
Extracellular Release of Mitochondrial DNA: Triggered by Cigarette Smoke and Detected in COPD

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Lung Parenchyma and Plasma Collection
2.2. Animal Experiments
2.3. Cigarette Smoke Extract (CSE) Preparation
2.4. Statistical Analysis
3. Results
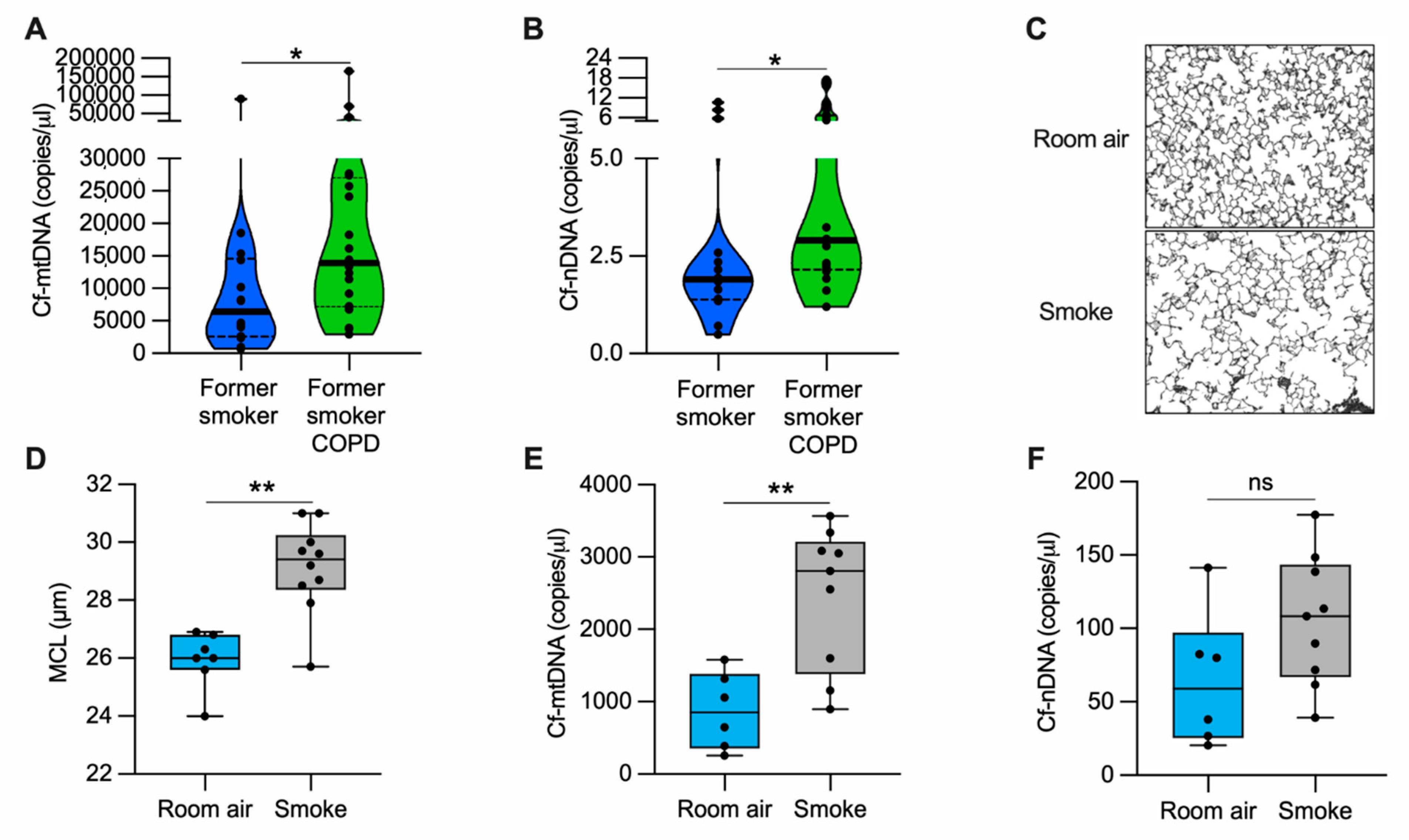
3.1. Cell Free-mtDNA and -nDNA Are Higher in Human Plasma from Former Smokers with COPD Than Former Smokers without COPD
3.2. Cell Free-mtDNA Levels Are Increased in the Serum of Mice with Alveolar Destruction Induced by Cigarette Smoke
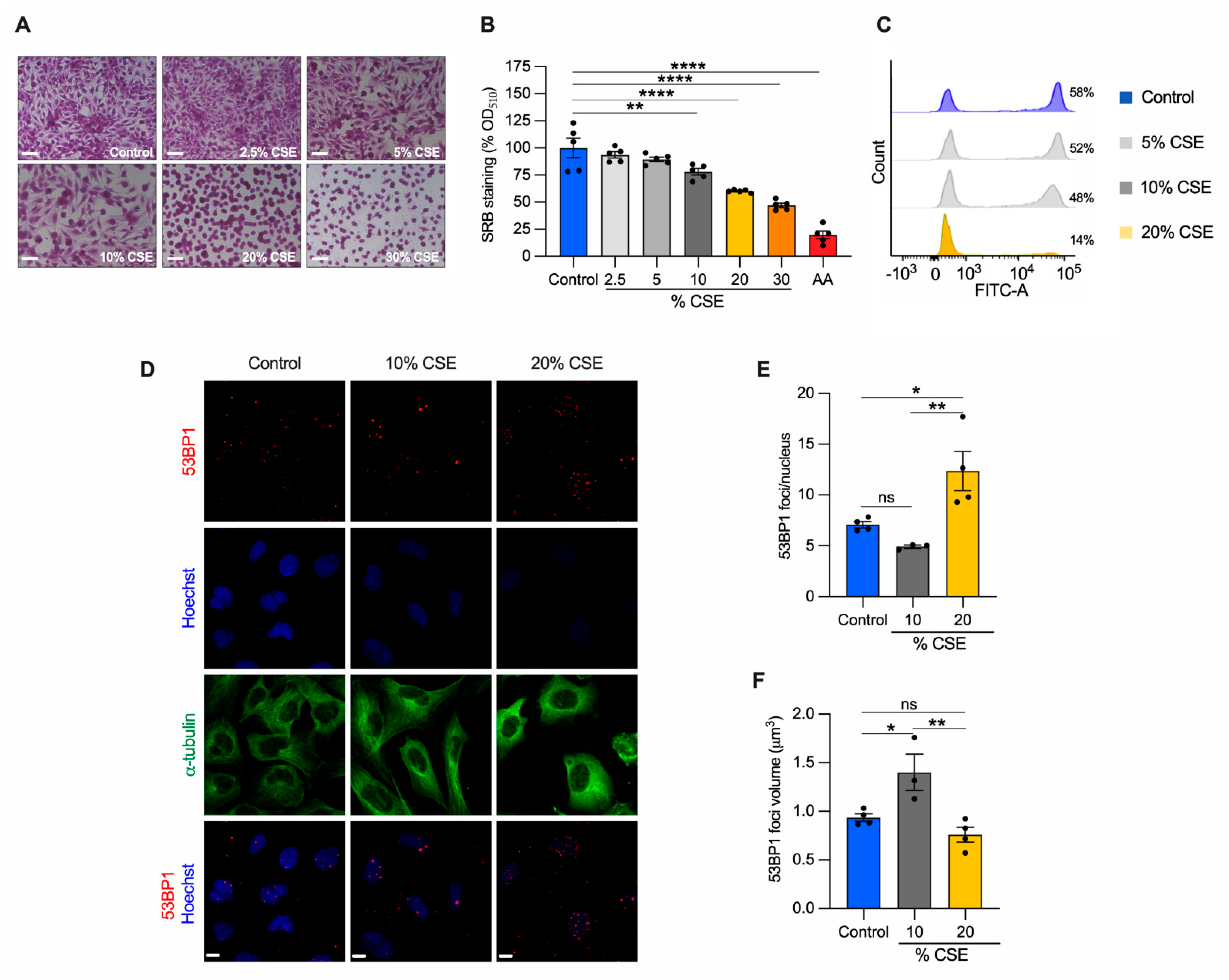
3.3. Cigarette Smoke Extract Inhibits Cell Proliferation and Promotes Cell Death in Bronchial Epithelial Cells
3.4. Mitochondrial DNA Is Released into the Extracellular Milieu via Vesicles following CSE Exposure
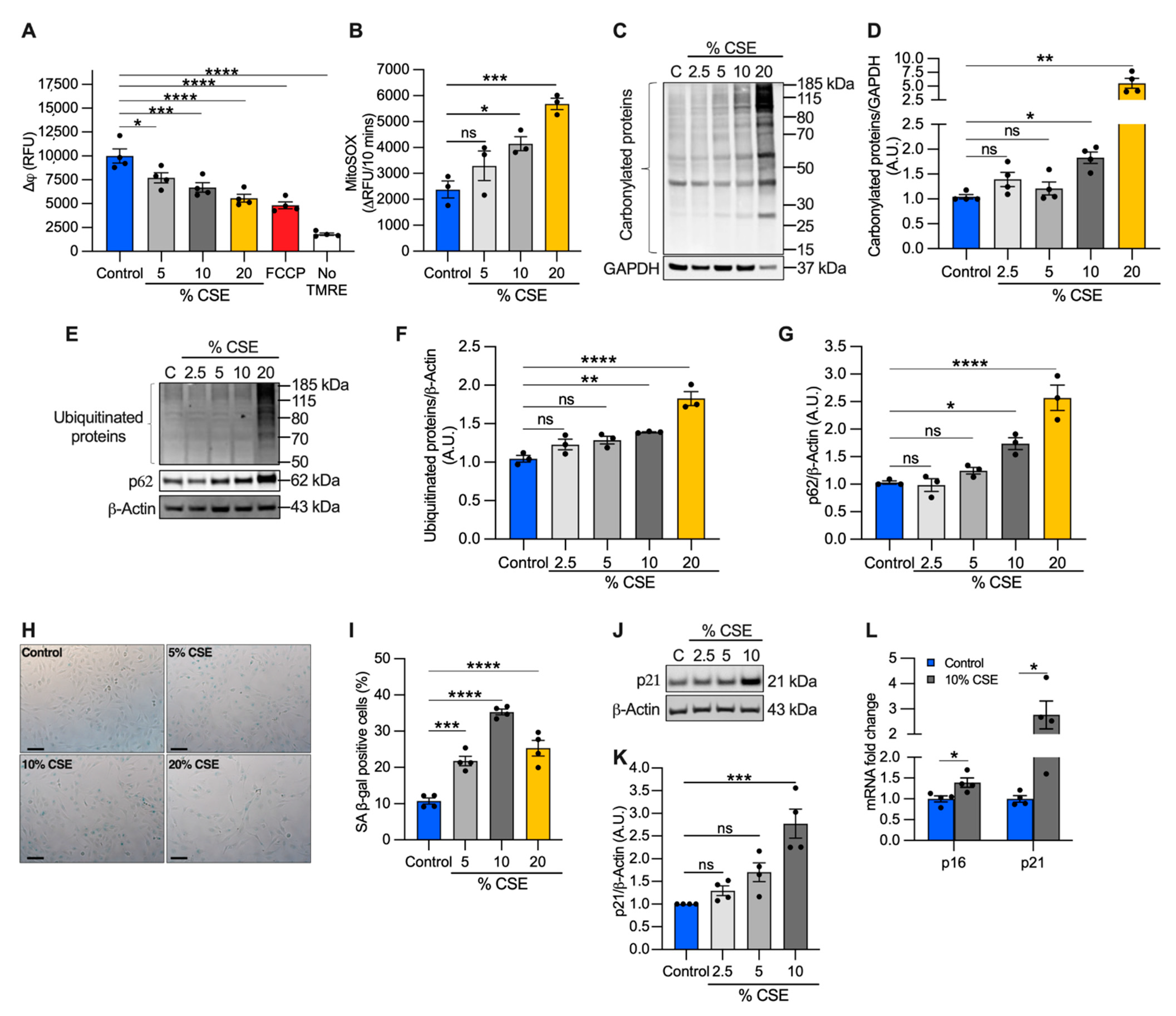
3.5. CSE Promotes Mitochondrial Membrane Depolarization, Superoxide Production, and Oxidative Stress
3.6. Exposure to a Sublethal Dose of CSE Promotes Cellular Senescence
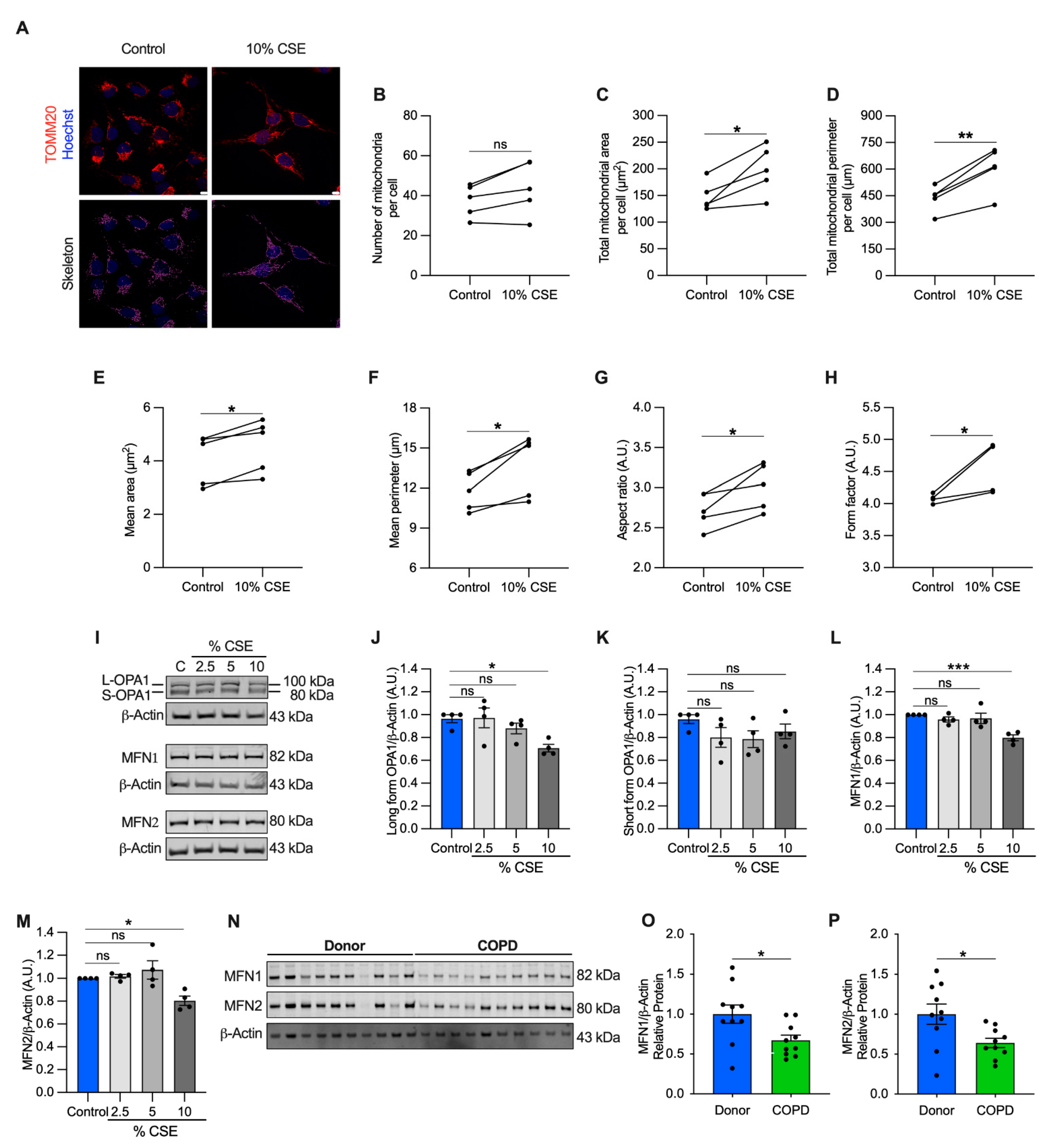
3.7. Mitochondrial Dynamics Are Altered in Cells Exposed to a Sublethal Dose of CSE and in COPD Lung Tissue
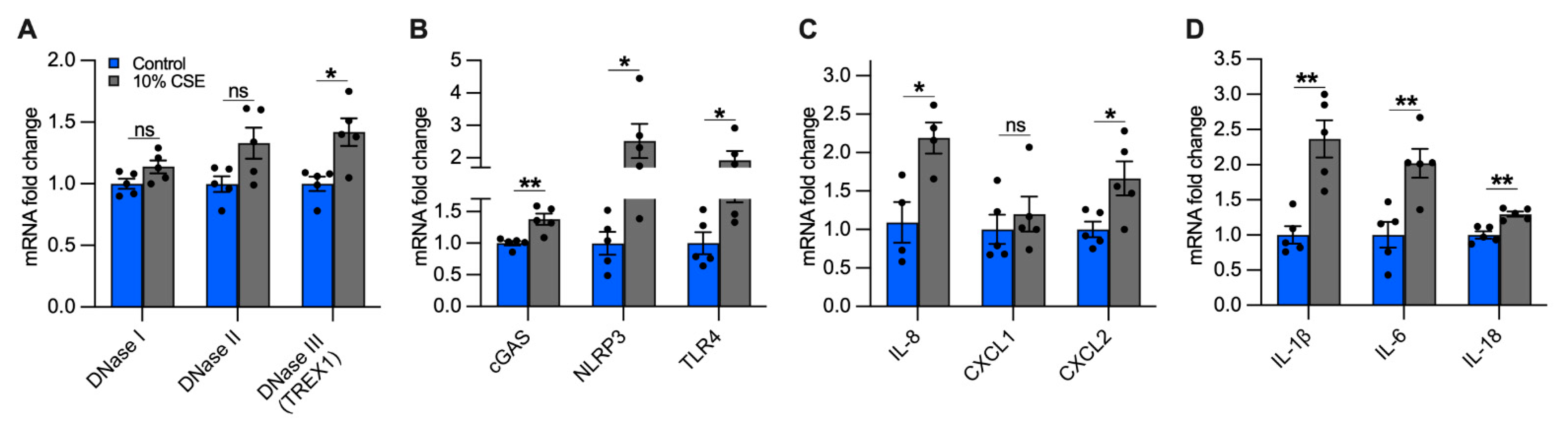
3.8. Exposure to a Sublethal Dose of CSE Upregulates the Expression of DNase III, cGAS, NLRP3, and TLR4
3.9. Exposure to a Sublethal Dose of CSE Upregulates the Expression of Proinflammatory Cytokines Involved in the Recruitment of Neutrophils and Macrophages
4. Result
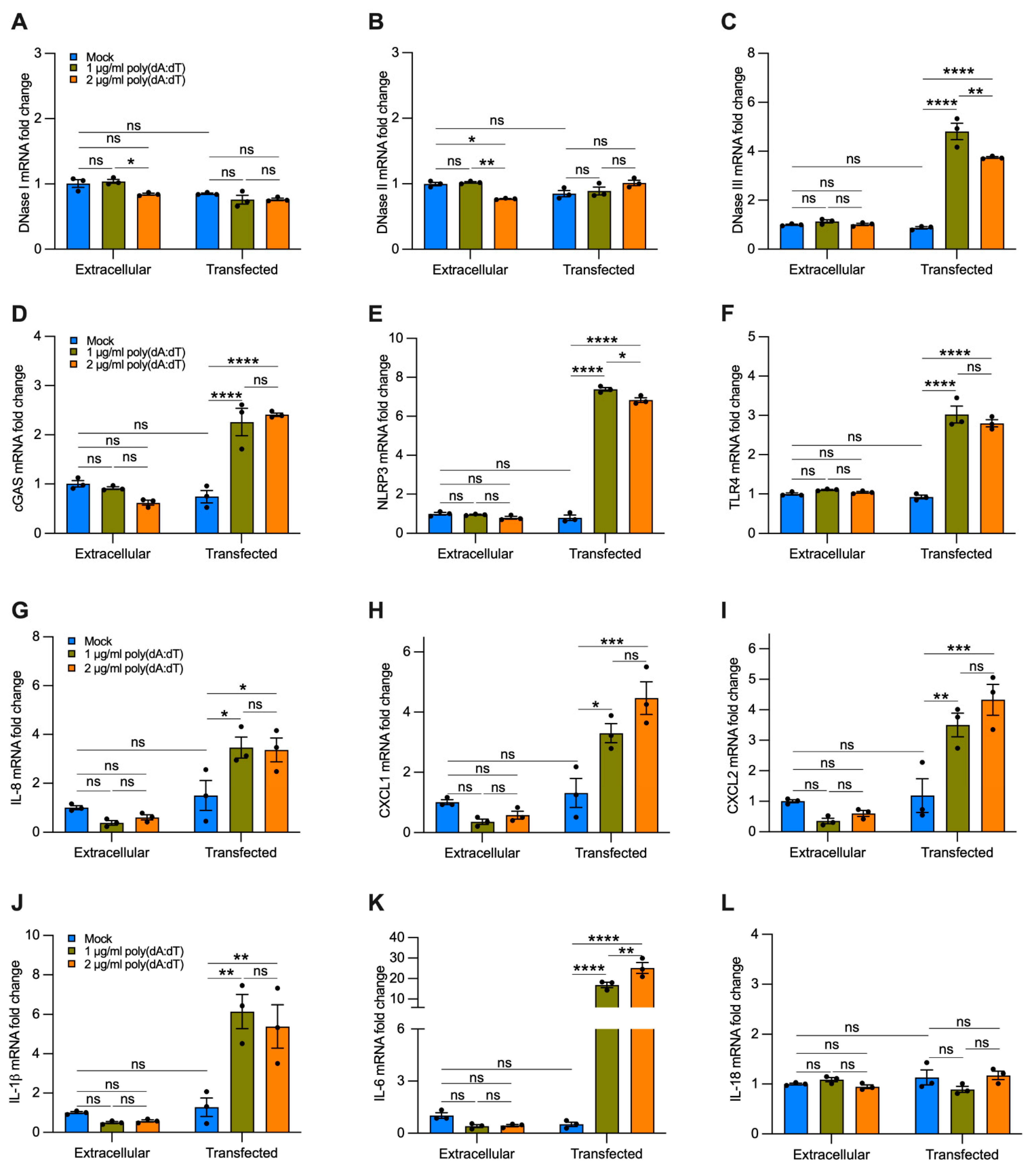
Transfection of Synthetic DNA to Mimic Delivery of cf-DNA by EVs Upregulates the Expression of DNase III, cGAS, NLRP3, TLR4, and Proinflammatory Cytokines Involved in the Recruitment of Neutrophils and Macrophages
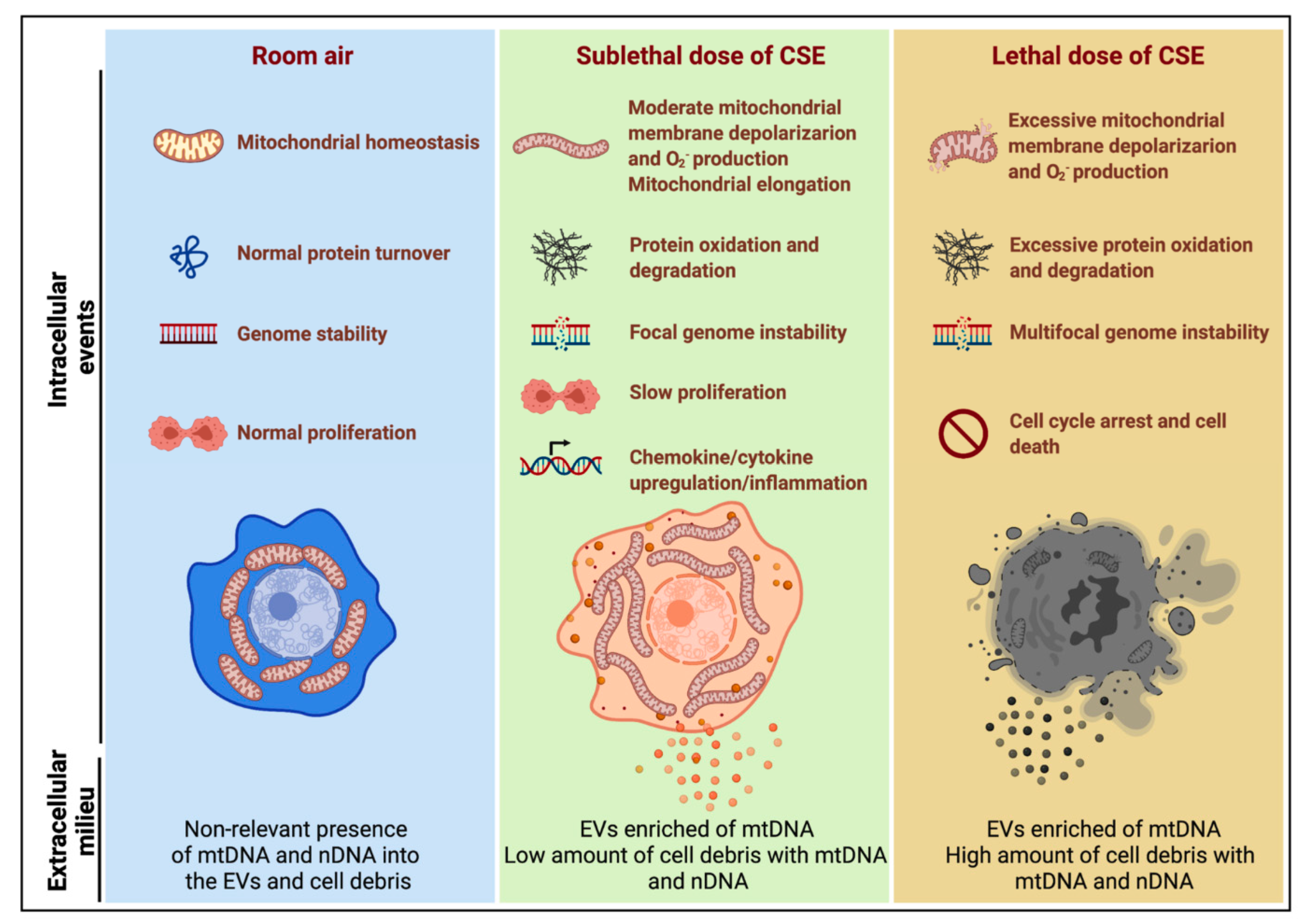
5. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mathers, C.D.; Loncar, D. Projections of Global Mortality and Burden of Disease from 2002 to 2030. PLoS Med. 2006, 3, e442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogelmeier, C.F.; Criner, G.J.; Martinez, F.J.; Anzueto, A.; Barnes, P.J.; Bourbeau, J.; Celli, B.R.; Chen, R.; Decramer, M.; Fabbri, L.M.; et al. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease 2017 Report. GOLD Executive Summary. Am. J. Respir. Crit. Care Med. 2017, 195, 557–582. [Google Scholar] [CrossRef]
- Cosio, M.G.; Saetta, M.; Agusti, A. Immunologic Aspects of Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2009, 360, 2445–2454. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. The Cytokine Network in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Cell Mol. Biol. 2009, 41, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R.F.; Zarrintan, S.; Brandenburg, S.M.; Kol, A.; De Bruin, H.G.; Jafari, S.; Dijk, F.; Kalicharan, D.; Kelders, M.; Gosker, H.R.; et al. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir. Res. 2013, 14, 97. [Google Scholar] [CrossRef] [Green Version]
- Van der Toorn, M.; Slebos, D.J.; de Bruin, H.G.; Leuvenink, H.G.; Bakker, S.J.; Gans, R.O.; Koëter, G.H.; van Oosterhout, A.J.; Kauffman, H.F. Cigarette smoke-induced blockade of the mitochondrial respiratory chain switches lung epithelial cell apoptosis into necrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L1211–L1218. [Google Scholar] [CrossRef] [Green Version]
- Mizumura, K.; Cloonan, S.; Nakahira, K.; Bhashyam, A.R.; Cervo, M.; Kitada, T.; Glass, K.; Owen, C.A.; Mahmood, A.; Washko, G.R.; et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J. Clin. Investig. 2014, 124, 3987–4003. [Google Scholar] [CrossRef] [Green Version]
- Cloonan, S.; Glass, K.; Laucho-Contreras, M.E.; Bhashyam, A.R.; Cervo, M.; Pabón, M.A.; Konrad, C.; Polverino, F.; Siempos, I.I.; Perez, E.; et al. Mitochondrial iron chelation ameliorates cigarette smoke–induced bronchitis and emphysema in mice. Nat. Med. 2016, 22, 163–174. [Google Scholar] [CrossRef]
- Giordano, L.; Farnham, A.; Dhandapani, P.K.; Salminen, L.; Bhaskaran, J.; Voswinckel, R.; Rauschkolb, P.; Scheibe, S.; Sommer, N.; Beisswenger, C.; et al. Alternative Oxidase Attenuates Cigarette Smoke-induced Lung Dysfunction and Tissue Damage. Am. J. Respir. Cell Mol. Biol. 2019, 60, 515–522. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Y.; Yeung, S.C.; Liang, Y.; Liang, X.; Ding, Y.; Ip, M.S.M.; Tse, H.F.; Mak, J.; Lian, Q. Mitochondrial Transfer of Induced Pluripotent Stem Cell–Derived Mesenchymal Stem Cells to Airway Epithelial Cells Attenuates Cigarette Smoke–Induced Damage. Am. J. Respir. Cell Mol. Biol. 2014, 51, 455–465. [Google Scholar] [CrossRef]
- Talhout, R.; Schulz, T.; Florek, E.; Van Benthem, J.; Wester, P.; Opperhuizen, A. Hazardous Compounds in Tobacco Smoke. Int. J. Environ. Res. Public Health 2011, 8, 613–628. [Google Scholar] [CrossRef] [PubMed]
- Scialò, F.; Fernández-Ayala, D.J.; Sanz, A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front. Physiol. 2017, 8, 428. [Google Scholar] [CrossRef]
- Pérez-Treviño, P.; Velásquez, M.; García, N. Mechanisms of mitochondrial DNA escape and its relationship with different metabolic diseases. Biochim. et Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165761. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef]
- Rongvaux, A. Innate immunity and tolerance toward mitochondria. Mitochondrion 2018, 41, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef]
- Li, Z.; Fan, E.K.; Liu, J.; Scott, M.J.; Li, Y.; Li, S.; Xie, W.; Billiar, T.R.; Wilson, M.A.; Jiang, Y.; et al. Cold-inducible RNA-binding protein through TLR4 signaling induces mitochondrial DNA fragmentation and regulates macrophage cell death after trauma. Cell Death Dis. 2017, 8, e2775. [Google Scholar] [CrossRef]
- Backer, J.M.; Weinstein, I.B. Mitochondrial DNA Is a Major Cellular Target for a Dihydrodiol-Epoxide Derivative of Benzo[a]pyrene. Science 1980, 209, 297–299. [Google Scholar] [CrossRef]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [Green Version]
- Ballinger, S.W.; Bouder, T.G.; Davis, G.S.; Judice, S.A.; Nicklas, J.A.; Albertini, R.J. Mitochondrial genome damage associated with cigarette smoking. Cancer Res. 1996, 56, 5692–5697. [Google Scholar] [PubMed]
- Gillespie, M.N.; Pastukh, V.M.; Zhang, L.; Ruchko, M.; Gorodnya, O.; Bardwell, G.C.; Tuder, R. Oxidative DNA damage in lung tissue from patients with COPD is clustered in functionally significant sequences. Int. J. Chronic Obstr. Pulm. Dis. 2011, 6, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Tedrow, J.; Nouraie, M.; Li, X.; Chandra, D.; Bon, J.; Kass, D.J.; Fuhrman, C.R.; Leader, J.K.; Duncan, S.R.; et al. Elevated plasma level of Pentraxin 3 is associated with emphysema and mortality in smokers. Thorax 2021, 76, 335–342. [Google Scholar] [CrossRef]
- Chen, D.; Gregory, A.D.; Li, X.; Wei, J.; Burton, C.L.; Gibson, G.; Scott, S.J.; Croix, C.M.S.; Zhang, Y.; Shapiro, S.D. RIP3-dependent necroptosis contributes to the pathogenesis of chronic obstructive pulmonary disease. JCI Insight 2021, 6, e144689. [Google Scholar] [CrossRef] [PubMed]
- Cottage, C.T.; Peterson, N.; Kearley, J.; Berlin, A.; Xiong, X.; Huntley, A.; Zhao, W.; Brown, C.; Migneault, A.; Zerrouki, K.; et al. Targeting p16-induced senescence prevents cigarette smoke-induced emphysema by promoting igf1/akt1 signaling in mice. Commun. Biol. 2019, 2, 307. [Google Scholar] [CrossRef] [Green Version]
- Vij, N.; Chandramani-Shivalingappa, P.; Van Westphal, C.; Hole, R.; Bodas, M. Cigarette smoke-induced autophagy impairment accelerates lung aging, COPD-emphysema exacerbations and pathogenesis. Am. J. Physiol. 2018, 314, C73–C87. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.-H.; Lam, H.C.; Jin, Y.; Kim, H.-P.; Cao, J.; Lee, S.-J.; Ifedigbo, E.; Parameswaran, H.; Ryter, S.W.; Choi, A.M.K. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc. Natl. Acad. Sci. USA 2010, 107, 18880–18885. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [Green Version]
- Panier, S.; Durocher, D. Push back to respond better: Regulatory inhibition of the DNA double-strand break response. Nat. Rev. Mol. Cell Biol. 2013, 14, 661–672. [Google Scholar] [CrossRef] [PubMed]
- De Gaetano, A.; Solodka, K.; Zanini, G.; Selleri, V.; Mattioli, A.V.; Nasi, M.; Pinti, M. Molecular mechanisms of mtdna-mediated inflammation. Cells 2021, 10, 2898. [Google Scholar] [CrossRef]
- Pajares, M.; Jiménez-Moreno, N.; Dias, I.H.; Debelec, B.; Vucetic, M.; Fladmark, K.E.; Basaga, H.; Ribaric, S.; Milisav, I.; Cuadrado, A. Redox control of protein degradation. Redox Biol. 2015, 6, 409–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Martín, P.; Komatsu, M. P62/sqstm1—steering the cell through health and disease. J. Cell Sci. 2018, 131, jcs222836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, S.; Hara, H.; Araya, J.; Takasaka, N.; Kojima, J.; Ito, S.; Minagawa, S.; Yumino, Y.; Ishikawa, T.; Numata, T.; et al. Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. OncoImmunology 2012, 1, 630–641. [Google Scholar] [CrossRef] [Green Version]
- Formosa, L.; Ryan, M.T. Mitochondrial fusion: Reaching the end of mitofusin’s tether. J. Cell Biol. 2016, 215, 597–598. [Google Scholar] [CrossRef]
- Maremanda, K.P.; Sundar, I.K.; Rahman, I. Role of inner mitochondrial protein OPA1 in mitochondrial dysfunction by tobacco smoking and in the pathogenesis of COPD. Redox Biol. 2021, 45, 102055. [Google Scholar] [CrossRef] [PubMed]
- Kosmider, B.; Lin, C.-R.; Karim, L.; Tomar, D.; Vlasenko, L.; Marchetti, N.; Bolla, S.; Madesh, M.; Criner, G.J.; Bahmed, K. Mitochondrial dysfunction in human primary alveolar type II cells in emphysema. EBioMedicine 2019, 46, 305–316. [Google Scholar] [CrossRef] [Green Version]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. Dna sensing by the cgas-sting pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef]
- Zhang, X.; Shan, P.; Jiang, G.; Cohn, L.; Lee, P.J. Toll-like receptor 4 deficiency causes pulmonary emphysema. J. Clin. Investig. 2006, 116, 3050–3059. [Google Scholar] [CrossRef]
- Barnes, P.J. Targeting cytokines to treat asthma and chronic obstructive pulmonary disease. Nat. Rev. Immunol. 2018, 18, 454–466. [Google Scholar] [CrossRef]
- Hoenderdos, K.; Condliffe, A. The Neutrophil in Chronic Obstructive Pulmonary Disease. Too Little, Too Late or Too Much, Too Soon? Am. J. Respir. Cell Mol. Biol. 2013, 48, 531–539. [Google Scholar] [CrossRef]
- Keatings, V.M.; Collins, P.D.; Scott, D.M.; Barnes, P.J. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am. J. Respir. Crit. Care Med. 1996, 153, 530–534. [Google Scholar] [CrossRef]
- Masubuchi, T.; Koyama, S.; Sato, E.; Takamizawa, A.; Kubo, K.; Sekiguchi, M.; Nagai, S.; Izumi, T. Smoke Extract Stimulates Lung Epithelial Cells to Release Neutrophil and Monocyte Chemotactic Activity. Am. J. Pathol. 1998, 153, 1903–1912. [Google Scholar] [CrossRef] [Green Version]
- Lappalainen, U.; Whitsett, J.A.; Wert, S.E.; Tichelaar, J.W.; Bry, K. Interleukin-1beta causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am. J. Respir. Cell Mol. Biol. 2005, 32, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Sapey, E.; Ahmad, A.; Bayley, D.; Newbold, P.; Snell, N.; Rugman, P.; Stockley, R.A. Imbalances Between Interleukin-1 and Tumor Necrosis Factor Agonists and Antagonists in Stable COPD. J. Clin. Immunol. 2009, 29, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Bucchioni, E.; Kharitonov, S.A.; Allegra, L.; Barnes, P.J. High levels of interleukin-6 in the exhaled breath condensate of patients with COPD. Respir. Med. 2003, 97, 1299–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, C.; Homer, R.J.; Zhu, Z.; Ward, N.; Flavell, R.A.; Geba, G.P.; Elias, J.A. Airway hyperresponsiveness and airway obstruction in transgenic mice. Morphologic correlates in mice overexpressing interleukin (il)-11 and il-6 in the lung. Am. J. Respir. Cell Mol. Biol. 2000, 22, 289–295. [Google Scholar] [CrossRef]
- Imaoka, H.; Hoshino, T.; Takei, S.; Kinoshita, T.; Okamoto, M.; Kawayama, T.; Kato, S.; Iwasaki, H.; Watanabe, K.; Aizawa, H. Interleukin-18 production and pulmonary function in COPD. Eur. Respir. J. 2008, 31, 287–297. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, T.; Kato, S.; Oka, N.; Imaoka, H.; Kinoshita, T.; Takei, S.; Kitasato, Y.; Kawayama, T.; Imaizumi, T.; Yamada, K.; et al. Pulmonary inflammation and emphysema: Role of the cytokines il-18 and il-13. Am. J. Respir. Crit. Care Med. 2007, 176, 49–62. [Google Scholar] [CrossRef]
- Avriel, A.; Rozenberg, D.; Raviv, Y.; Heimer, D.; Bar-Shai, A.; Gavish, R.; Sheynin, J.; Douvdevani, A. Prognostic utility of admission cell-free DNA levels in patients with chronic obstructive pulmonary disease exacerbations. Int. J. Chronic Obstr. Pulm. Dis. 2016, 11, 3153–3161. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.Z.; Hoffman, K.L.; Schiffer, K.T.; Oromendia, C.; Rice, M.C.; Barjaktarevic, I.; Peters, S.P.; Putcha, N.; Bowler, R.P.; Wells, J.M.; et al. Association of plasma mitochondrial DNA with COPD severity and progression in the SPIROMICS cohort. Respir. Res. 2021, 22, 126. [Google Scholar] [CrossRef]
- Zhang, W.Z.; Rice, M.C.; Hoffman, K.L.; Oromendia, C.; Barjaktarevic, I.; Wells, J.M.; Hastie, A.T.; Labaki, W.W.; Cooper, C.B.; Comellas, A.P.; et al. Association of urine mitochondrial DNA with clinical measures of COPD in the SPIROMICS cohort. JCI Insight 2020, 5, e133984. [Google Scholar] [CrossRef] [Green Version]
- Lehrich, B.M.; Liang, Y.; Khosravi, P.; Federoff, H.J.; Fiandaca, M.S. Fetal Bovine Serum-Derived Extracellular Vesicles Persist within Vesicle-Depleted Culture Media. Int. J. Mol. Sci. 2018, 19, 3538. [Google Scholar] [CrossRef] [Green Version]
- Lehrich, B.M.; Liang, Y.; Fiandaca, M.S. Foetal bovine serum influence on in vitro extracellular vesicle analyses. J. Extracell. Vesicles 2021, 10, e12061. [Google Scholar] [CrossRef]
- Benedikter, B.J.; Volgers, C.; Van Eijck, P.H.; Wouters, E.F.; Savelkoul, P.H.; Reynaert, N.L.; Haenen, G.R.; Rohde, G.G.; Weseler, A.R.; Stassen, F.R. Cigarette smoke extract induced exosome release is mediated by depletion of exofacial thiols and can be inhibited by thiol-antioxidants. Free. Radic. Biol. Med. 2017, 108, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Sanders, D.A. Sulfhydryl Involvement in Fusion Mechanisms. Subcell. Biochem. 2002, 34, 483–514. [Google Scholar] [CrossRef]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [Green Version]
- Nicolás-Ávila, J.A.; Lechuga-Vieco, A.V.; Esteban-Martínez, L.; Sánchez-Díaz, M.; García, E.D.; Santiago, D.J.; Rubio-Ponce, A.; Li, J.L.; Balachander, A.; Quintana, J.A.; et al. A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell 2020, 183, 94–109.e23. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef] [Green Version]
- Aarreberg, L.D.; Esser-Nobis, K.; Driscoll, C.; Shuvarikov, A.; Roby, J.A.; Gale, M. Interleukin-1β induces mtdna release to activate innate immune signaling via cgas-sting. Mol. Cell 2019, 74, 801–815.e806. [Google Scholar] [CrossRef] [PubMed]
- Heijink, I.H.; Pouwels, S.D.; Leijendekker, C.; De Bruin, H.G.; Zijlstra, G.J.; Van Der Vaart, H.; Hacken, N.H.T.T.; Van Oosterhout, A.J.M.; Nawijn, M.C.; van der Toorn, M. Cigarette Smoke–Induced Damage-Associated Molecular Pattern Release from Necrotic Neutrophils Triggers Proinflammatory Mediator Release. Am. J. Respir. Cell Mol. Biol. 2015, 52, 554–562. [Google Scholar] [CrossRef] [PubMed]
- King, P.T.; Sharma, R.; O’Sullivan, K.M.; Callaghan, J.; Dousha, L.; Thomas, B.; Ruwanpura, S.; Lim, S.; Farmer, M.W.; Jennings, B.R.; et al. Deoxyribonuclease 1 reduces pathogenic effects of cigarette smoke exposure in the lung. Sci. Rep. 2017, 7, 12128. [Google Scholar] [CrossRef] [PubMed]
- Giordano, L.; Aneja, M.K.; Sommer, N.; Alebrahimdehkordi, N.; Seraji, A.; Weissmann, N.; Rudolph, C.; Plank, C.; Jacobs, H.T.; Szibor, M. Alternative oxidase encoded by sequence-optimized and chemically-modified RNA transfected into mammalian cells is catalytically active. Gene Ther. 2021, 1–10. [Google Scholar] [CrossRef]
- Aghapour, M.; Remels, A.H.V.; Pouwels, S.D.; Bruder, D.; Hiemstra, P.S.; Cloonan, S.M.; Heijink, I.H. Mitochondria: At the crossroads of regulating lung epithelial cell function in chronic obstructive pulmonary disease. Am. J. Physiol. Cell. Mol. Physiol. 2020, 318, L149–L164. [Google Scholar] [CrossRef]
- Alter, P.; Baker, J.R.; Dauletbaev, N.; Donnelly, L.E.; Pistenmaa, C.; Schmeck, B.; Washko, G.; Vogelmeier, C.F. Update in chronic obstructive pulmonary disease 2019. Am. J. Respir. Crit. Care Med. 2020, 202, 348–355. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giordano, L.; Gregory, A.D.; Pérez Verdaguer, M.; Ware, S.A.; Harvey, H.; DeVallance, E.; Brzoska, T.; Sundd, P.; Zhang, Y.; Sciurba, F.C.; et al. Extracellular Release of Mitochondrial DNA: Triggered by Cigarette Smoke and Detected in COPD. Cells 2022, 11, 369. https://doi.org/10.3390/cells11030369
Giordano L, Gregory AD, Pérez Verdaguer M, Ware SA, Harvey H, DeVallance E, Brzoska T, Sundd P, Zhang Y, Sciurba FC, et al. Extracellular Release of Mitochondrial DNA: Triggered by Cigarette Smoke and Detected in COPD. Cells. 2022; 11(3):369. https://doi.org/10.3390/cells11030369
Chicago/Turabian StyleGiordano, Luca, Alyssa D. Gregory, Mireia Pérez Verdaguer, Sarah A. Ware, Hayley Harvey, Evan DeVallance, Tomasz Brzoska, Prithu Sundd, Yingze Zhang, Frank C. Sciurba, and et al. 2022. "Extracellular Release of Mitochondrial DNA: Triggered by Cigarette Smoke and Detected in COPD" Cells 11, no. 3: 369. https://doi.org/10.3390/cells11030369
APA StyleGiordano, L., Gregory, A. D., Pérez Verdaguer, M., Ware, S. A., Harvey, H., DeVallance, E., Brzoska, T., Sundd, P., Zhang, Y., Sciurba, F. C., Shapiro, S. D., & Kaufman, B. A. (2022). Extracellular Release of Mitochondrial DNA: Triggered by Cigarette Smoke and Detected in COPD. Cells, 11(3), 369. https://doi.org/10.3390/cells11030369