaluate the clinical efficacy and safety of erdafitinib in Asian advanced cholangiocarcinoma (CCA) patients with FGFR alterations. J. Clin. Oncol. 2019, 37, 4117. [Google Scholar] [CrossRef]
- Guagnano, V.; Furet, P.; Spanka, C.; Bordas, V.; Le Douget, M.; Stamm, C.; Brueggen, J.; Jensen, M.R.; Schnell, C.; Schmid, H.; et al. Discovery of 3-(2,6-Dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamino]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), A Potent and Selective Inhibitor of the Fibroblast Growth Factor Receptor Family of Receptor Tyrosine Kinase. J. Med. Chem. 2011, 54, 7066–7083. [Google Scholar] [CrossRef]
- Nogova, L.; Sequist, L.V.; Garcia, J.M.P.; Andre, F.; Delord, J.-P.; Hidalgo, M.; Schellens, J.H.; Cassier, P.A.; Camidge, D.R.; Schuler, M.; et al. Evaluation of BGJ398, a Fibroblast Growth Factor Receptor 1-3 Kinase Inhibitor, in Patients with Advanced Solid Tumors Harboring Genetic Alterations in Fibroblast Growth Factor Receptors: Results of a Global Phase I, Dose-Escalation and Dose-Expansion Study. J. Clin. Oncol. 2017, 35, 157–165. [Google Scholar] [CrossRef]
- Javle, M.M.; Roychowdhury, S.; Kelley, R.K.; Sadeghi, S.; Macarulla, T.; Waldschmidt, D.T.; Goyal, L.; Borbath, I.; El-Khoueiry, A.B.; Yong, W.-P.; et al. Final results from a phase II study of infigratinib (BGJ398), an FGFR-selective tyrosine kinase inhibitor, in patients with previously treated advanced cholangiocarcinoma harboring an FGFR2 gene fusion or rearrangement. J. Clin. Oncol. 2021, 39, 265. [Google Scholar] [CrossRef]
- Makawita, S.; Abou-Alfa, G.K.; Roychowdhury, S.; Sadeghi, S.; Borbath, I.; Goyal, L.; Cohn, A.; Lamarca, A.; Oh, D.-Y.; Macarulla, T.; et al. Infigratinib in patients with advanced cholangiocarcinoma with FGFR2 gene fusions/translocations: The PROOF 301 trial. Futur. Oncol. 2020, 16, 2375–2384. [Google Scholar] [CrossRef]
- Rizzo, A.; Ricci, A.D.; Brandi, G. Futibatinib, an investigational agent for the treatment of intrahepatic cholangiocarcinoma: Evidence to date and future perspectives. Expert Opin. Investig. Drugs 2021, 30, 317–324. [Google Scholar] [CrossRef]
- Sootome, H.; Fujita, H.; Ito, K.; Ochiiwa, H.; Fujioka, Y.; Ito, K.; Miura, A.; Sagara, T.; Ito, S.; Ohsawa, H.; et al. Futibatinib Is a Novel Irreversible FGFR 1–4 Inhibitor That Shows Selective Antitumor Activity against FGFR-Deregulated Tumors. Cancer Res. 2020, 80, 4986–4997. [Google Scholar] [CrossRef] [PubMed]
- Bahleda, R.; Meric-Bernstam, F.; Goyal, L.; Tran, B.; He, Y.; Yamamiya, I.; Benhadji, K.; Matos, I.; Arkenau, H.-T. Phase I, first-in-human study of futibatinib, a highly selective, irreversible FGFR1–4 inhibitor in patients with advanced solid tumors. Ann. Oncol. 2020, 31, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F.; Arkenau, H.; Tran, B.; Bahleda, R.; Kelley, R.; Hierro, C.; Ahn, D.; Zhu, A.; Javle, M.; Winkler, R.; et al. Efficacy of TAS-120, an irreversible fibroblast growth factor receptor (FGFR) inhibitor, in cholangiocarcinoma patients with FGFR pathway alterations who were previously treated with chemotherapy and other FGFR inhibitors. Ann. Oncol. 2018, 29, v100. [Google Scholar] [CrossRef]
- Goyal, L.; Meric-Bernstam, F.; Hollebecque, A.; Valle, J.W.; Morizane, C.; Karasic, T.B.; Abrams, T.A.; Furuse, J.; He, Y.; Soni, N.; et al. FOENIX-CCA2: A phase II, open-label, multicenter study of futibatinib in patients (pts) with intrahepatic cholangiocarcinoma (iCCA) harboring FGFR2 gene fusions or other rearrangements. J. Clin. Oncol. 2020, 38, 108. [Google Scholar] [CrossRef]
- Goyal, L.; Meric-Bernstam, F.; Hollebecque, A.; Morizane, C.; Valle, J.W.; Karasic, T.B.; Abrams, T.A.; Kelley, R.K.; Cassier, P.A.; Furuse, J.; et al. Updated results of the FOENIX-CCA2 trial: Efficacy and safety of futibatinib in intrahepatic cholangiocarcinoma (iCCA) harboring FGFR2 fusions/rearrangements. J. Clin. Oncol. 2022, 40, 4009. [Google Scholar] [CrossRef]
- Walden, D.; Eslinger, C.; Bekaii-Saab, T. Pemigatinib for adults with previously treated, locally advanced or metastatic cholangiocarcinoma with FGFR2 fusions/rearrangements. Ther. Adv. Gastroenterol. 2022, 15, 175628482211153. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Pemigatinib: First Approval. Drugs 2020, 80, 923–929. [Google Scholar] [CrossRef]
- Romero, D. Benefit from pemigatinib in cholangiocarcinoma. Nat. Rev. Clin. Oncol. 2020, 17, 337. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.C.C.; Koblish, H.; Wu, L.; Bowman, K.; Diamond, S.; DiMatteo, D.; Zhang, Y.; Hansbury, M.; Rupar, M.; Wen, X.; et al. INCB054828 (pemigatinib), a potent and selective inhibitor of fibroblast growth factor receptors 1, 2, and 3, displays activity against genetically defined tumor models. PLoS ONE 2020, 15, e0231877. [Google Scholar] [CrossRef] [Green Version]
- Subbiah, V.; Barve, M.; Iannotti, N.O.; Gutierrez, M.; Smith, D.C.; Roychowdhury, S.; Papadopoulos, K.P.; Mettu, N.; Edenfield, W.J.; Morgensztern, D.; et al. Abstract A078: FIGHT-101: A phase 1/2 study of pemigatinib, a highly selective fibroblast growth factor receptor (FGFR) inhibitor, as monotherapy and as combination therapy in patients with advanced malignancies. Mol. Cancer Ther. 2019, 18, A078. [Google Scholar] [CrossRef]
- Bekaii-Saab, T.S.; Valle, J.W.; Van Cutsem, E.; Rimassa, L.; Furuse, J.; Ioka, T.; Melisi, D.; Macarulla, T.; Bridgewater, J.; Wasan, H.; et al. FIGHT-302: First-line pemigatinib vs gemcitabine plus cisplatin for advanced cholangiocarcinoma with FGFR2 rearrangements. Futur. Oncol. 2020, 16, 2385–2399. [Google Scholar] [CrossRef] [PubMed]
- Mahipal, A.; Tella, S.H.; Kommalapati, A.; Anaya, D.; Kim, R. FGFR2 genomic aberrations: Achilles heel in the management of advanced cholangiocarcinoma. Cancer Treat. Rev. 2019, 78, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Silverman, I.M.; Hollebecque, A.; Friboulet, L.; Owens, S.; Newton, R.C.; Zhen, H.; Féliz, L.; Zecchetto, C.; Melisi, D.; Burn, T.C. Clinicogenomic Analysis of FGFR2-Rearranged Cholangiocarcinoma Identifies Correlates of Response and Mechanisms of Resistance to Pemigatinib. Cancer Discov. 2021, 11, 326–339. [Google Scholar] [CrossRef] [PubMed]
- Pearson, A.; Smyth, E.; Babina, I.S.; Herrera-Abreu, M.T.; Tarazona, N.; Peckitt, C.; Kilgour, E.; Smith, N.R.; Geh, C.; Rooney, C.; et al. High-Level Clonal FGFR Amplification and Response to FGFR Inhibition in a Translational Clinical Trial. Cancer Discov. 2016, 6, 838–851. [Google Scholar] [CrossRef] [Green Version]
- Goyal, L.; Saha, S.K.; Liu, L.Y.; Siravegna, G.; Leshchiner, I.; Ahronian, L.G.; Lennerz, J.K.; Vu, P.; Deshpande, V.; Kambadakone, A.; et al. Polyclonal Secondary FGFR2 Mutations Drive Acquired Resistance to FGFR Inhibition in Patients with FGFR2 Fusion–Positive Cholangiocarcinoma. Cancer Discov. 2017, 7, 252–263. [Google Scholar] [CrossRef] [Green Version]
- Cowell, J.K.; Qin, H.; Hu, T.; Wu, Q.; Bhole, A.; Ren, M. Mutation in the FGFR1 tyrosine kinase domain or inactivation of PTEN is associated with acquired resistance to FGFR inhibitors in FGFR1-driven leukemia/lymphomas. Int. J. Cancer 2017, 141, 1822–1829. [Google Scholar] [CrossRef] [Green Version]
- Datta, J.; Damodaran, S.; Parks, H.; Ocrainiciuc, C.; Miya, J.; Yu, L.; Gardner, E.P.; Samorodnitsky, E.; Wing, M.R.; Bhatt, D.; et al. Akt Activation Mediates Acquired Resistance to Fibroblast Growth Factor Receptor Inhibitor BGJ398. Mol. Cancer Ther. 2017, 16, 614–624. [Google Scholar] [CrossRef] [Green Version]
- Goyal, L.; Shi, L.; Liu, L.Y.; De La Cruz, F.F.; Lennerz, J.K.; Raghavan, S.; Leschiner, I.; Elagina, L.; Siravegna, G.; Ng, R.W.; et al. TAS-120 Overcomes Resistance to ATP-Competitive FGFR Inhibitors in Patients with FGFR2 Fusion–Positive Intrahepatic Cholangiocarcinoma. Cancer Discov. 2019, 9, 1064–1079. [Google Scholar] [CrossRef] [Green Version]
- Su, N.; Jin, M.; Chen, L. Role of FGF/FGFR signaling in skeletal development and homeostasis: Learning from mouse models. Bone Res. 2014, 2, 14003. [Google Scholar] [CrossRef] [Green Version]
- Lacouture, M.E.; Sibaud, V.; Anadkat, M.J.; Kaffenberger, B.; Leventhal, J.; Guindon, K.; Abou-Alfa, G. Dermatologic Adverse Events Associated with Selective Fibroblast Growth Factor Receptor Inhibitors: Overview, Prevention, and Management Guidelines. Oncologist 2021, 26, e316–e326. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chmiel, P.; Gęca, K.; Rawicz-Pruszyński, K.; Polkowski, W.P.; Skórzewska, M. FGFR Inhibitors in Cholangiocarcinoma—A Novel Yet Primary Approach: Where Do We Stand Now and Where to Head Next in Targeting This Axis? Cells 2022, 11, 3929. https://doi.org/10.3390/cells11233929
Chmiel P, Gęca K, Rawicz-Pruszyński K, Polkowski WP, Skórzewska M. FGFR Inhibitors in Cholangiocarcinoma—A Novel Yet Primary Approach: Where Do We Stand Now and Where to Head Next in Targeting This Axis? Cells. 2022; 11(23):3929. https://doi.org/10.3390/cells11233929
Chicago/Turabian StyleChmiel, Paulina, Katarzyna Gęca, Karol Rawicz-Pruszyński, Wojciech P. Polkowski, and Magdalena Skórzewska. 2022. "FGFR Inhibitors in Cholangiocarcinoma—A Novel Yet Primary Approach: Where Do We Stand Now and Where to Head Next in Targeting This Axis?" Cells 11, no. 23: 3929. https://doi.org/10.3390/cells11233929
APA StyleChmiel, P., Gęca, K., Rawicz-Pruszyński, K., Polkowski, W. P., & Skórzewska, M. (2022). FGFR Inhibitors in Cholangiocarcinoma—A Novel Yet Primary Approach: Where Do We Stand Now and Where to Head Next in Targeting This Axis? Cells, 11(23), 3929. https://doi.org/10.3390/cells11233929