1. Introduction
In dermatology, a youthful appearance is attained through treatments promoting skin rejuvenation. However, cutaneous ageing will occur by environmental and intrinsic physiological factors [
1]. To maintain its integrity and elasticity, this organ is especially good at regenerating itself at each layer: epidermis, dermis and hypodermis. Dermis decaying composition, structure and organization is the cause of wrinkles, skin sagging and other signs of skin ageing. In children and adults, it is rich in connective tissue, which is composed of fibroblasts and extracellular matrix (ECM) [
2]. The ECM is gel-like, and it is comprised largely of proteins, such as collagen, elastin and laminin, and associated with proteoglycans and glycosaminoglycans. Its composition is ideal to support cellular migration, proliferation and adhesion [
3,
4].
Upon ageing, the tissue draws mitochondrial dysfunction, deregulated nutrient sensing, epigenetic alterations, telomere attrition, genomic instability and altered intercellular communication [
5,
6]. These events lead to senescent cells and stem cell exhaustion. Fibroblasts produce less ECM while increasing the transcription of metalloproteinase (MMP) and decreasing tissue inhibitors of metalloproteinase synthesis (TIMP) [
7]. The low turnover of the ECM causes a loss of dermal elasticity.
Another reason for elasticity loss is damage to the ECM. Oxidative stress is a pivotal biochemical event inducing degradation and disorganization in the ECM [
1]. UV radiations are common initiators of reactive oxygen species (ROS) generation in skin, which cause cell and ECM damage, targeting the collagen network and elastin (elastosis) in a process called photoaging. In the skin, keratinocytes and fibroblasts are the leading producers of mitochondrial ROS; therefore, ageing-induced mitochondrial dysfunction also endangers fibroblasts and fibroblast-produced ECM.
During cutaneous ageing, senescent and radical-damaged cells trigger inflammation and erythema [
8,
9,
10,
11,
12,
13]. Inflamed cells produce growth factors, pro-inflammatory cytokines and chemokines, whose role is to chemoattract and activate cells of the innate immune system. On site, phagocytes and natural killers eliminate damaged cells and molecules. However, uncontrolled inflammation can produce counter-effective results. Tissue damage can be due to an overly stimulated innate immune system and an exaggerated production of chemokines and cytokines. This often occurs in older skin, as tissue-specific immune surveillance decreases with people’s age [
7].
The field of regenerative medicine is therefore in constant search of new antioxidant and immunomodulatory therapeutics for skin regeneration [
14,
15]. Rejuvenation therapies that have been investigated include direct applications of mesenchymal stem cells, raising quite a few safety issues [
16,
17]. A novel “cell-free” therapy with similar results concerns the use of MSC-derived extracellular vesicles [
18,
19,
20,
21,
22,
23,
24]. Vesicles of nanometric proportions (NVs) are a physiological part of complex intercellular signaling [
25] A double lipid layer surrounds a bioactive cargo loaded in the cell of origin and delivered in the recipient cell as a message. The outer layer mirrors the cell-type-specific membrane, while cargo molecules are cytosolic components, mainly nucleic acids and proteins, that trigger the recipient cells. Extracellular vesicles have been categorized based on their size [
26]. The most commonly studied are exosomes, vesicles of 50–150 nm in diameter originating from the cytosolic multi vesicular body (MVB) in mammalian cells [
27]. Exosomes have been investigated as diagnostic and therapeutic tools and as drug-delivery systems with cell-specific target capabilities [
28,
29,
30]. In a study by Xu et al. (2020), MSCs-NVs were subcutaneously injected in photoaged mice, resulting in lower ROS generation and increased cellular proliferation of fibroblasts [
31]. In recent years, investigators have discovered that NVs also play a role in vegetable communication.
Plant-derived NVs have been successfully isolated and characterized from several edible fruits and vegetables, including lemon [
32], strawberry [
33], grape [
34], ginger [
35] and broccoli [
36]. Interestingly, several investigations showed how plant-derived NVs can interact and influence mammalian cells, indicating the possibility of cross-kingdom communications [
37,
38,
39]. Several NVs have exhibited antioxidant and anti-inflammatory properties. An in vivo study by Deng et al. (2017) observed broccoli-derived NVs’ anti-inflammatory action by preventing dendritic cell activation and protecting mice against colitis [
36]. Citrus-derived NVs observed in vitro relieved MSCs from oxidative stress [
32], while in vivo they inhibited cancer cell proliferation and cell viability through the activation of TRAIL-mediated apoptosis and reduction of pro-angiogenic cytokines IL-6 and IL-8 [
40].
Apples, one of the most consumed fruits globally, are rich in polyphenols, whose exact composition is variety-dependent [
41,
42]. Polyphenols are anti-cancer, anti-inflammatory and antioxidant molecules [
43]. Thanks to their high content in polyphenols, and to the famous saying, apples are already perceived as a healthy fruit by the general population. Apple-derived nanovesicles (ADNVs) have been isolated and characterized. Their range is approximately around 130 nm in diameter. They have exhibited anti-inflammatory activity in canonically activated THP-1-derived macrophages by the regulation of miR-146a [
44]. On the strength of these findings, the research community is investigating plant-derived NV potential and exploiting them for several applications [
45].
This study aimed to investigate ADNVs’ effect on human cells involved in skin ageing and reparation. ADNVs were associated with well-known cosmetic delivery systems to investigate possible applicability issues, all the while looking for the molecular markers predictive of ADNVs’ effect on human cell lines.
2. Materials and Methods
2.1. EV Isolation and Quantification
Apple-derived nanovesicles (ADNVs) were isolated from DOP Golden delicious variety apples (Malus domestica sp.) cultivated in Val Di Non, Trentino (Italy). Briefly, three apples of approximately 250 g each were washed thoroughly and smashed into a pulp. The pulp was subjected to a series of centrifugations at increasing speed (650× g, 3000× g and 10,000× g). After each spin, the pellet was discharged and the supernatant retained. The supernatant fraction was then filtered with 0.22 μm syringe filters (GVS S.p.A, Rome, Italy) and centrifuged at 15,000× g with Ultracentrifuge Optima L-70 (Beckman Coulter Inc., Pasadena, CA, USA) type 70 Ti rotor to remove smaller particles and debris. The supernatant was further centrifuged at 110,000× g. The pellet, resulting from this last centrifugation, was resuspended in 1 mL of PBS (Thermo Fisher Scientific, Waltham, MA, USA) and was used as the ADNV fraction in all following experiments. The ADNV fraction was conserved at −80 °C until use.
2.2. ADNV Quantification and Size Characterization
Following isolation, ADNVs in PBS were quantified through tunable resistive pulse sensing (qNANO Gold, Izon Science Ltd., Cambridge, MA, USA). The analysis also provided data on size distribution of particles in the ADNV fraction. The nanopore (NP150, Izon Science Ltd., Cambridge, MA, USA) was stretched 49 mm wide using a digital caliber. The preparation of the instrument was performed with reagents provided by the manufacturer following the manufacturer’s instructions. Each sample was analyzed at two pressure points, 10 atm and 20 atm. During each measurement, the particle rate was maintained above 200 particles/min, and the total particle count surpassed 500 particles. Calibration particle (CPC200, Izon Science Ltd., Cambridge, MA, USA) measurements were taken at both 10 atm and 20 atm and were used for calibrating each sample measurement during data analysis. The protein content was also established through Bradford assay with Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA) following manufacturer’s instructions. Absorbance was measured at 570 nm using multilabel plate reader Victor 3 (Perkin Elmer, Milano, Italy).
2.3. SEM Imaging of ADNVs
To assess ADNV morphology, electronic imaging techniques were put into practice. After isolation, ADNVs were resuspended in 1 mL of 2% glutaraldehyde in phosphate buffer for fixation. The fixed fraction particles were allowed to gravity settle for 1 h on a clean cover slide at room temperature (25 °C). For observation under scanning electron microscopy (SEM), the sample was dehydrated by washes with increasing ethanol concentrations (50%, 70%, 80%, 90%, 100%). The coverslip was mounted on proper support and sputter-coated with gold following standard procedures. Imaging was performed under high vacuum condition using secondary electron detector with SEM Zeiss EVO 40 (Zeiss, Oberkochen, Germany).
2.4. MeHA and HY Release Assays
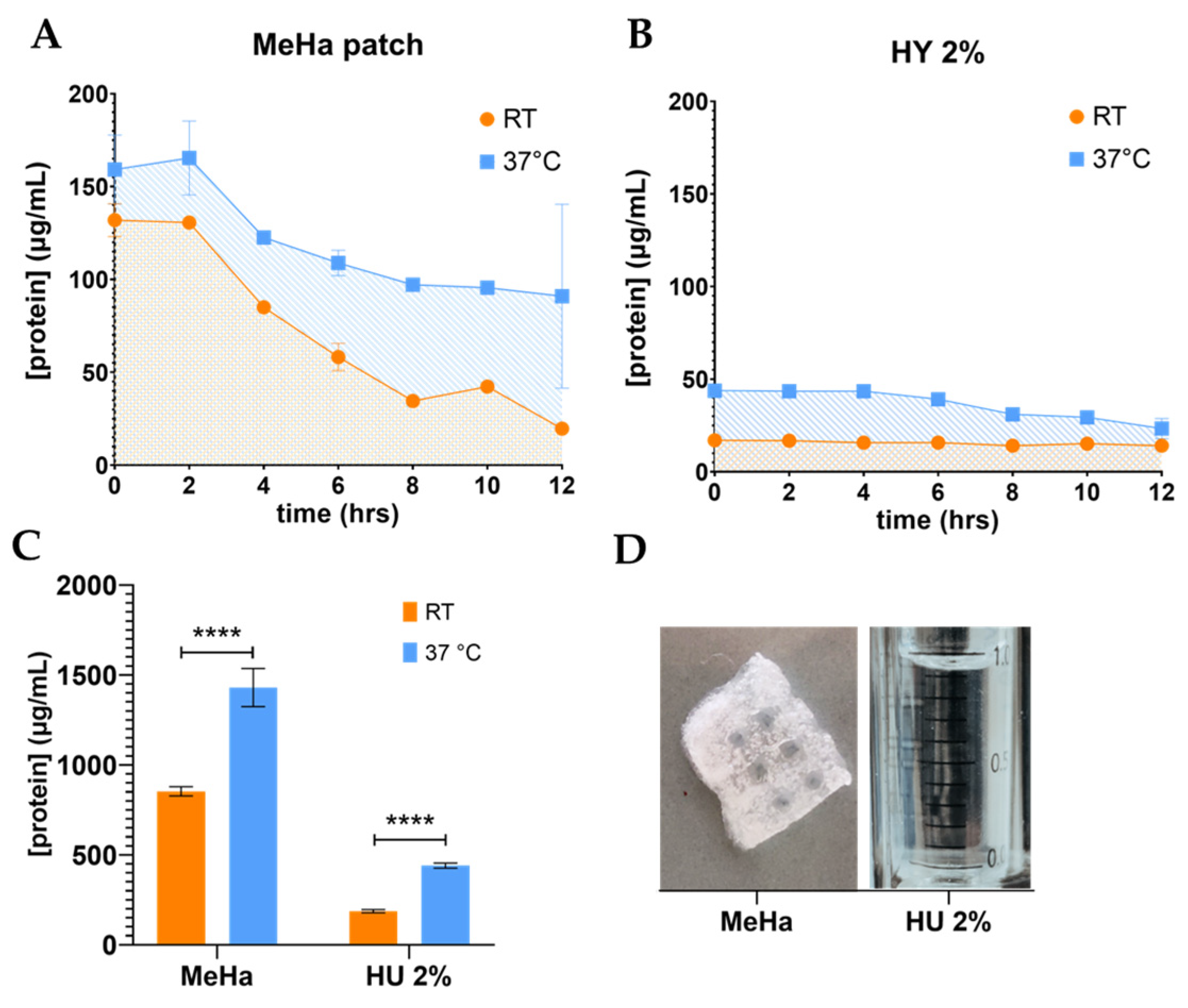
A gel-like solution of 2% hyaluronic acid (HA 2%) was used as the base for the addition of ADNVs. To create the final formulation, 20 μL of the ANDV fraction was mixed with 1 mL of Hyaluronic acid 2% gel for a final concentration of 500 μg/mL. 3D-printed methacrylate hyaluronic acid patches (MeHA) were re-hydrated with 500 μL of ADNV solution (500 μg/mL) [
46]. ADNVs’ release from the hyaluronic acid formulation and MeHA was monitored by measuring the protein concentration in the release reservoir. Briefly, 0.1 mL of hyaluronic acid w/ ADNVs was placed in a 48 multi-well and covered with 0.1 mL of PBS, which constituted the reservoir. Replicates of the experiment were kept at room temperature (25 °C) or at physiological temperature (37 °C). Every two hours over a period of 12 h, the reservoir was removed and replaced by 0.1 mL of fresh PBS. Each fraction was kept at 4 °C until all were collected. Similarly, a 0.5 × 0.5 mm square MeHA w/ ADNVs was placed in a 48 multi-well and covered with 0.1 mL of fresh PBS as reservoir. The analysis was carried on as described for HY w/ ADNVs. Protein quantification was performed on each reservoir fraction with Bradford assay, as described in the
Section 3.1. All experiments were performed in triplicates.
2.5. Cell Culture and Treatment
Human primary dermal fibroblasts were purchased from Resnova (Rome, Italy, from old donor). Fibroblasts were maintained as monolayer cultures in 75 mL flasks (Corning, VWR) in an incubator at 37 °C, 5% CO2 and 80% humidity. Cells were passaged every 72 h, dissociated with TrypLETM (Thermo Fisher Scientific, Waltham, MA, USA) and spun at 265× g. Medium was aspirated, and cells were resuspended in pre-warmed DMEM (Euroclone S.p.a, Milan, Italy) containing 10% FBS (Euroclone S.p.a, Milan, Italy). Once 70% confluency was reached, inflammation was induced in fibroblasts by the addition of TNFα (10 ng/mL) (Thermo Fisher Scientific, Waltham, MA, USA) to complete DMEM, for six hours. Cells cultured both w/ or w/o TNFα were treated with ADNVs for 24 h and also added to complete DMEM (protein concentration 200 μg/mL). For RT-qPCR experiment were used two ADNV concentrations: 1× (200 μg/mL) and 0.5× (100 μg/mL) for 48 h.
2.6. Migration Assay-Scratch Test
Cells were cultured to 100% confluency on a 6-well plate either w/ or w/o TNFα, as mentioned above. The scratch test was performed as described by Chun Chi Liang and Ann Y. Park et al. (2007) [
47]. Briefly, with a 200 μm tip, a straight scratch was performed in the middle of the well, removing cells in that area. Wells were then washed three times with PBS to remove debris, and new medium was added to the cells for treated and control conditions (w/ or w/o ADNVs, respectively). The scratches were marked to ensure that images were taken in the same position and, thus, cell migration progress could be monitored evenly. Images were acquired with EVOS cell imaging system inverted microscope (Thermo Fisher Scientific, Waltham, MA, USA) every 2 h for 12 h. For each condition, the experiment was performed in triplicate, each comprised of twelve replicates.
2.7. Cell Viability-MTT Assay
MTT assay was performed on NCTC L929 cells after ADNV incubation. Briefly, all samples were incubated with 1 mL of 0.5 mg/mL MTT [3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide] solution in PBS for 3 h at 37 °C. The MTT solution was then removed, and the formazan cell content was extracted with 0.5 mL of 10% DMSO. OD values were recorded at 570 nm for each sample in duplicate, using a multilabel plate reader (Victor 3, Perkin Elmer, Milano, Italy) [
48]. This experiment was performed three times independently.
2.8. Evaluation of Reactive Oxygen Species Production
MitoSOXTM fluorescent probe (Thermo Fisher Scientific, Waltham, MA, USA) superoxide indicator was used to assess the level of oxidative stress in fibroblasts w/ and w/o TNFα, w/ or w/o ADNVs. After culture and treatment, cells were washed with PBS. The assay was carried out according to manufacturer’s instructions. After incubation with the fluorescent dye, cells were resuspended in 100 μL of PBS. Red fluorescence was read with TaliTM image-based cytometer (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA), with 13 fields capture. Fluorescence unit (RFU) threshold was applied at 1000 RFU for all samples. All experiments were carried out in triplicate.
2.9. RNA Extraction and RT-qPCR
Fibroblasts cultured w/ or w/o TNFα and w/ or w/o ADNVs underwent lysis. Total RNA was extracted using Total RNA Purification Plus Kit (Norgen Biotek Corp., Thorold, ON, Canada) following manufacturer’s instructions for cells growing in monolayer. The extracted RNA quality and concentration was verified with NanoDrop One (Thermo Fisher Scientific, Waltham, MA, USA). RNA was then stored at −80 °C until use. First strand cDNA was synthetized from total RNA. For each sample, 1200 ng of RNA were reverse-transcribed with SensiFAST cDNA Synthesis Kit (Meridian Bioscience, Boston, MA, USA) in a final volume of 20 μL. Forward and reverse primers for genes of interest (GOI) and housekeeping genes (HKG) (
Table 1) were added to sample’s cDNA and SensiFAST SYBR No-ROX master mix (Meridian Bioscience, USA) according to manufacturer’s instructions. Real-time quantitative PCR was performed with GDS Rotor-Gene
® Q Thermocycler (Qiagen, London, UK). Thermal cycling and fluorescence detection were carried out as follows: PCR activation step at 95 °C for two minutes, followed by 40 repeating cycles of denaturation (95 °C for 5 s), annealing (60 °C for 10 s) and extension (70 °C for 20 s). A melting step was added at the end, with temperatures gradually increasing from 72 °C to 95 °C over five minutes. Three biological replicates were used for each sample, and three technical replicates were measured.
2.10. Total RNA Sequencing
Total RNA was quantitatively and qualitatively evaluated using NanoDrop 2000 (Thermo Fisher Scientific, Waltham, MA, USA) and Agilent Bioanalyzer 2100 (Agilent, Santa Clara, CA, USA), respectively. Libraries were generated with 1 μg of total RNA by TruSeq Sample Preparation RNA Kit (Illumina Inc., San Diego, CA, USA) according to the manufacturer’s protocol. The quality and quantity were analyzed with Bioanalyzer 2100 (Agilent Technologies, Berlin, Germany) on DNA 1000 Chip. The libraries were quantified using Qubit dsDNA BR Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA) on Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA). Sequencing was performed on Novaseq 6000 sequencer (Illumina Inc., San Diego, CA, USA) according to the manufacturer’s protocol. Illumina BCLFASTQ v2.20 software was used for de-multiplexing and the production of FASTQ files. Raw files were subsequently quality checked with FASTQC software (
http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc; accessed on 15 October 2022), and sequences with low quality score or including adaptor dimers were discarded from the analysis. The resulting set of selected reads were aligned onto the complete human genome using Splices Transcripts Alignment to a Reference algorithm STAR version 2.7.3 using hg38 Genome Assembly and Genecode.v35 as gene definition. The resulting mapped reads were used as input for feature count functions of Rsubread packages and used as gene counts for differential expression analysis using Deseq2 package [
49]. Reads for fibroblasts grown w/o TNFα were confronted with fibroblasts grown w/o TNFα and w/o ADNVs, while fibroblasts treated w/ TNFα were confronted with fibroblasts treated w/ both TNFα and ADNVs. Differentially expressed genes (DEGs) were selected for log
2(FC) < −1 or >1 and Adjp-value < 0.05.
2.11. Bioinformatic and Statistical Analysis
All results were represented as mean, with an indication of the standard error (SE) obtained from at least three independent replicas of the experiment. Significant difference between groups was determined by ANOVA analysis of variance and multiple comparisons post hoc Bonferroni test with Prism 8.03 software (GraphPad Software Inc, Boston, MA, USA). Statistical significance is labelled as follows: * p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001. Images from the migration test were analyzed with ImageJ software, the distance between cells was measured with the Measure tool, and the difference through time was calculated with Microsoft Excel. All datasets from RNA sequencing were analyzed with Qiagen Ingenuity Pathway Analysis (IPA) software core analysis. Through IPA, all DEGs were categorized in canonical pathways. IPA also predicted possible master regulators and downstream disease and functions. “Upstream regulators” were ranked based on their significance (p-value) and predicted state of activation/inhibition (z-Score). IPA assorts plausible downstream effects derived from (i) the activation/inhibition of upstream regulators and (ii) the DRGs expression rate in “Diseases and Functions”. Upstream and downstream predictions and DEGs were combined in functional networks. At each prediction, IPA attributes a z-Score, which was cut off at z-Score < −2 or >+2. Heatmaps were used to demonstrate the expression patterns of these DEGs. Data were cross-checked with KEGG database for interpretation and visual aid.
4. Discussion
Vegetable-derived nanovesicles are a novelty in the field of regenerative medicine. Nonetheless, much is yet to be discovered on the changes they induce in human cells. In this study, we focused on apple-derived nanovesicles as bioactive principles for cosmetic applications and their role in skin rejuvenation. RNA sequencing was used to comprehensively characterize the effect of ADNVs on human primary dermal fibroblasts, which are first responders during tissue regeneration.
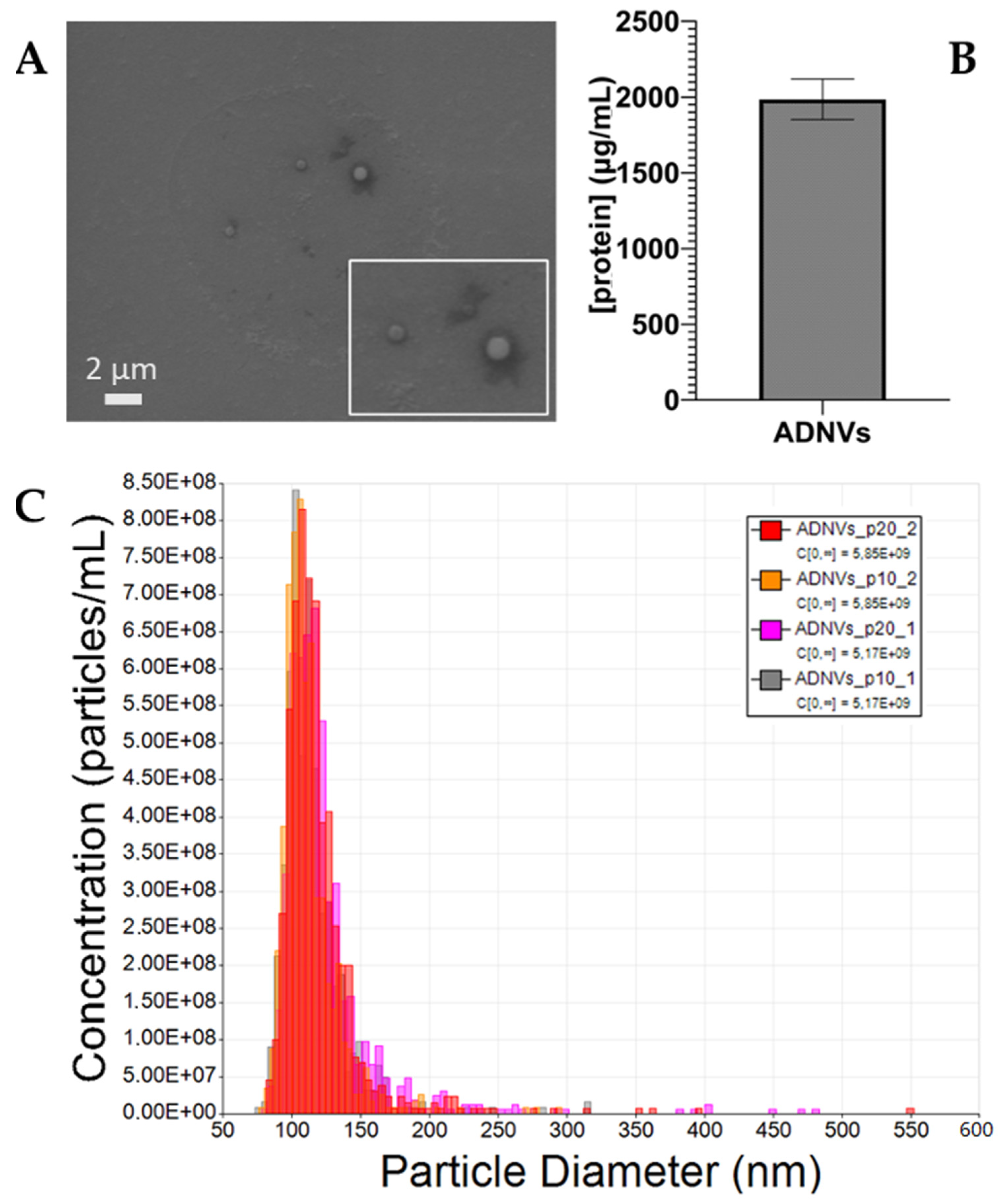
The Golden delicious apple variety was chosen for its popularity among producers and consumers. Here, ADNVs were physically characterized through TRPS and electron microscopy imaging. The findings were regarded comparable to previously published studies [
32,
34,
50,
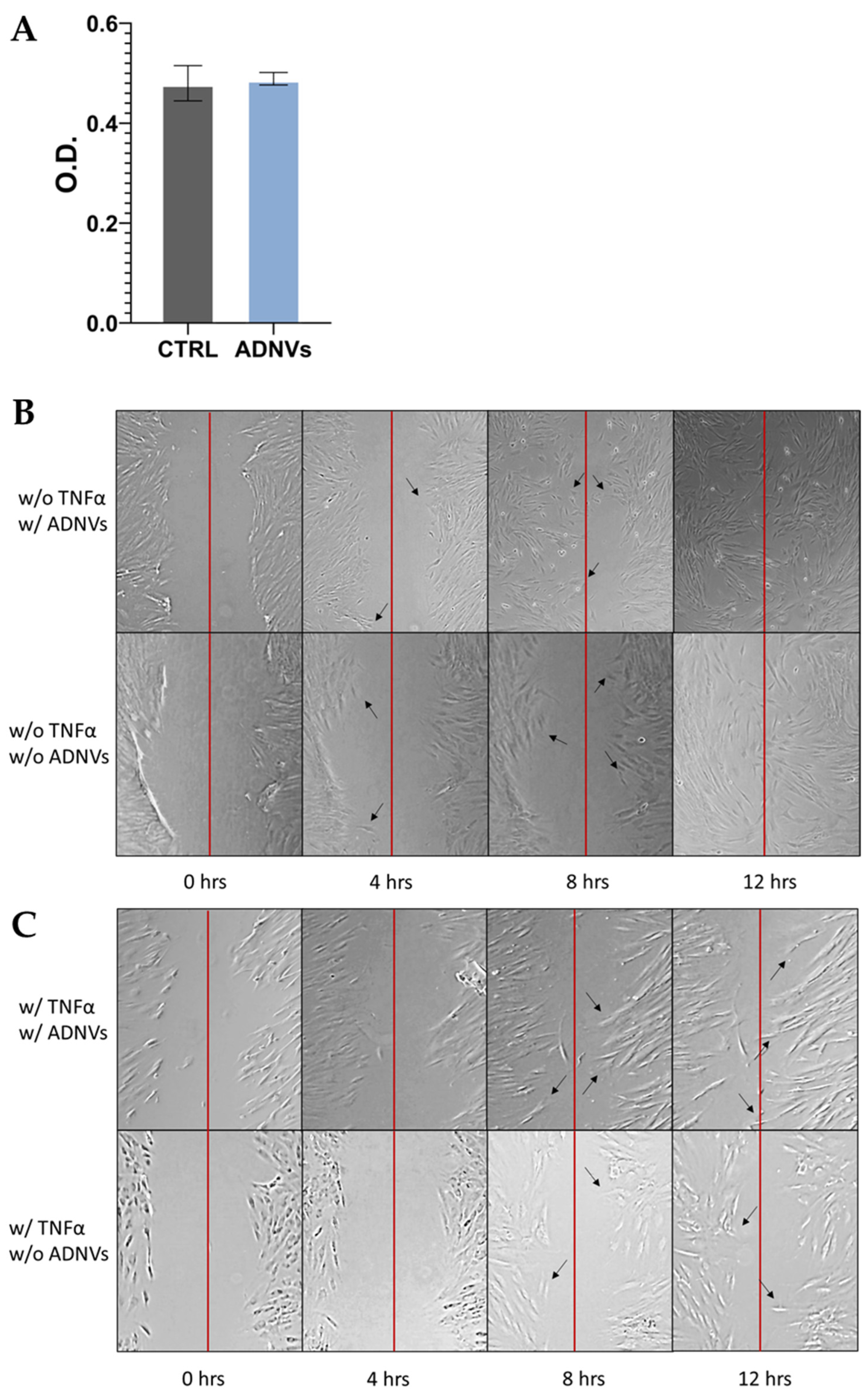
51]. The dimension and shape of ADNVs were parallel to other vegetable-derived nanovesicles and the ones already described for apples. Cytotoxicity assays were employed to provide data on ADNV harmfulness to human cells (
Figure 3A). It was established that ADNVs are not harmful to human cells by the results of cell vitality assay on both inflamed and non-inflamed fibroblasts, as per previously published data [
44].
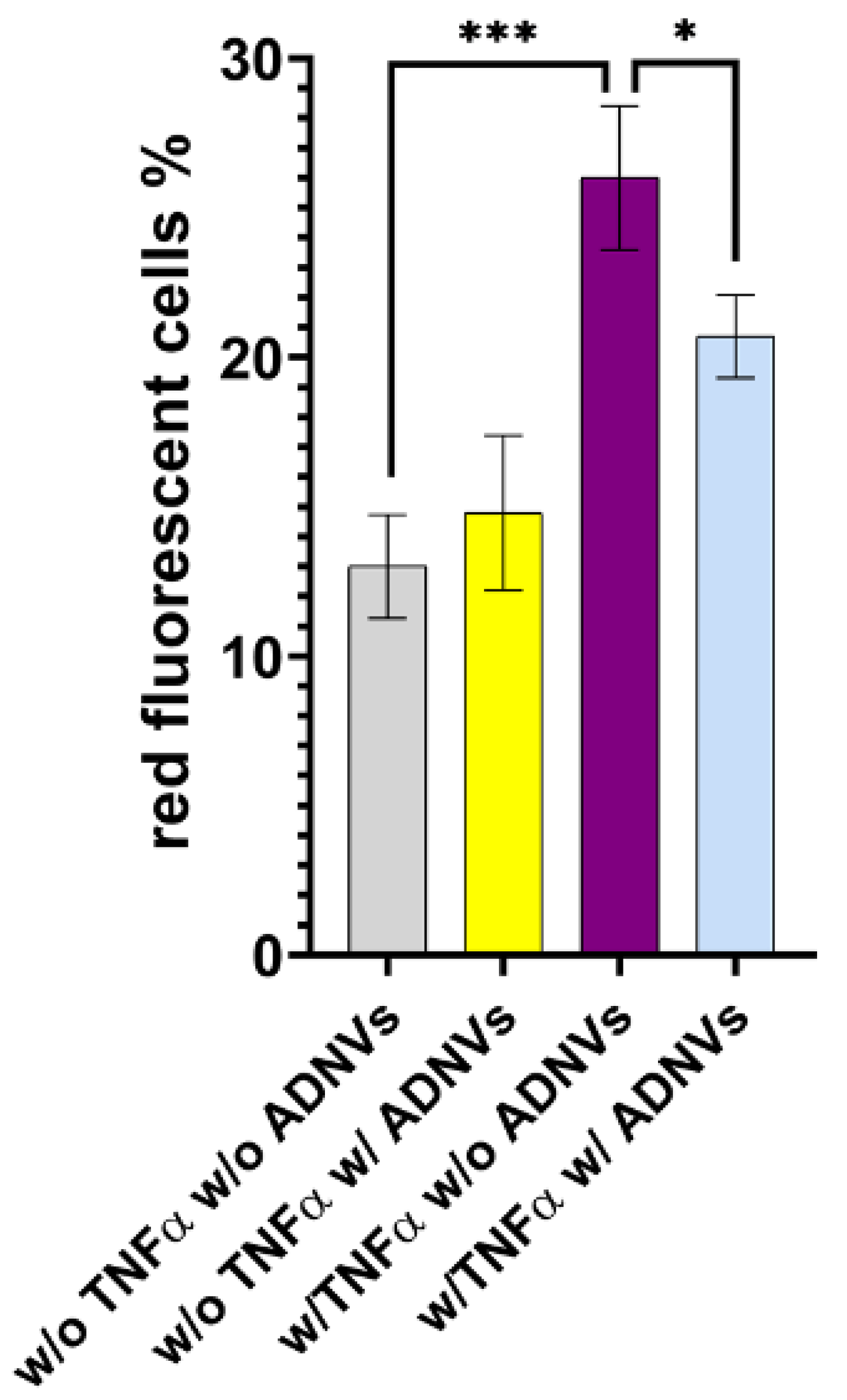
After establishing their safety, investigators explored ADNVs’ role in skin preservation. Two main events associated with intrinsic skin ageing are the decrease in the replicative ability of cells (cellular senescence) and the increased degradation of the ECM due to ROS activity [
8,
9]. Fibroblasts in the dermis are the greatest producers of collagen and other dermal proteins that constitute the ECM. Therefore, fibroblast migration and ECM production are indispensable for tissue regeneration and healing. This study established that ADNVs do not affect fibroblast migration significantly, both in a situation of inflammation and non-inflammation (
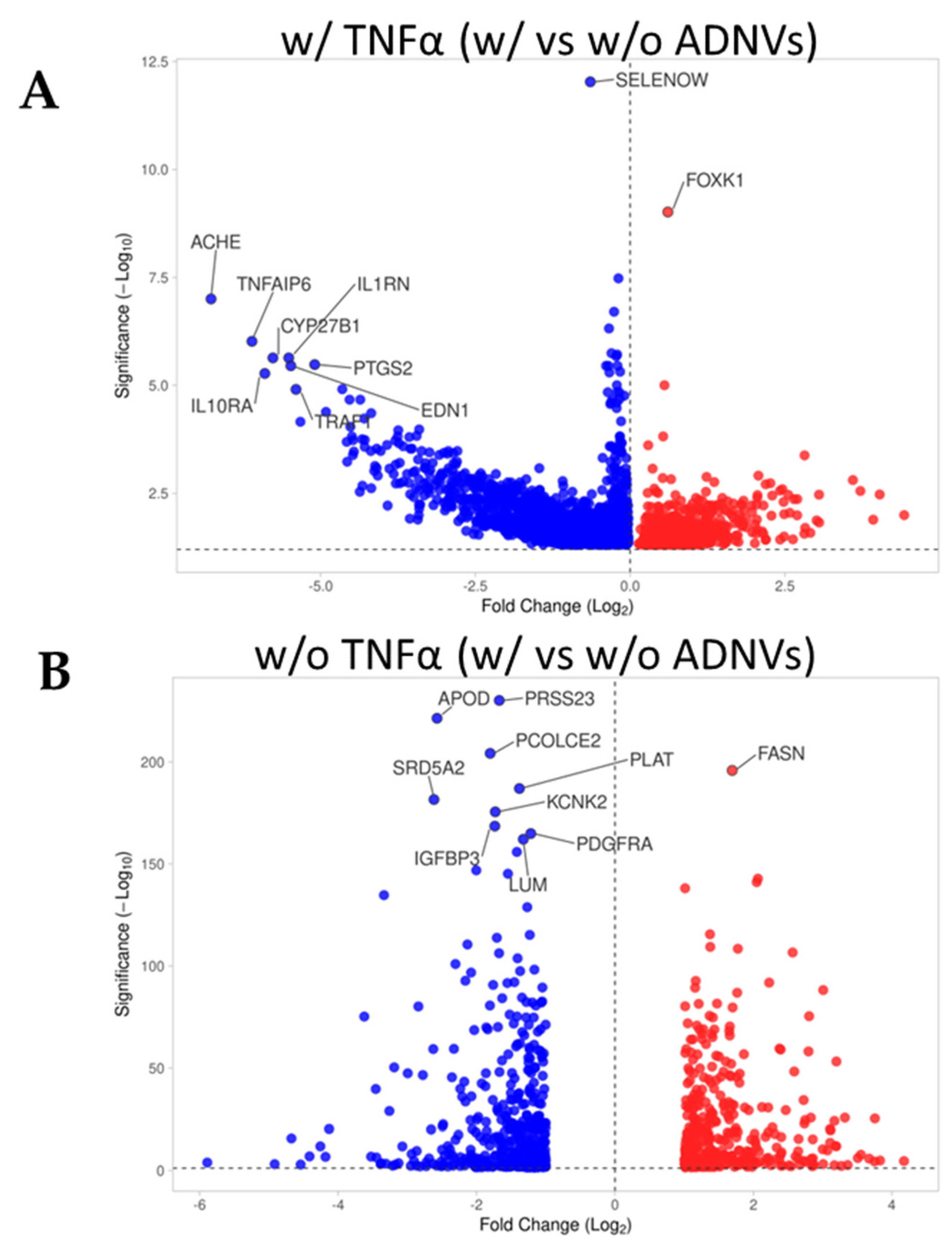
Figure 3B,C). However, RNA sequencing analysis has helped to describe the metabolic changes induced by ADNVs in fibroblasts w/ or w/o the influence of pro-inflammatory factors. Here, fibroblasts have responded more strongly to ADNVs when previously subjected to pro-inflammatory treatment (w/ TNFα) (
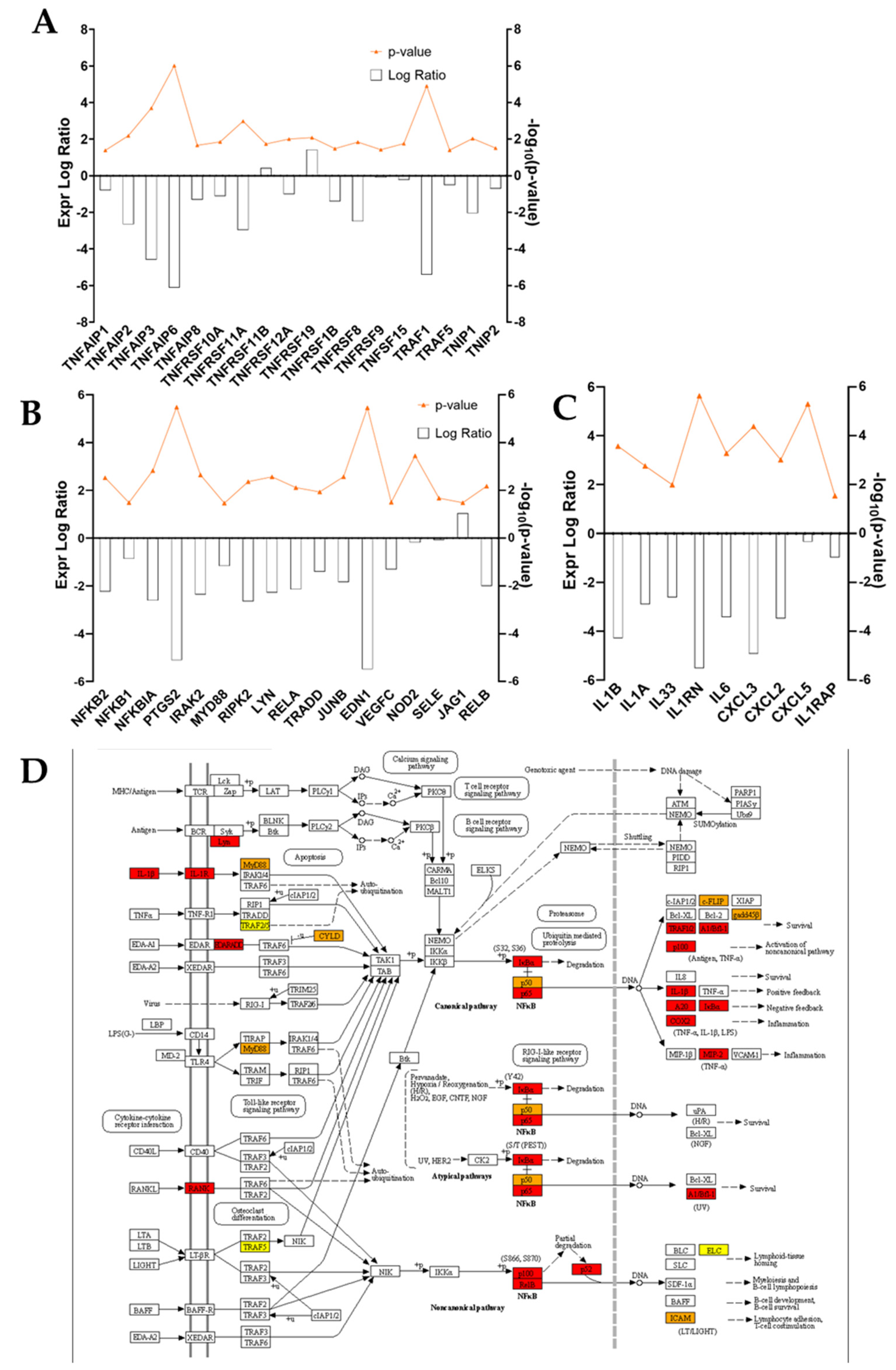
Figure 4). In such conditions, fibroblasts grown in an ADNV-spiked medium responded through the downregulation of genes involved in IL-1β signaling and TNFα signaling pathways (
Figure 6A–C). An abnormal expression or activity of pro-inflammatory cytokines and factors is expected in non-healing wounds, as described by several authors through time [
52,
53]. Thus, the inactivation of IL-1β and TNFα signaling pathways is evidence of lower inflammation in ADNV-treated fibroblasts [
54]. In physiological conditions, IL-1β and TNFα promote the activation of the canonical NF-κB signaling pathway: NF-κB transcription factor, translocating into the nucleus, increments positive feedback to IL-1, signaling the transcription of pro-inflammatory cytokines (IL-8 and IL-6) and chemokines (CXCL5 and CXCL4) [
9,
55,
56]. In accordance with this premise, proteins associated with NF-κB signaling were decreased by ADNV treatment (
Figure 6D). Prostaglandin-Endoperoxide Synthase 2 (PTGS2) is a crucial gene in prostaglandin E2 synthesis. One mechanism of skin erythema activation by ROS is through increased prostaglandin E2 levels [
10]. Thus, the downregulation of PTGS2 contributes to the overall picture of ADNVs’ anti-ageing effect. This is also confirmed by the diseases and functions ranking of IPA, which indicates an overall decrease in inflammatory response and inflammatory diseases (
Table 5).
Unsurprisingly, IL-1β and TNFα have been pinpointed as deactivated regulators of an ADNV-induced response in inflamed fibroblasts by IPA (
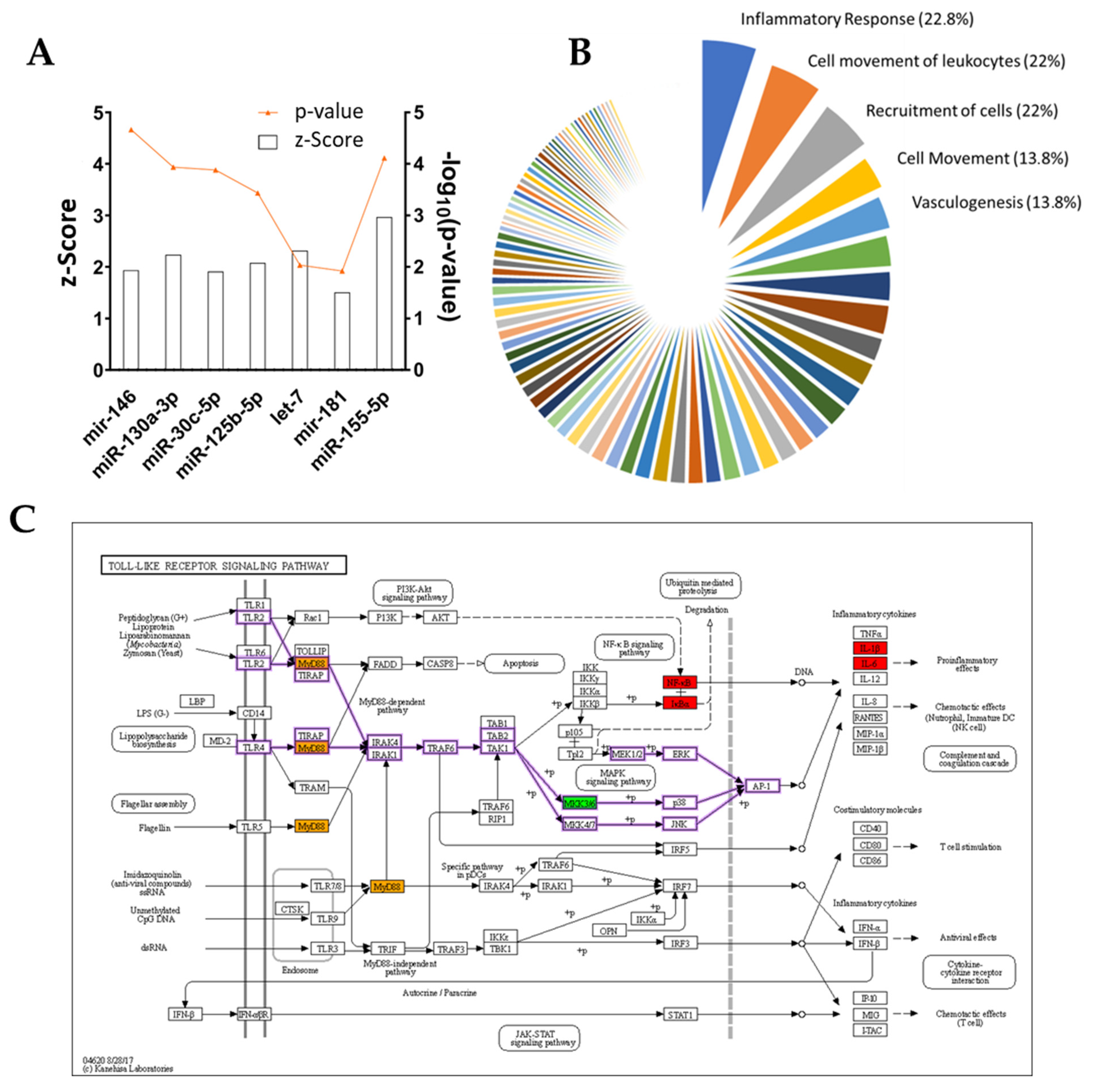
Table 4). A few miRNAs were also included in the list of possible upstream regulators, such as miR125b-5p and mir-146 family (
Figure 8A). As a previous study uncovered [
44], these miRNAs are key regulators for anti-inflammatory behavior in THP-1-derived macrophages treated with ADNVs. Their positive regulation in both fibroblasts and macrophages adds another layer of complexity to the extent that ADNVs can reduce pro-inflammatory signals in human cells. Moreover, IPA software recognized a prominent TLR4-regulated network in fibroblasts’ ADNV response (
Table 6). TRL4 belongs to the Toll-like receptors family and acts via MYD88, TIRAP and TRAF6 (factors involved in the IL-1 receptor and TNFα signaling pathways) to activate NF-κB signaling and mediate the innate immune response (
Figure 8C) [
57]. The network’s identified targets are all the DEGs involved in the inflammatory response (MYD88, PTGS2, IL1B and IL1A) as it is its final downstream function regulation. Interestingly, miRNA 146a and 125a are also involved in the negative regulation of TLR4, as described by several previous investigations [
58,
59,
60]. As such, the TLR4-regulated cellular response to ADNVs was considered one possible response mechanism of macrophage-derived THP-1 to ADNVs [
44].
In summary, all evidence points to a clear mechanism of action provoked by ADNVs in fibroblasts w/ TNFα, resulting in the negative regulation of NF-κB pathways and, overall, a suppressed pro-inflammatory response. Given the causal relationship between ROS-induced skin damage and inflammation, we can deduce that ADNV-induced reduction in inflammatory signaling could counteract the effects of oxidative-stress-induced ageing. Other predicted networks reinforce the connection between ADNVs action and the anti-inflammatory effect (
Figure 8B). Several of them target “Cell movement”, which plays a crucial role in regeneration; wound healing is given by a cells’ capacity to migrate and produce ECM, and the cells of the immune systems are required to migrate to the site of action. As determined by scratch assay, migration progress was not delayed by ADNV treatment (both w/ and w/o TNFα). However, to establish a quantitative significant difference between treated and control, a more extended study needs to be conducted.
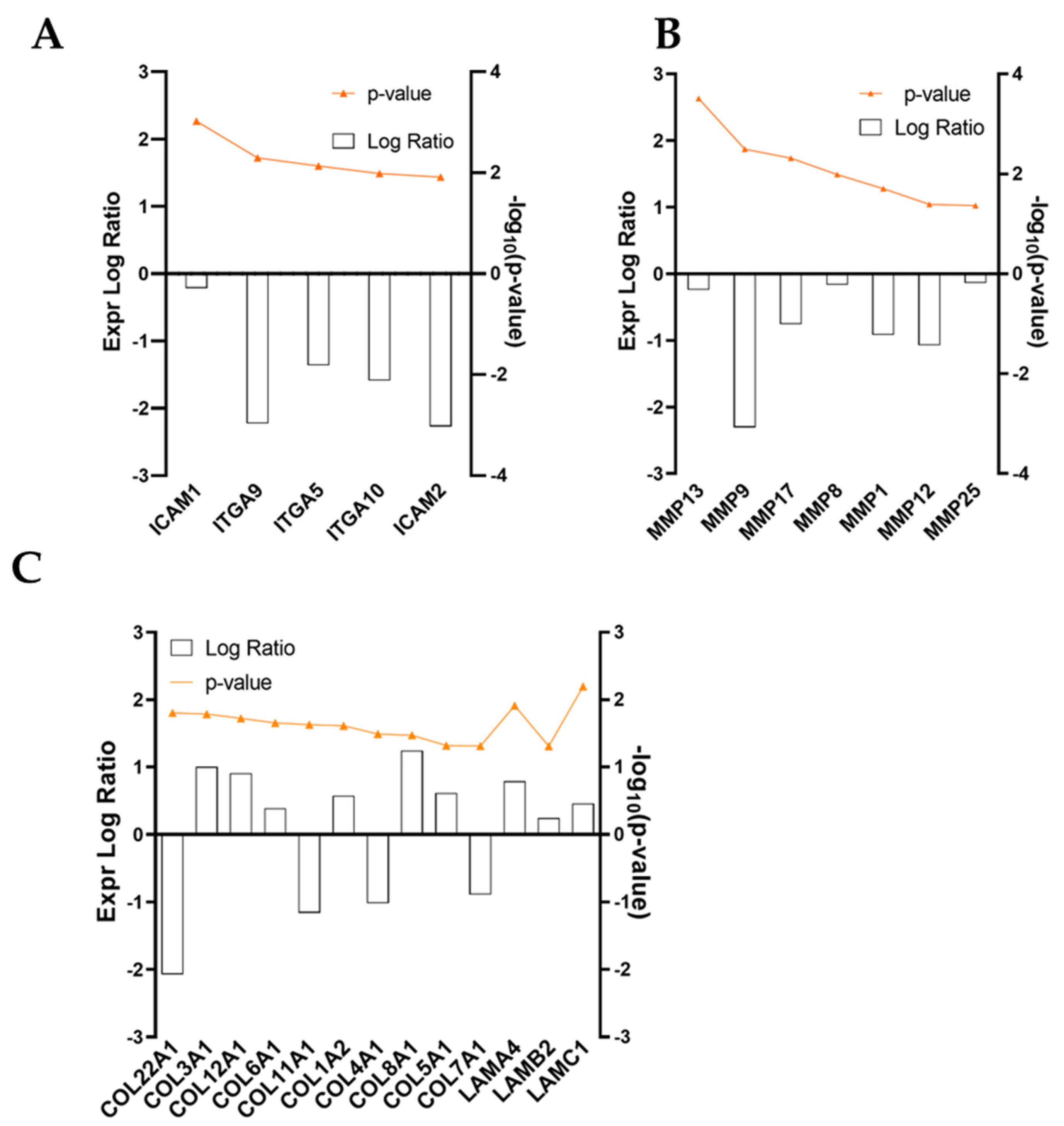
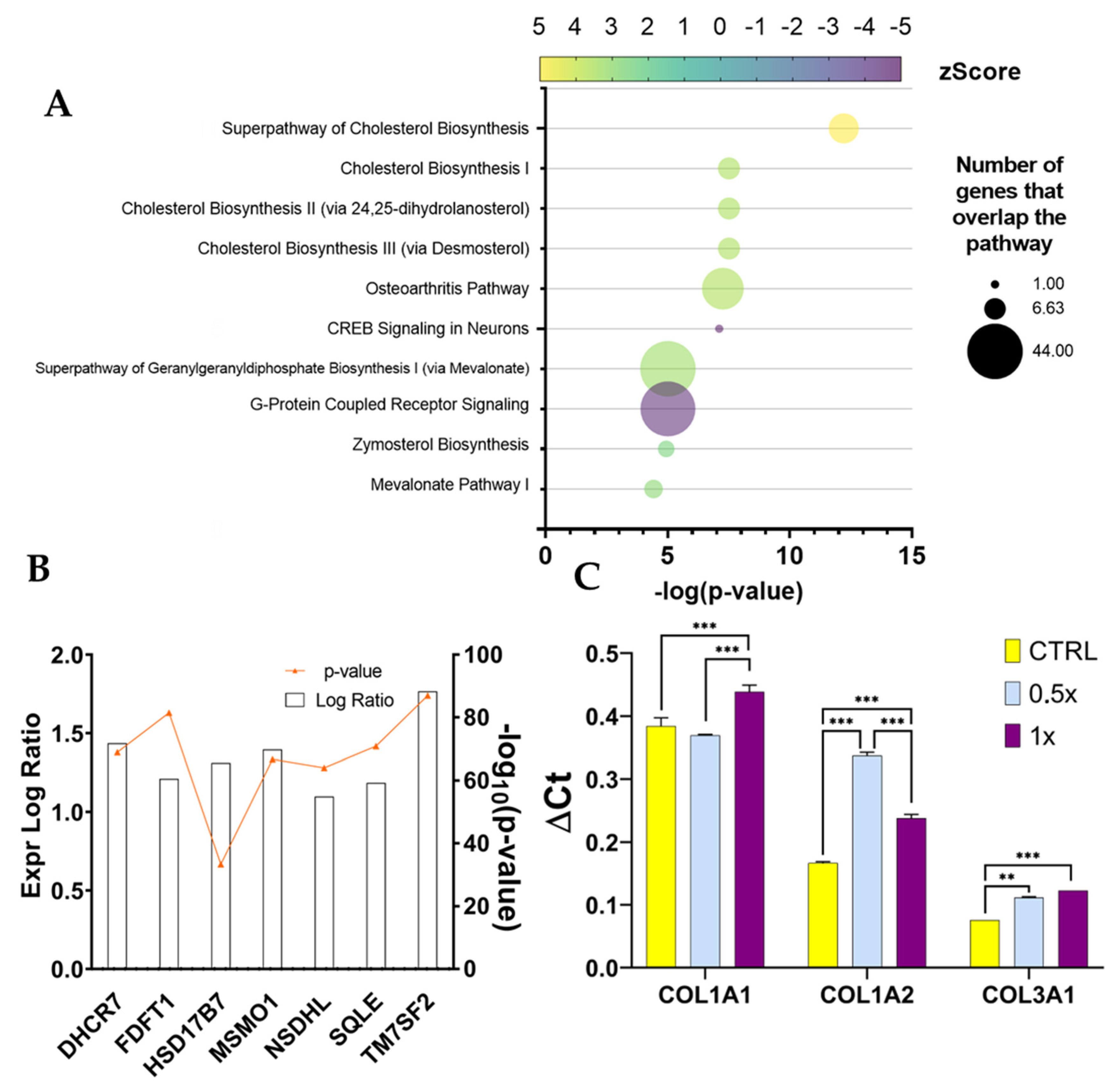
Aside from molding the inflammation response, ADNVs’ effect has been surveyed concerning the changes in ECM production by fibroblasts w/ TNFα. Collagen fibers are structural proteins that play critical roles in providing tensile strength to the skin [
17]. However, collagen is a frequent target of modifications by ROS due to its slow turnover rates [
61]. For this reason, monitoring its production rate is essential for assessing the ability of fibroblasts to supply new matrices. Type I collagen is most abundant in the dermal ECM associated with type III collagen. Type IV is also involved in skin health, as it is found in the basal lamina. This investigation observed an ADNV-induced increase in collagen type I α2 and collagen type III chain α1, contrasted by a decrease in collagen type I α1 chain and collagen type IV α1 chain (
Figure 5C). In addition to an accelerated matrix turnover, ADNVs promote mitochondrial fitness in inflamed fibroblasts by decreasing oxidative stress and ROS production (
Figure 10). These preliminary data indicate the role of ADNVs in the preservation and maintenance of the ECM.
Under stressful conditions, activated NF-κB in dermal fibroblasts also represses collagen synthesis. This is achieved by increasing the MMP/TIMP balance [
8]. MMPs are extracellular endopeptidases that differ greatly in substrate specificity and, thus, are collectively capable of breaking down several components of the ECM [
62]. Thus, NF-κB signaling plays a well-established and essential role in ECM maintenance and, consequentially, in photoaging and intrinsic ageing. Indeed, MMP1, MMP8 and MMP9 are downregulated by ADNVs (
Figure 5B). Metalloproteinase 1 and 8 both target interstitial collagen type I, II and III, while Metalloproteinase 9 is specialized in collagen type IV and V. Thus, together they would be able to break down and remodel the ECM with significant changes. Furthermore, MMPs have a part in other pathophysiological processes; particularly, MMP9 is considered a cancer biomarker [
63,
64].
In all, it is possible to say that the repression of the NF-κB pathway, in addition to the upregulation of collagen chains, laminin chains and the downregulation of pivotal MMPs, poses positive premises for ADNV treatment to induce ECM restoration. Given the centrality of its role, collagen synthesis was chosen as a marker to establish a correlation between quantity and effect in ADNV treatment. While COL3A1 expression is linearly dose-dependent, COL1A2 expression tendency is in accordance with the hormetic model. The lower dosage (100 μg/mL) stimulates COL1A2 expression in a stronger manner compared to higher dosage (200 μg/mL). However, higher dosage still stimulates a significant response compared to control conditions. Thus, collagen chain I a2 could be one of the most reliable markers for ADNV activity moving forward.
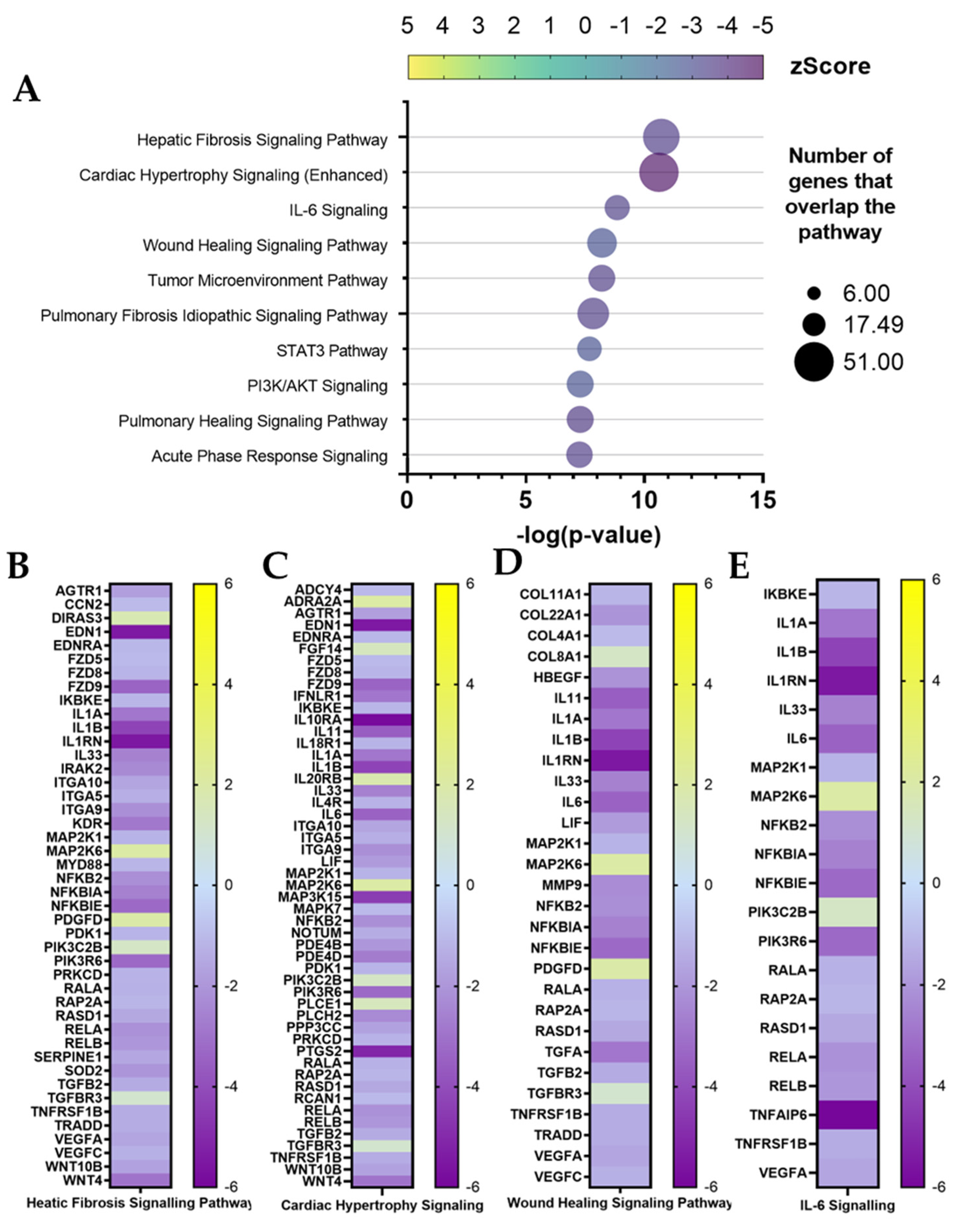
Another consideration can be made starting from RNA-seq analysis data. The inactivation of canonical pathways for fibrosis-related pathologies could indicate a diminished fibrotic healing mechanism by ADNV treatment (
Figure 7A–C). ICAM1 is considered an indicator of fibrosis-inducing fibroblasts [
65] as it can interact with integrins presented by infiltrated leukocytes. The interactions result in the activation of both fibroblasts and leukocytes, increasing the fibrotic response and cytokine IL-6 and IL-8 production. In this work, ICAM1, IL-6 and IL-8 were found downregulated by ADNVs in fibroblasts w/ TNFα, which could partially explain the results of the canonical pathway.
As already mentioned, fibroblasts grown in normal conditions (w/o TNFα) did not react as strongly to ADNVs. The results point to an increase in cholesterol biosynthesis tied to the expression of a handful of genes coding for the expression of cholesterol biosynthesis enzymes (
Figure 9A). In the literature, the stimulation of cholesterol biosynthesis in dermal fibroblasts is loosely associated with bio-protection. An in vitro study by Chee et al. (2021) on naked mole-rat-derived cells linked cholesterol metabolism inhibition to increased oxidative stress [
66]. However, data are scarce on cholesterol’s role in skin-derived fibroblasts. The master regulator identified by IPA, MAP2K5 (
Table 7), is also involved in lipid metabolism regulation, together with ERK5 [
67]. MAP2K5 activates MAPK7 that, in combination with MMP9, is associated with positive tumor-inducing signaling in cancer patients [
63,
68]. However, in healthy cells, MAPK2K5 is involved in several cellular processes, such as proliferation, differentiation, transcription regulation and development. IPA has also predicted the inhibition of WNT3A, involved in the canonical WNT signaling pathway that results in the activation of transcription factors of the TCF/LEF family. Overall, significant data are scarce in this dataset (w/o TNFα), and as such, the authors were not able to determine a significant and valid mechanism of function of ADNVs in this setting.
Lastly, this work found it appropriate to investigate ADNVs’ applicability in cosmetic formulations and medical dressing. Hyaluronic acid is a natural polysaccharide, which has been used in various forms and with numerous functionalization [
69,
70,
71,
72,
73]. From topical applications to drug delivery, it has been adopted for biomedical and cosmetic solutions. Alone, it has demonstrated pharmacological activity against inflammation [
74] and promising results in anti-ageing [
75]. In the right formulation, it can easily encapsulate nanometric particles, and it is widely used for controlled drug release [
46,
76]. Therefore, HA was chosen to be combined with ADNVs to focus on and increase each other’s anti-inflammatory activities. In both hydrogel (HY 2%) and mesh form (MeHA patches), ADNVs were successfully encapsulated and released (
Figure 2A–D). Therefore, they are both valid alternatives, although they differ in release rates. In conclusion, this solution can bring ADNVs to the site where tissue regeneration is needed to maximize their potential.







 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}