
Niclosamide Ethanolamine Salt Alleviates Idiopathic Pulmonary Fibrosis by Modulating the PI3K-mTORC1 Pathway

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. BLM-Induced IPF Mouse Model
2.2. Cell Culture and Reagents
2.3. Cell Viability and Cell Death Assays
2.4. Wound-Healing Assays
2.5. Western Blot Analysis
2.6. RNA Isolation and Quantitative Real-Time PCR
2.7. Immunofluorescence Confocal Microscopy
2.8. Cell Cycle Detection
2.9. Histology and Immunohistochemistry
2.10. Hydroxyproline Assay
2.11. Gene Expression Array Reanalysis
2.12. IPF Patient Lung Tissue
2.13. Statistical Analysis
3. Results
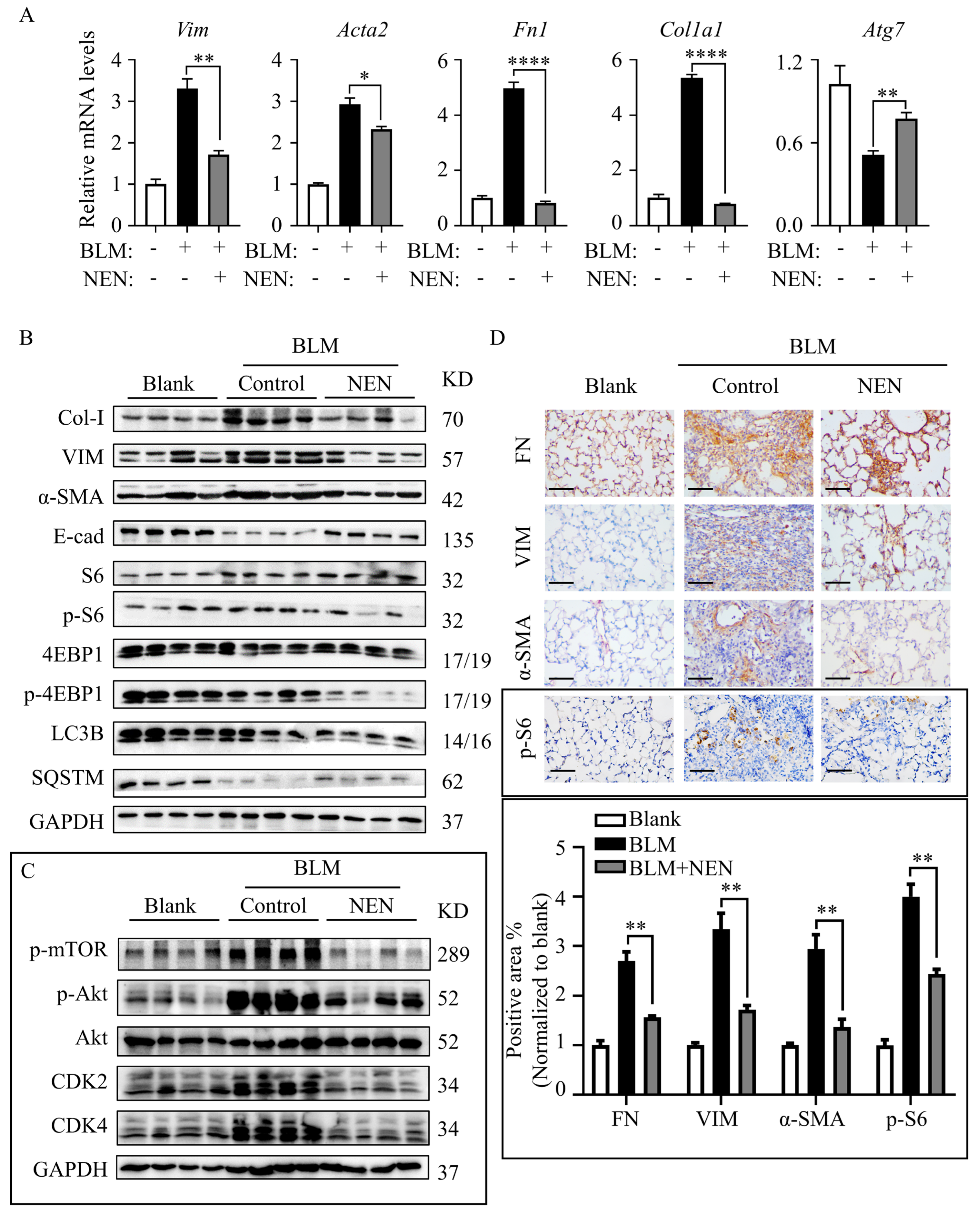
3.1. NEN Attenuated IPF in BLM-Induced Mice
3.2. NEN Inhibited Cell Viability and Migration in TGF-β1-Induced A549 Cells and DHLF-IPF Cells
3.3. NEN Reversed Elevations in EMT and ECM Markers in TGF-β1-Induced A549 and DHLF-IPF Cells
3.4. Antifibrotic Effect of NEN Mediated by PI3K/mTORC1 Pathway Blockade and Cell Cycle Arrest
3.5. NEN Prevented BLM-Induced EMT and ECM by Inhibiting the mTORC1 Signalling Pathway in Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NEN | niclosamide ethanolamine salt. |
| mTOR | mammalian target of rapamycin. |
| IPF | idiopathic pulmonary fibrosis. |
| EMT | epithelial–mesenchymal transition. |
| ECM | extracellular matrix. |
| TGF-β1 | transforming growth factor-β. |
| BLM | bleomycin. |
| DHLF-IPF | diseased human lung fibroblasts, idiopathic pulmonary fibrosis. |
| FVC | forced vital capacity. |
| Cdyn | dynamic compliance. |
| E-cad | E-cadherin. |
| VIM | vimentin. |
| α-SMA | alpha-smooth muscle actin. |
| FN | fibronectin. |
| Col-I | type I collagen. |
References
- Luppi, F.; Kalluri, M.; Faverio, P.; Kreuter, M.; Ferrara, G. Idiopathic pulmonary fibrosis beyond the lung: Understanding disease mechanisms to improve diagnosis and management. Respir. Res. 2021, 22, 109. [Google Scholar] [CrossRef] [PubMed]
- Fois, A.G.; Sotgiu, E.; Scano, V.; Negri, S.; Mellino, S.; Zinellu, E.; Pirina, P.; Pintus, G.; Carru, C.; Mangoni, A.A.; et al. Effects of Pirfenidone and Nintedanib on Markers of Systemic Oxidative Stress and Inflammation in Patients with Idiopathic Pulmonary Fibrosis: A Preliminary Report. Antioxidants 2020, 9, 1064. [Google Scholar] [CrossRef] [PubMed]
- Fois, A.G.; Posadino, A.M.; Giordo, R.; Cossu, A.; Agouni, A.; Rizk, N.M.; Pirina, P.; Carru, C.; Zinellu, A.; Pintus, G. Antioxidant Activity Mediates Pirfenidone Antifibrotic Effects in Human Pulmonary Vascular Smooth Muscle Cells Exposed to Sera of Idiopathic Pulmonary Fibrosis Patients. Oxid. Med. Cell. Longev. 2018, 2018, 2639081. [Google Scholar] [CrossRef]
- Chanda, D.; Otoupalova, E.; Smith, S.R.; Volckaert, T.; De Langhe, S.P.; Thannickal, V.J. Developmental pathways in the pathogenesis of lung fibrosis. Mol. Asp. Med. 2019, 65, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.H.G.; Paliogiannis, P.; Nasrallah, G.K.; Giordo, R.; Eid, A.H.; Fois, A.G.; Zinellu, A.; Mangoni, A.A.; Pintus, G. Emerging cellular and molecular determinants of idiopathic pulmonary fibrosis. Cell. Mol. Life Sci. 2021, 78, 2031–2057. [Google Scholar] [CrossRef]
- Marmai, C.; Sutherland, R.E.; Kim, K.K.; Dolganov, G.M.; Fang, X.; Kim, S.S.; Jiang, S.; Golden, J.A.; Hoopes, C.W.; Matthay, M.A.; et al. Alveolar epithelial cells express mesenchymal proteins in patients with idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L71–L78. [Google Scholar] [CrossRef]
- King, T.E.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Jhanwar-Uniyal, M.; Wainwright, J.V.; Mohan, A.L.; Tobias, M.E.; Murali, R.; Gandhi, C.D.; Schmidt, M.H. Diverse signaling mechanisms of mTOR complexes: mTORC1 and mTORC2 in forming a formidable relationship. Adv. Biol. Regul. 2019, 72, 51–62. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Plate, M.; Guillotin, D.; Chambers, R.C. The promise of mTOR as a therapeutic target pathway in idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2020, 29, 200269. [Google Scholar] [CrossRef]
- Oshi, M.; Tokumaru, Y.; Mukhopadhyay, S.; Yan, L.; Matsuyama, R.; Endo, I.; Takabe, K. Annexin A1 Expression Is Associated with Epithelial-Mesenchymal Transition (EMT), Cell Proliferation, Prognosis, and Drug Response in Pancreatic Cancer. Cells 2021, 10, 653. [Google Scholar] [CrossRef]
- Lukey, P.T.; Harrison, S.A.; Yang, S.; Man, Y.; Holman, B.F.; Rashidnasab, A.; Azzopardi, G.; Grayer, M.; Simpson, J.K.; Bareille, P.; et al. A randomised, placebo-controlled study of omipalisib (PI3K/mTOR) in idiopathic pulmonary fibrosis. Eur. Respir. J. 2019, 53, 1801992. [Google Scholar] [CrossRef]
- Vancheri, C.; Kreuter, M.; Richeldi, L.; Ryerson, C.J.; Valeyre, D.; Grutters, J.C.; Wiebe, S.; Stansen, W.; Quaresma, M.; Stowasser, S.; et al. Nintedanib with Add-on Pirfenidone in Idiopathic Pulmonary Fibrosis. Results of the INJOURNEY Trial. Am. J. Respir. Crit. Care Med. 2018, 197, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.S.; Xin, H.R.; Qiu, R.L.; Deng, Z.L.; Deng, F.; Yan, Z.J. Niclosamide: Drug repurposing for human chondrosarcoma treatment via the caspase-dependent mitochondrial apoptotic pathway. Am. J. Transl. Res. 2020, 12, 3688–3701. [Google Scholar]
- Pearson, R.D.; Hewlett, E.L. Niclosamide therapy for tapeworm infections. Ann. Intern. Med. 1985, 102, 550–551. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, J.Y.; Choi, J.H.; Kim, J.H.; Lee, C.J.; Singh, P.; Sarkar, S.; Baek, J.H.; Nam, J.S. Inhibition of LEF1-Mediated DCLK1 by Niclosamide Attenuates Colorectal Cancer Stemness. Clin. Cancer Res. 2019, 25, 1415–1429. [Google Scholar] [CrossRef] [Green Version]
- Gyamfi, J.; Lee, Y.H.; Min, B.S.; Choi, J. Niclosamide reverses adipocyte induced epithelial-mesenchymal transition in breast cancer cells via suppression of the interleukin-6/STAT3 signalling axis. Sci. Rep. 2019, 9, 11336. [Google Scholar] [CrossRef]
- Chae, H.D.; Cox, N.; Dahl, G.V.; Lacayo, N.J.; Davis, K.L.; Capolicchio, S.; Smith, M.; Sakamoto, K.M. Niclosamide suppresses acute myeloid leukemia cell proliferation through inhibition of CREB-dependent signaling pathways. Oncotarget 2018, 9, 4301–4317. [Google Scholar] [CrossRef]
- Li, Y.; Li, P.K.; Roberts, M.J.; Arend, R.C.; Samant, R.S.; Buchsbaum, D.J. Multi-targeted therapy of cancer by niclosamide: A new application for an old drug. Cancer Lett. 2014, 349, 8–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, H.; Zhang, Y.; Zeng, X.; Shulman, G.I.; Jin, S. Niclosamide ethanolamine-induced mild mitochondrial uncoupling improves diabetic symptoms in mice. Nat. Med. 2014, 20, 1263–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Zeng, X.; Wang, J.; Fu, Z.; Wang, J.; Liu, M.; Ren, D.; Yu, B.; Zheng, L.; Hu, X.; et al. Immunomodulation by mesenchymal stem cells in treating human autoimmune disease-associated lung fibrosis. Stem Cell Res. Ther. 2016, 7, 63. [Google Scholar] [CrossRef] [Green Version]
- Di Gregorio, J.; Robuffo, I.; Spalletta, S.; Giambuzzi, G.; De Iuliis, V.; Toniato, E.; Martinotti, S.; Conti, P.; Flati, V. The Epithelial-to-Mesenchymal Transition as a Possible Therapeutic Target in Fibrotic Disorders. Front. Cell Dev. Biol. 2020, 8, 607483. [Google Scholar] [CrossRef]
- Wang, H.; Xie, Q.; Ou-Yang, W.; Zhang, M. Integrative analyses of genes associated with idiopathic pulmonary fibrosis. J. Cell. Biochem. 2019, 120, 8648–8660. [Google Scholar] [CrossRef]
- Goldmann, T.; Zissel, G.; Watz, H.; Drömann, D.; Reck, M.; Kugler, C.; Rabe, K.F.; Marwitz, S. Human alveolar epithelial cells type II are capable of TGFβ-dependent epithelial-mesenchymal-transition and collagen-synthesis. Respir. Res. 2018, 19, 138. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Bueno, G.; Peinado, H.; Molina, P.; Olmeda, D.; Cubillo, E.; Santos, V.; Palacios, J.; Portillo, F.; Cano, A. The morphological and molecular features of the epithelial-to-mesenchymal transition. Nat. Protoc. 2009, 4, 1591–1613. [Google Scholar] [CrossRef]
- Mack, M. Inflammation and fibrosis. Matrix Biol. J. Int. Soc. Matrix Biol. 2018, 68–69, 106–121. [Google Scholar] [CrossRef] [PubMed]
- Lodyga, M.; Hinz, B. TGF-β1—A truly transforming growth factor in fibrosis and immunity. Semin. Cell Dev. Biol. 2020, 101, 123–139. [Google Scholar] [CrossRef]
- El-Baz, L.M.F.; Shoukry, N.M.; Hafez, H.S.; Guzy, R.D.; Salem, M.L. Fibroblast Growth Factor 2 Augments Transforming Growth Factor Beta 1 Induced Epithelial-mesenchymal Transition in Lung Cell Culture Model. Iran. J. Allergy Asthma Immunol. 2020, 19, 348–361. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Pan, C.; Tang, C.; Tan, W.; Zhang, W.; Guan, J. miR-184 targets TP63 to block idiopathic pulmonary fibrosis by inhibiting proliferation and epithelial-mesenchymal transition of airway epithelial cells. Lab. Invest. 2021, 101, 142–154. [Google Scholar] [CrossRef]
- Chen, W.; Mook, R.A., Jr.; Premont, R.T.; Wang, J. Niclosamide: Beyond an antihelminthic drug. Cell. Signal. 2018, 41, 89–96. [Google Scholar] [CrossRef]
- Boyapally, R.; Pulivendala, G.; Bale, S.; Godugu, C. Niclosamide alleviates pulmonary fibrosis in vitro and in vivo by attenuation of epithelial-to-mesenchymal transition, matrix proteins & Wnt/β-catenin signaling: A drug repurposing study. Life Sci. 2019, 220, 8–20. [Google Scholar] [CrossRef]
- Jewell, J.L.; Russell, R.C.; Guan, K.-L. Amino acid signalling upstream of mTOR. Nat. Rev. Mol. Cell Biol. 2013, 14, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Saito, M.; Mitani, A.; Ishimori, T.; Miyashita, N.; Isago, H.; Mikami, Y.; Noguchi, S.; Tarui, M.; Nagase, T. Active mTOR in Lung Epithelium Promotes Epithelial-Mesenchymal Transition and Enhances Lung Fibrosis. Am. J. Respir. Cell Mol. Biol. 2020, 62, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, H.V.; Eley, J.D.; Guillotin, D.; Plate, M.; Nanthakumar, C.B.; Martufi, M.; Peace, S.; Joberty, G.; Poeckel, D.; Good, R.B.; et al. The mTORC1/4E-BP1 axis represents a critical signaling node during fibrogenesis. Nat. Commun. 2019, 10, 6. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.; Zhang, M.; Leng, C.; Blokzijl, T.; Jansen, B.H.; Dijkstra, G.; Faber, K.N. Pirfenidone Inhibits Cell Proliferation and Collagen I Production of Primary Human Intestinal Fibroblasts. Cells 2020, 9, 775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balgi, A.D.; Fonseca, B.D.; Donohue, E.; Tsang, T.C.; Lajoie, P.; Proud, C.G.; Nabi, I.R.; Roberge, M. Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling. PLoS ONE 2009, 4, e7124. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Yang, M.; Yuan, Y.; Li, X.; Kuang, E. Niclosamide inhibits lytic replication of Epstein-Barr virus by disrupting mTOR activation. Antivir. Res. 2017, 138, 68–78. [Google Scholar] [CrossRef]
- Gremke, N.; Polo, P.; Dort, A.; Schneikert, J.; Elmshauser, S.; Brehm, C.; Klingmuller, U.; Schmitt, A.; Reinhardt, H.C.; Timofeev, O.; et al. mTOR-mediated cancer drug resistance suppresses autophagy and generates a druggable metabolic vulnerability. Nat. Commun. 2020, 11, 4684. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liu, R.; Wu, J.; Li, X. Self-eating: Friend or foe? The emerging role of autophagy in fibrotic diseases. Theranostics 2020, 10, 7993–8017. [Google Scholar] [CrossRef]
- Cabrera, S.; Maciel, M.; Herrera, I.; Nava, T.; Vergara, F.; Gaxiola, M.; Lopez-Otin, C.; Selman, M.; Pardo, A. Essential role for the ATG4B protease and autophagy in bleomycin-induced pulmonary fibrosis. Autophagy 2015, 11, 670–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Wei, S.; Li, Z.; Lin, C.; Zhu, Z.; Sun, D.; Bai, R.; Qian, J.; Gao, X.; Chen, G.; et al. Autophagic flux blockage in alveolar epithelial cells is essential in silica nanoparticle-induced pulmonary fibrosis. Cell Death Dis. 2019, 10, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Fang, S.; Wang, W.; Cheng, Y.; Zhang, Y.; Liao, H.; Yao, H.; Chao, J. Macrophage-derived MCPIP1 mediates silica-induced pulmonary fibrosis via autophagy. Part. Fibre Toxicol. 2016, 13, 55. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Ren, X.R.; Piao, H.; Zhao, S.; Osada, T.; Premont, R.T.; Mook, R.A., Jr.; Morse, M.A.; Lyerly, H.K.; Chen, W. Niclosamide-induced Wnt signaling inhibition in colorectal cancer is mediated by autophagy. Biochem. J. 2019, 476, 535–546. [Google Scholar] [CrossRef]
- Liu, Y.; Luo, X.; Shan, H.; Fu, Y.; Gu, Q.; Zheng, X.; Dai, Q.; Xia, F.; Zheng, Z.; Liu, P.; et al. Niclosamide Triggers Non-Canonical LC3 Lipidation. Cells 2019, 8, 248. [Google Scholar] [CrossRef] [Green Version]
- Rangarajan, S.; Kurundkar, A.; Kurundkar, D.; Bernard, K.; Sanders, Y.Y.; Ding, Q.; Antony, V.B.; Zhang, J.; Zmijewski, J.; Thannickal, V.J. Novel Mechanisms for the Antifibrotic Action of Nintedanib. Am. J. Respir. Cell Mol. Biol. 2016, 54, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Meng, L.; Li, X.; Li, D.; Liu, Q.; Chen, Y.; Li, X.; Bu, W.; Sun, H. Regulation of FN1 degradation by the p62/SQSTM1-dependent autophagy-lysosome pathway in HNSCC. Int. J. Oral. Sci. 2020, 12, 34. [Google Scholar] [CrossRef]
- Li, M.; Khambu, B.; Zhang, H.; Kang, J.H.; Chen, X.; Chen, D.; Vollmer, L.; Liu, P.Q.; Vogt, A.; Yin, X.M. Suppression of lysosome function induces autophagy via a feedback down-regulation of MTOR complex 1 (MTORC1) activity. J. Biol. Chem. 2013, 288, 35769–35780. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pei, X.; Zheng, F.; Li, Y.; Lin, Z.; Han, X.; Feng, Y.; Tian, Z.; Ren, D.; Cao, K.; Li, C. Niclosamide Ethanolamine Salt Alleviates Idiopathic Pulmonary Fibrosis by Modulating the PI3K-mTORC1 Pathway. Cells 2022, 11, 346. https://doi.org/10.3390/cells11030346
Pei X, Zheng F, Li Y, Lin Z, Han X, Feng Y, Tian Z, Ren D, Cao K, Li C. Niclosamide Ethanolamine Salt Alleviates Idiopathic Pulmonary Fibrosis by Modulating the PI3K-mTORC1 Pathway. Cells. 2022; 11(3):346. https://doi.org/10.3390/cells11030346
Chicago/Turabian StylePei, Xiaolin, Fangxu Zheng, Yin Li, Zhoujun Lin, Xiao Han, Ya Feng, Zhenhuan Tian, Dunqiang Ren, Ke Cao, and Chenggang Li. 2022. "Niclosamide Ethanolamine Salt Alleviates Idiopathic Pulmonary Fibrosis by Modulating the PI3K-mTORC1 Pathway" Cells 11, no. 3: 346. https://doi.org/10.3390/cells11030346
APA StylePei, X., Zheng, F., Li, Y., Lin, Z., Han, X., Feng, Y., Tian, Z., Ren, D., Cao, K., & Li, C. (2022). Niclosamide Ethanolamine Salt Alleviates Idiopathic Pulmonary Fibrosis by Modulating the PI3K-mTORC1 Pathway. Cells, 11(3), 346. https://doi.org/10.3390/cells11030346