1. Introduction
MicroRNA (miRNA) is a single-stranded small molecule RNA with a length of about 19–25 nucleotides that is encoded by endogenous genes [
1,
2]. MiRNAs are linked to and play a crucial part in many vital human body processes, such as cell proliferation, differentiation, immunity, and metabolism [
3]. As a result, miRNAs have received increased attention, particularly in the field of associations between miRNAs and complex human diseases. Overexpression and downregulation of miRNA expression in humans have been linked to a variety of complex diseases, according to research [
4,
5]. Upregulation of miR-17-5p expression, for example, has a greater effect on pancreatic cancer cell proliferation and significantly increases the number of invading cells [
6]. When compared to normal breast tissue, abnormal expression of miRNAs such as mir-125b, mir-145, mir-21, and mir-155 causes human breast cancer [
7]. Cressatti et al. [
8] discovered that miR-153 and miR-223 could be used as biomarkers for Parkinson’s disease (PD) diagnosis through paired regulation of
-synuclein. MiR-34, miR-124a, -146, miR-187, miR-199a-5p, miR-203, miR-210, and miR-383 dysregulation all have a negative impact on pancreatic
-cell viability and function, which leads to uncontrolled proliferation of insulin-secreting cells and the development of diabetes [
9,
10]. In conclusion, miRNAs have been shown to be inextricably linked to the emergence of many human complex diseases, making the prediction of potential miRNA-disease association (MDA) a promising area of research. It can help researchers comprehend the pathological mechanisms of complex diseases, which can be beneficial in both the treatment and diagnosis of complex diseases.
Traditional biological wet experiments, such as anchored polymerase chain reaction and reverse transcription polymerase chain reaction, were used in the early years to identify the relationship between miRNAs and diseases, but they all have drawbacks such as complicated experiments, long time periods, and high costs [
11,
12,
13]. Several studies in the field of bioinformatics have been developed in recent years, such as drug–drug interactions [
14], drug–target interactions [
15], lncRNA–disease association prediction [
16], and lncRNA–miRNA interaction [
17]. Each of these studies has added to our understanding of computational approaches for predicting miRNA–disease connections. Many superior computational methods for predicting potential miRNA–disease associations have been proposed as more biological data sets have been collected, which not only saves significant money and time but also provides researchers with a new perspective to further validate the predicted potential associations. These MDA prediction computational approaches can be roughly categorized into three categories [
18]: machine learning-based prediction models, deep learning-based prediction models, and matrix transformation-based prediction models.
Machine learning has been widely applied in all areas, and numerous machine learning models for predicting MDA have produced positive results. As there are not enough known miRNA–disease connections, existing prediction models perform poorly, Zhou et al. [
19] presented a new model combining gradient boosting decision tree and logistic regression (GBDT-LR) to rank miRNA candidates for diseases. The model can extract features and then score them using logistic regression. Peng et al. [
20] proposed a new prediction model called Ensemble of Kernel Ridge Regression-based MiRNA-Disease Association prediction (EKRRMDA), which used KRR to build two classifiers in miRNA space and disease space, respectively, and combined them with ensemble learning to improve model prediction accuracy. Liu et al. [
21] created a computational model for the SMALF by learning potential features from the original miRNA–disease association matrix and then predicting unknown miRNA–disease associations using XGBoost. Tang et al. [
22] developed an ensemble learning method (PMDFI) based on higher-order feature interactions to predict potential miRNA–disease associations. It uses stacked autoencoders to learn higher-order features from the similarity matrix and then uses an integrated model combining multiple random forests with logistic regression to predict an association. Liu et al. [
23] proposed an autoencoder-based deep forest ensemble learning model (DFELMDA), which was further validated through case studies of colon, breast, and lung tumors with varying disease types. Both PMDFI and DFELMDA use automatic encoders, but as they do not consider graph structure information, they cannot learn the miRNA and disease feature representation well. Although machine learning-based methods have demonstrated good performance, they typically require domain knowledge to build sample features.
With the advent of Deep Learning, many methods of end-to-end computing have been developed, and this novel prediction method predicts better than earlier traditional machine learning methods. Xuan et al. [
24] developed CNNMDA, a deep learning method that uses two convolutional neural networks to efficiently learn the potential relationship between miRNAs and diseases (CNN). Li et al. [
25] created a GAEMDA model that takes miRNA and disease similarity as feature information, aggregates it using a graph neural network-based encoder to generate a low-dimensional representation of the nodes, and finally predicts it using a bilinear decoder. Zhou et al. [
26] proposed a deep self-coding multicore learning approach (DAEMKL) the following year, which uses multicore learning to build miRNA-disease heterogeneous networks and then uses regression models to learn their feature representations. Li et al. [
27] designed a computational framework based on graph attention network fusion of multi-source information (GATMDA). It utilized the graph attention network to aggregate information from neighbors with different weights to extract nonlinear features of diseases and miRNAs, and then predicted MDA by efficiently fusing linear and nonlinear features of diseases and miRNAs through a random forest algorithm. Han et al. [
28] proposed that LAGCN build a heterogeneous network by integrating miRNA similarity, disease similarity, and miRNA-disease association information, and then use the attention mechanism to synthesize multiple CNNs to learn miRNA and disease embedding. Although deep learning-based methods can learn feature representations automatically and improve model prediction performance to some extent, they require a large number of training samples and do not incorporate graph structure information, making it difficult to capture neighborhood information in the network.
Furthermore, in recent years, several MDA prediction algorithms based on matrix transformation have appeared. Yu et al. [
29] proposed a prediction model based on matrix completion and label propagation (MCLPMDA). It used matrix completion to reconstruct a new miRNA and disease similarity matrix based on the miRNA-disease association matrix, and then used the label propagation algorithm to predict MDA. Gao et al. [
30] proposed the Nearest Profile-based Collaborative Matrix Factorization (NPCMF) algorithm, which uses L2,1-norm to complete the unknown association, using miRNA and disease nearest neighbor information to construct similarity functions and thus find new MDAs. Chen et al. [
31] proposed the neighborhood constraint matrix completion algorithm (NCMCMDA), which combined neighborhood constraints with matrix completion for assisted prediction before transforming the prediction task into an optimization problem that could be solved by a rapid iterative algorithm. Yin et al. [
32] created a new computational model called Logistic Weighted Profile-based Collaborative Matrix Factorization by combining two methods, weighted profile and collaborative matrix factorization (LWPCMF). The findings show that LWPCMF can accurately predict potential MDA. Although the matrix transformation-based method overcomes the problem of feature representation using vectors in high-dimensional space, its results are highly dependent on the initial solution selection, and it often fails to converge, which is time-consuming.
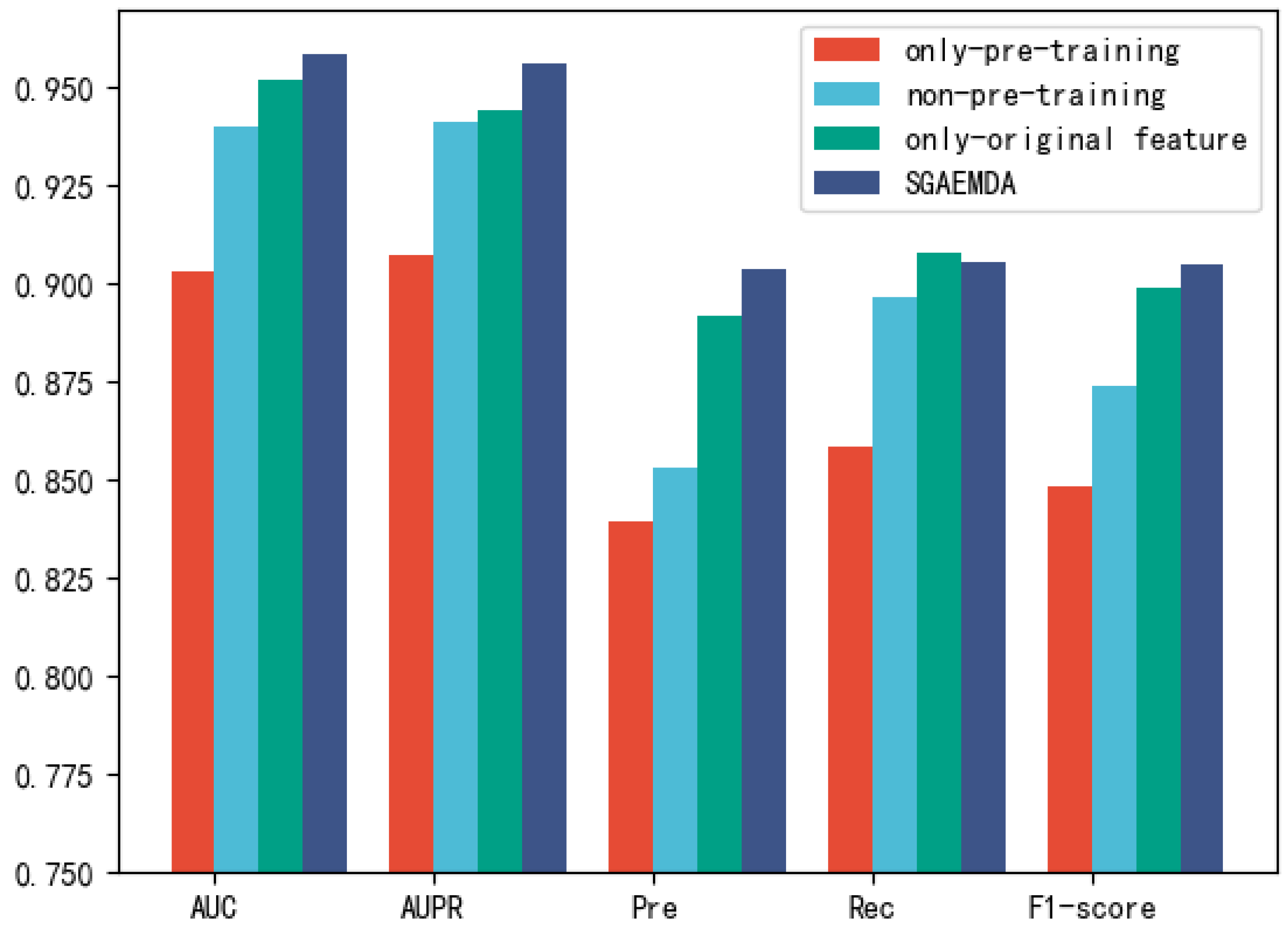
Although the models presented above predicted MDA well, they do have certain limitations. In recent years, autoencoders have been widely used in various fields [
33,
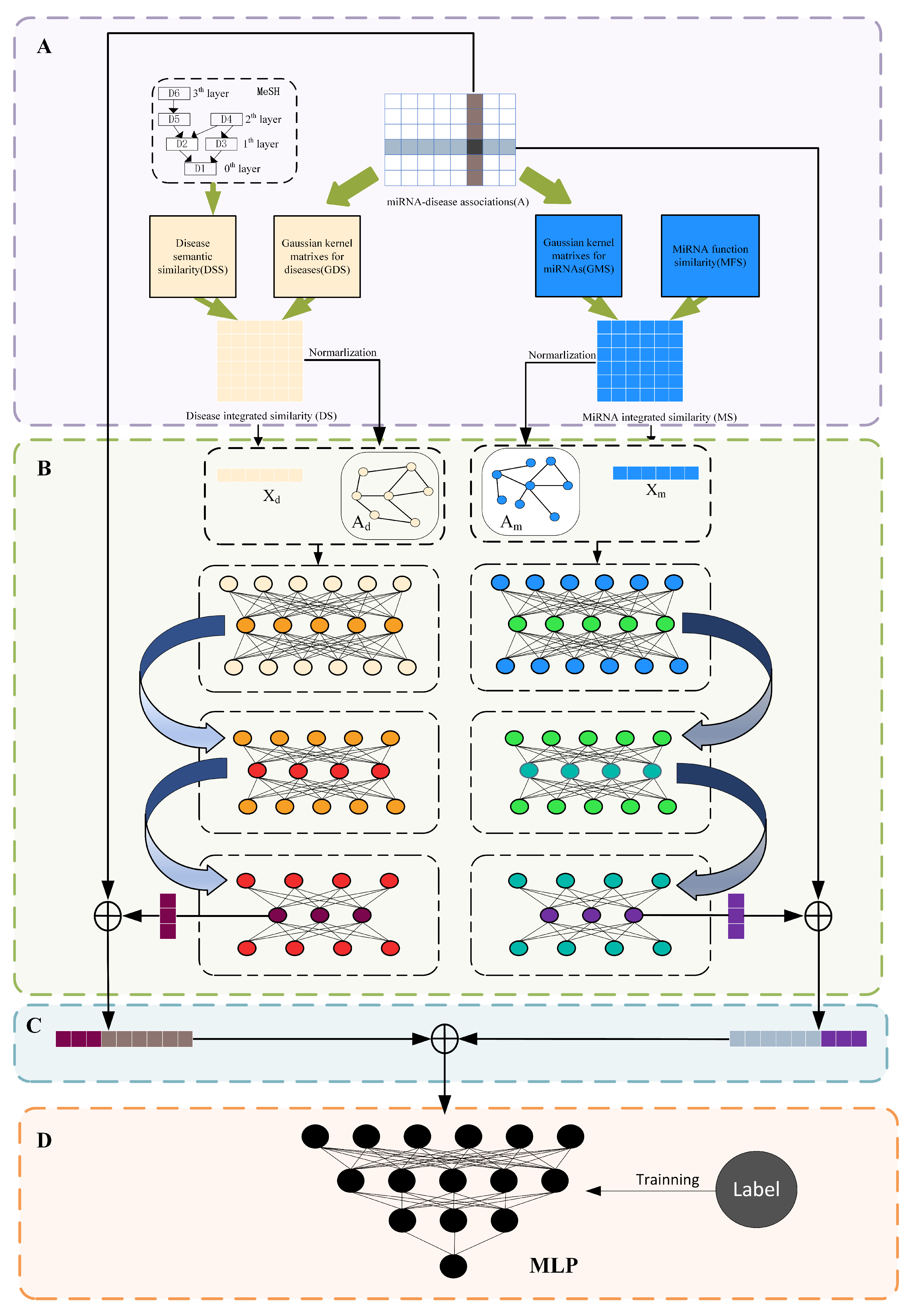
34] to efficiently learn the feature representation of miRNAs and diseases without losing the graph structure topology information, we propose a stacked graph autoencoder-based miRNA-disease association prediction algorithm (SGAEMDA), as shown in
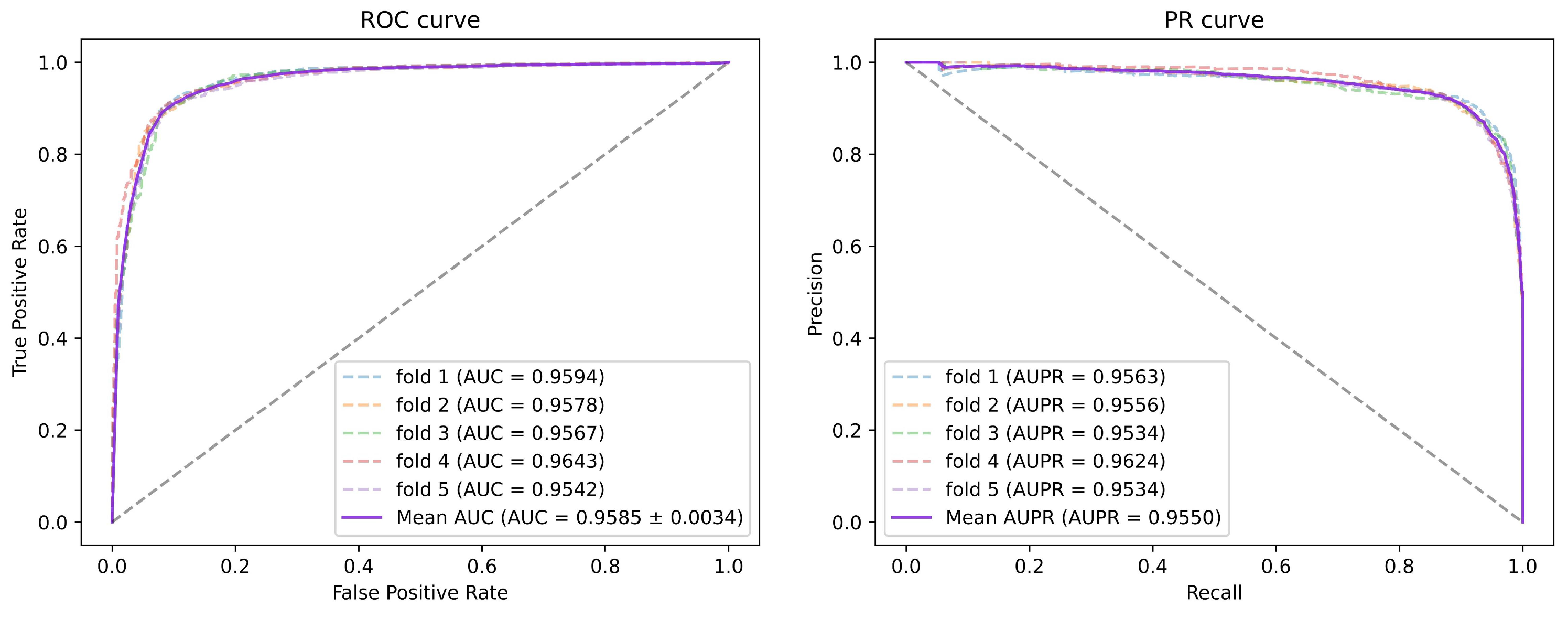
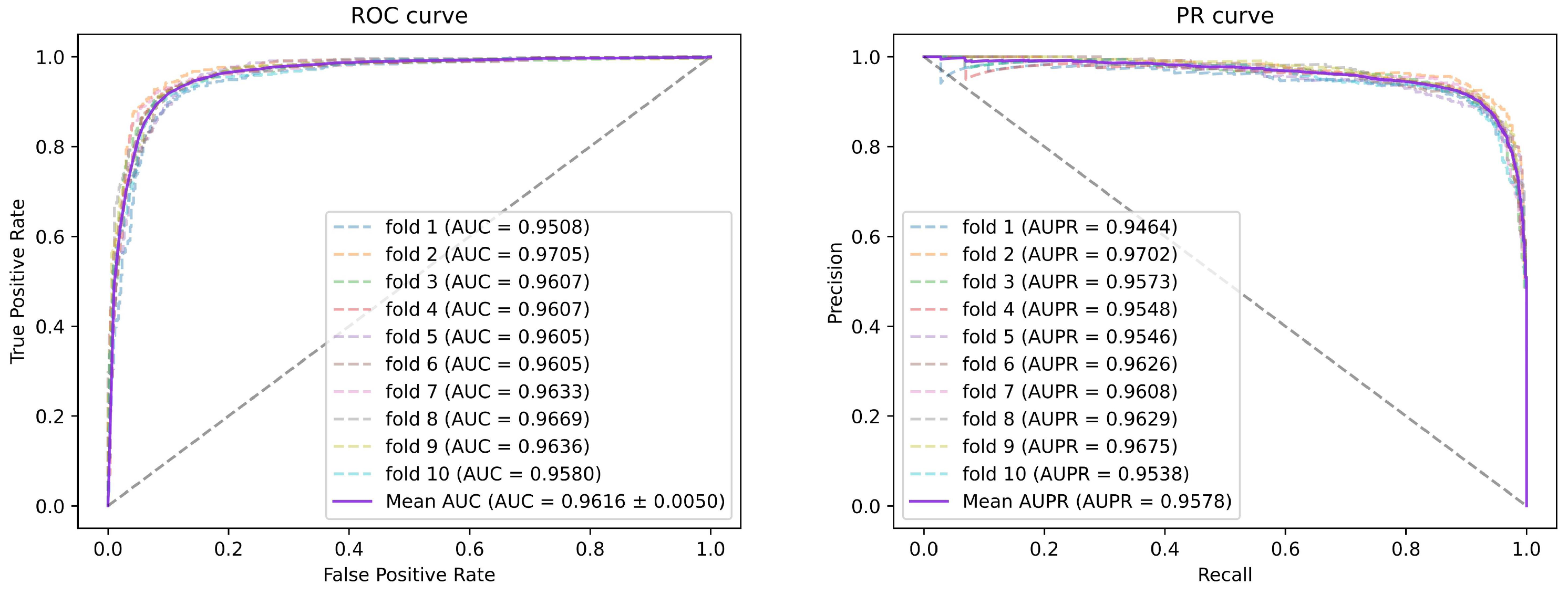
Figure 1. All miRNA features were then concatenated with disease features as miRNA-disease pair features. We employed 5-fold and 10-fold cross-validation to evaluate the prediction performance of our method. As a consequence, the AUCs of SGAEMDA in 5-fold and 10-fold cross-validation were 0.9585 and 0.9616, respectively, much higher than the other baseline methods. In addition, to demonstrate SGAEMDA’s performance, we conducted case studies on brain neoplasms, breast neoplasms, colon neoplasms, and kidney neoplasms. According to the findings, the bulk of our predicted possible miRNA-disease associations were verified by the dbDEMC and miRCancer databases. This paper’s significant contributions are summarized as follows.
- (1)
We integrated both association information and similarity information to construct the initial features and could better learn the potential information in miRNA-disease pairs.
- (2)
We propose a stacked graph autoencoder prediction framework. Unlike previous stacked autoencoders, which used layer-by-layer training, the stacked graph autoencoder uses multi-layer collaborative unsupervised training. It is capable of effectively extracting potential, deep, and unknown feature information from the similarity network to compensate for the shortcomings of previous models’ prediction results, which are biased toward miRNAs and diseases with known associations.
- (3)
We use a multilayer perceptron (MLP) for prediction of the final results, which has high fault tolerance and can learn feature information from miRNA-disease pairs rapidly and efficiently to improve model prediction performance.
Figure 1.
SGAEMDA flowchart. (A) Construction of initial features and data processing. (B) Pre-training to extract low-dimensional similarity features of miRNA and disease. (C) Fusion of learned miRNA and disease features to generate miRNA-disease pair feature vector. (D) Association prediction score by MLP.
Figure 1.
SGAEMDA flowchart. (A) Construction of initial features and data processing. (B) Pre-training to extract low-dimensional similarity features of miRNA and disease. (C) Fusion of learned miRNA and disease features to generate miRNA-disease pair feature vector. (D) Association prediction score by MLP.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}