
p53 Promotes Cytokine Expression in Melanoma to Regulate Drug Resistance and Migration


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Cell Treatment
2.2. Reagent and Antibodies
2.3. Lentiviral Production and Infection
2.4. Generation of p53ko Lines
2.5. Preparation of Cellular Lysates and Western Blotting
2.6. RNA Extraction and qPCR
2.7. TCGA Analysis
2.8. mRNA Sequencing and Analysis
2.9. Si-RNA Mediated Gene Silencing
2.10. Transwell Migration Assay
2.11. Cytokine Expression Analysis
2.12. Immunostaining and Microscopy
2.13. Cell-Survival Analysis
2.14. Statistical Analysis of qPCR, Western Blots, Cell Survival and Cytokine Expression
3. Results
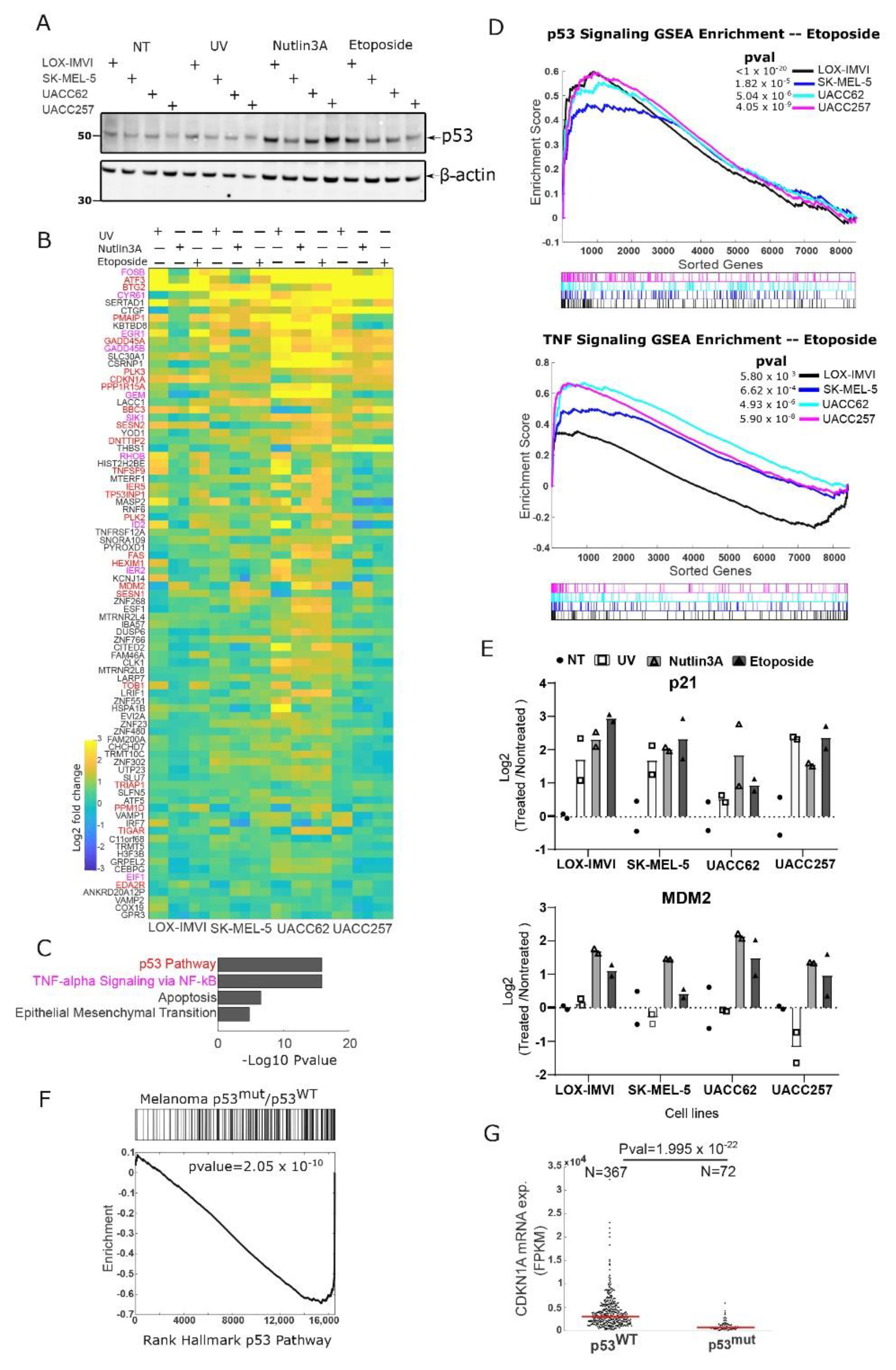
3.1. Melanoma Cell Lines Retain a Functional p53
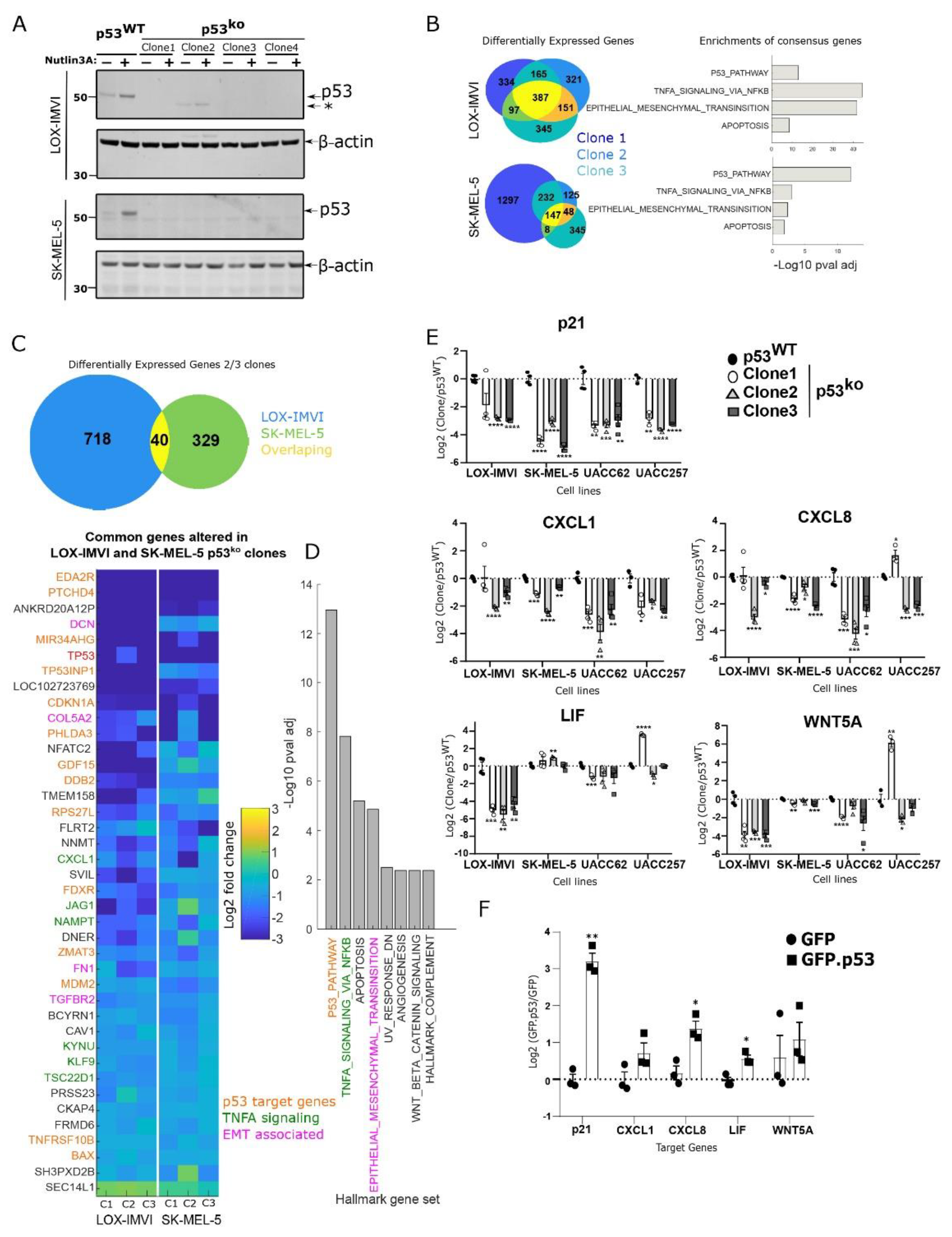
3.2. Loss of p53 in Melanoma Cell Lines Results in Reduced p53 and NF-kB Signaling
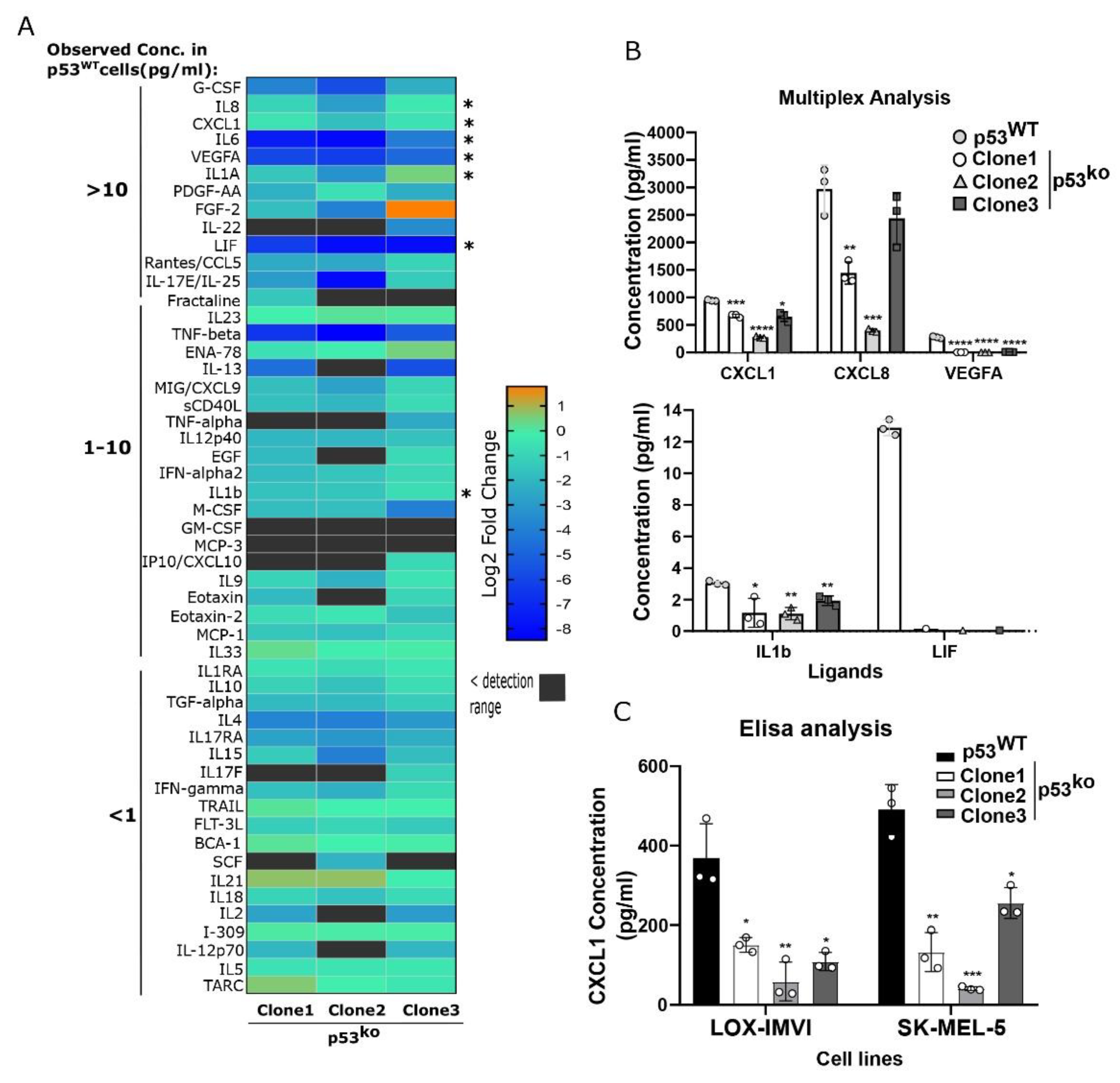
3.3. p53 Loss Reduces Secretion of Cytokines
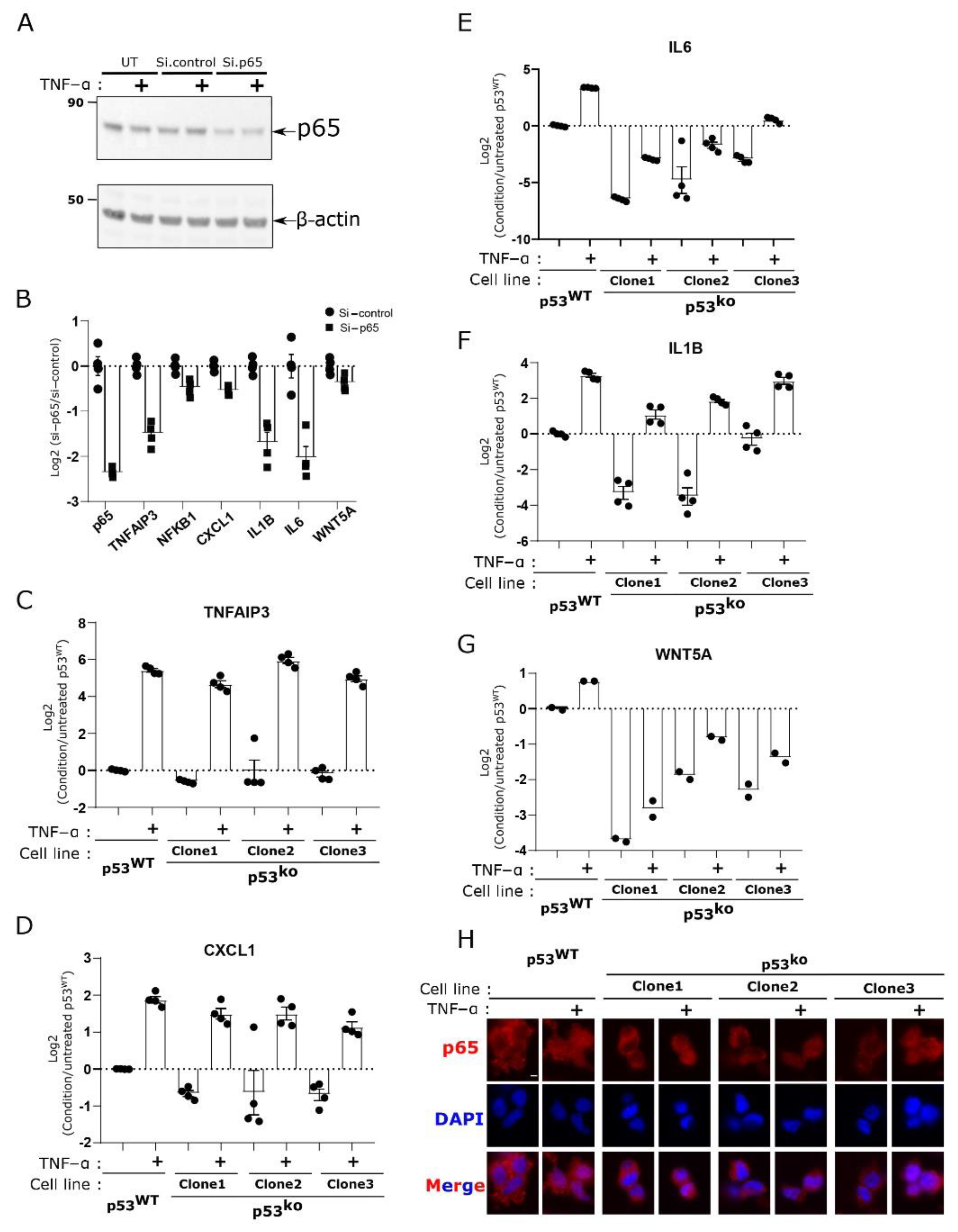
3.4. Key Cytokines Reduced by p53 Loss Are Regulated by NF-kB
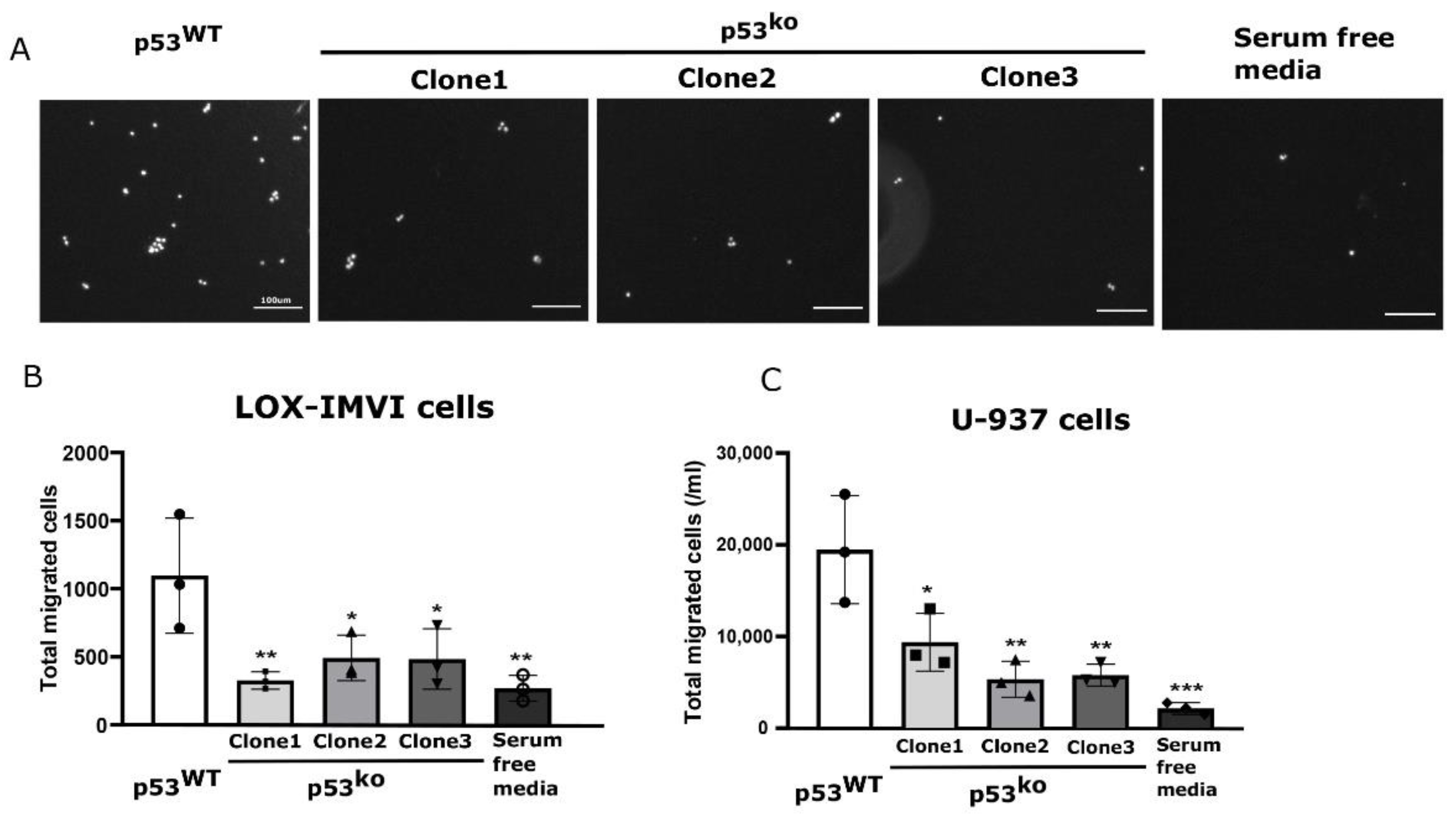
3.5. Loss of p53 Results in Reduced Cell Migration and Sensitivity to BRAF Inhibitor
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, J.; Zhang, C.; Feng, Z. Tumor suppressor p53 and its gain-of-function mutants in cancer. Acta Biochim. Biophys. Sin. 2014, 46, 170–179. [Google Scholar] [CrossRef] [Green Version]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.-P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutation in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Essner, R.; Kuo, C.T.; Wang, H.; Wen, D.R.; Turner, R.R.; Nguyen, T.; Hoon, D. Prognostic implications of p53 overexpression in cutaneous melanoma from sun-exposed and nonexposed sites. Cancer 1998, 82, 309–316. [Google Scholar] [CrossRef]
- Kim, D.W.; Haydu, L.E.; Joon, A.; Bassett, R.L.; Siroy, A.E.; Tetzlaff, M.T.; Routbort, M.J.; Amaria, R.N.; Wargo, J.A.; McQuade, J.L.; et al. Clinicopathological features and clinical outcomes associated with TP53 and BRAFNon-V600 mutations in cutaneous melanoma patients. Cancer 2017, 123, 1372–1381. [Google Scholar] [CrossRef] [Green Version]
- Li, F.P.; Fraumeni, J.F., Jr. Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann. Int. Med. 1969, 71, 747–752. [Google Scholar] [CrossRef] [Green Version]
- Bougeard, G.; Renaux-Petel, M.; Flaman, J.-M.; Charbonnier, C.; Fermey, P.; Belotti, M.; Gauthier-Villars, M.; Stoppa-Lyonnet, D.; Consolino, E.; Brugières, L.; et al. Revisiting Li-Fraumeni Syndrome From TP53 Mutation Carriers. J. Clin. Onco. 2015, 33, 2345–2352. [Google Scholar] [CrossRef]
- Webster, M.R.; Fane, M.E.; Alicea, G.M.; Basu, S.; Kossenkov, A.V.; Marino, G.E.; Douglass, S.M.; Kaur, A.; Ecker, B.L.; Gnanapradeepan, K.; et al. Paradoxical role for Wild-type p53 in driving therapy resistance in melanoma. Mol. Cell 2020, 77, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Hafner, A.; Kublo, L.; Tsabar, M.; Lahav, G.; Stewart-Ornstein, J. Identification of universal and cell-type specific p53 DNA binding. BMC Mol. Cell Biol. 2020, 21, 5. [Google Scholar] [CrossRef] [Green Version]
- Arandkar, S.; Furth, N.; Elisha, Y.; Nataraj, N.B.; van der Kuip, H.; Yarden, Y.; Aulitzky, W.; Ulitsky, I.; Geiger, B.; Oren, M. Altered p53 functionality in cancer-associated fibroblasts contributes to their cancer-supporting features. Proc. Natl. Acad. Sci. USA 2018, 115, 6410–6415. [Google Scholar] [CrossRef] [Green Version]
- Lowe, J.M.; Menendez, D.; Bushel, P.R.; Shatz, M.; Kirk, E.L.; Troester, M.A.; Garantziotis, S.; Fessler, M.B.; Resnick, M.A. p53 and NF-kB Co-regulate Pro-inflammatory Gene Responses in Human Macrophages. Cancer Res. 2014, 74, 2182–2192. [Google Scholar] [CrossRef] [Green Version]
- Herrero, A.B.; Rojas, E.A.; Misiewicz-krzemiska, I.; Krzeminski, P.; Gutierrez, N.C. Molecular mechanism of p53 deregulation in Cancer: An overview in multiple myeloma. Int. J. Mol. Sci. 2016, 17, 2003. [Google Scholar] [CrossRef]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutation in the p53 tumor suppressor gene. Important milestones at the various steps of tumorigenesis. Genes Cancer 2011, 4, 466–474. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; You, D.; Jeong, Y.; Yoon, S.Y.; Kim, S.A.; Kim, S.W.; Nam, S.J.; Lee, J.E. WNT5A augments cell invasiveness by inducing CXCL8 in HER2-positive breast cancer cells. Cytokine 2020, 135, 155213. [Google Scholar] [CrossRef]
- Xu, G.; Wang, H.; Li, W.; Xue, Z.; Luo, Q. Leukemia inhibitory factor inhibits the proliferation of gastric cancer by inducing G1-phase arrest. J. Cell Physiol. 2019, 234, 3613–3620. [Google Scholar] [CrossRef]
- Ishigami, K.; Nosho, K.; Koide, H.; Kanno, S.; Mitsuhashi, K.; Igarashi, H.; Shitani, M.; Motoya, M.; Kimura, Y.; Hasegawa, T.; et al. MicroRNA-31 reflects IL-6 expression in cancer tissue and is related with poor prognosis in bile duct cancer. Carcinogenesis 2018, 39, 1127–1134. [Google Scholar] [CrossRef]
- Zeng, A.; Yin, J.; Li, Y.; Li, R.; Wang, Z.; Zhou, X.; Jin, X.; Shen, F.; Yan, W.; You, Y. miR-129-5p targets Wnt5a to block PKC/ERK/NF-kB and JNK pathways in glioblastoma. Cell Death Dis. 2018, 9, 394. [Google Scholar] [CrossRef] [Green Version]
- Zhou, A.; Scoggin, S.; Gaynor, R.B.; William, N.S. Identification of NF-kb regulatied genes induced by TNFalpha utilizing expression profiling and RNA interference. Oncogene 2003, 22, 2054–2064. [Google Scholar] [CrossRef] [Green Version]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Lim, W.K.; Lyashenko, E.; Califano, A. Master regulators used as breast cancer metastasis classifier. Pac. Symp. Biocomput. 2009, 14, 504–515. [Google Scholar]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Gene Set Enrichment Analysis. Molecular Signatures Database. Available online: https://www.gsea-msigdb.org/gsea/msigdb/ (accessed on 1 August 2021).
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1a-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonist of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [Green Version]
- Acharyya, S.; Oskarsson, T.; Vanharanta, S.; Malladi, S.; Kim, J.; Morris, P.G.; Manova-Todorova, K.; Leversha, M.; Hogg, N.; Seshan, V.E.; et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 2012, 150, 165–178. [Google Scholar] [CrossRef] [Green Version]
- Konieczkowski, D.J.; Johannessen, C.M.; Abudayyeh, O.; Kim, J.W.; Cooper, Z.A.; Piris, A.; Frederick, D.T.; Barzily-Rokni, M.; Straussman, R.; Haq, R.; et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014, 4, 816–827. [Google Scholar] [CrossRef] [Green Version]
- Orgaz, J.L.; Crosas-Molist, E.; Sadok, A.; Perdrix-Rosell, A.; Maiques, O.; Rodriguez-Hernandez, I.; Monger, J.; Mele, S.; Georgouli, M.; Bridgeman, V.; et al. Myosin II reactivation and cytoskeletal remodeling as a hallmark and a vulnerability in melanoma therapy resistance. Cancer Cell 2020, 37, 85–103. [Google Scholar] [CrossRef]
- Germain, M.; Affar, E.B.; D’Aamours, D.; Dixit, V.M.; Salvesen, G.S.; Poirier, G.G. Cleavage of automodified poly(ADP-ribose) polymerase during apoptosis. Evidence for involvement of caspase-7. J. Biol. Chem. 1999, 74, 28379–28384. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, S.H.; Desnoyers, S.; Ottaviano, Y.; Davidson, N.E.; Poirier, G.G. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: An early marker of chemotherapy-induced apoptosis. Cancer Res. 1993, 53, 3976–3985. [Google Scholar]
- Dhawan, P.; Richmond, A. Role of CXCL1 in tumorigenesis of melanoma. J. Leukoc. Biol. 2002, 72, 9–18. [Google Scholar]
- Bald, T.; Quast, T.; Landsberg, J.; Rogava, M.; Glodde, N.; Lopez-Ramos, D.; Kohlmeyer, J.; Riesenberg, S.; Van Den Boorn-Konijnenberg, D.; Hömig-Hölzel, C.; et al. Ultraviolet-radiation-induced inflammation promotes angiotropism and metastasis in melanoma. Nature 2014, 507, 109–113. [Google Scholar] [CrossRef]
- Ryan, K.M.; Ernst, M.K.; Rice, N.R.; Vousden, K.H. Role of NF-kb in p53-mediated programmed cell death. Nature 2000, 404, 892–897. [Google Scholar] [CrossRef]
- Pavlakis, E.; Stiewe, T. p53’s extended reach: The mutant p53 secretome. Biomolecules 2020, 10, 307. [Google Scholar] [CrossRef] [Green Version]
- Schneider, G.; Henrich, A.; Greiner, G.; Wolf, V.; Lovas, A.; Wieczorek, M.; Wagner, T.; Reichardt, S.; Von Werder, A.; Schmid, R.M.; et al. Cross talk between stimulated NF-kb and the tumor suprressor p53. Oncogene 2010, 29, 2795–2806. [Google Scholar] [CrossRef] [Green Version]
- Avery-Kiejda, K.A.; Bowden, N.A.; Croft, A.J.; Scurr, L.L.; Kairupan, C.F.; Ashton, K.A.; Talseth-Palmer, B.A.; Rizos, H.; Zhang, X.D.; Scott, R.J.; et al. p53 in human melanoma fails to regulate target genes associated with apoptosis and the cell cycle and may contribute to proliferation. BMC Cancer 2011, 11, 203. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandya, P.; Kublo, L.; Stewart-Ornstein, J. p53 Promotes Cytokine Expression in Melanoma to Regulate Drug Resistance and Migration. Cells 2022, 11, 405. https://doi.org/10.3390/cells11030405
Pandya P, Kublo L, Stewart-Ornstein J. p53 Promotes Cytokine Expression in Melanoma to Regulate Drug Resistance and Migration. Cells. 2022; 11(3):405. https://doi.org/10.3390/cells11030405
Chicago/Turabian StylePandya, Pinakin, Lyubov Kublo, and Jacob Stewart-Ornstein. 2022. "p53 Promotes Cytokine Expression in Melanoma to Regulate Drug Resistance and Migration" Cells 11, no. 3: 405. https://doi.org/10.3390/cells11030405
APA StylePandya, P., Kublo, L., & Stewart-Ornstein, J. (2022). p53 Promotes Cytokine Expression in Melanoma to Regulate Drug Resistance and Migration. Cells, 11(3), 405. https://doi.org/10.3390/cells11030405