
Adoptive Cellular Therapy for Multiple Myeloma Using CAR- and TCR-Transgenic T Cells: Response and Resistance


Abstract
:1. Cellular Therapy in Multiple Myeloma
2. CAR-T Cell Therapy in MM and the Evolving Generations of CARs
2.1. General Aspects of CAR Constructs
2.2. Potential Target Structures for CAR-Therapy in MM
2.3. Targeting BCMA in MM CAR-Treatment

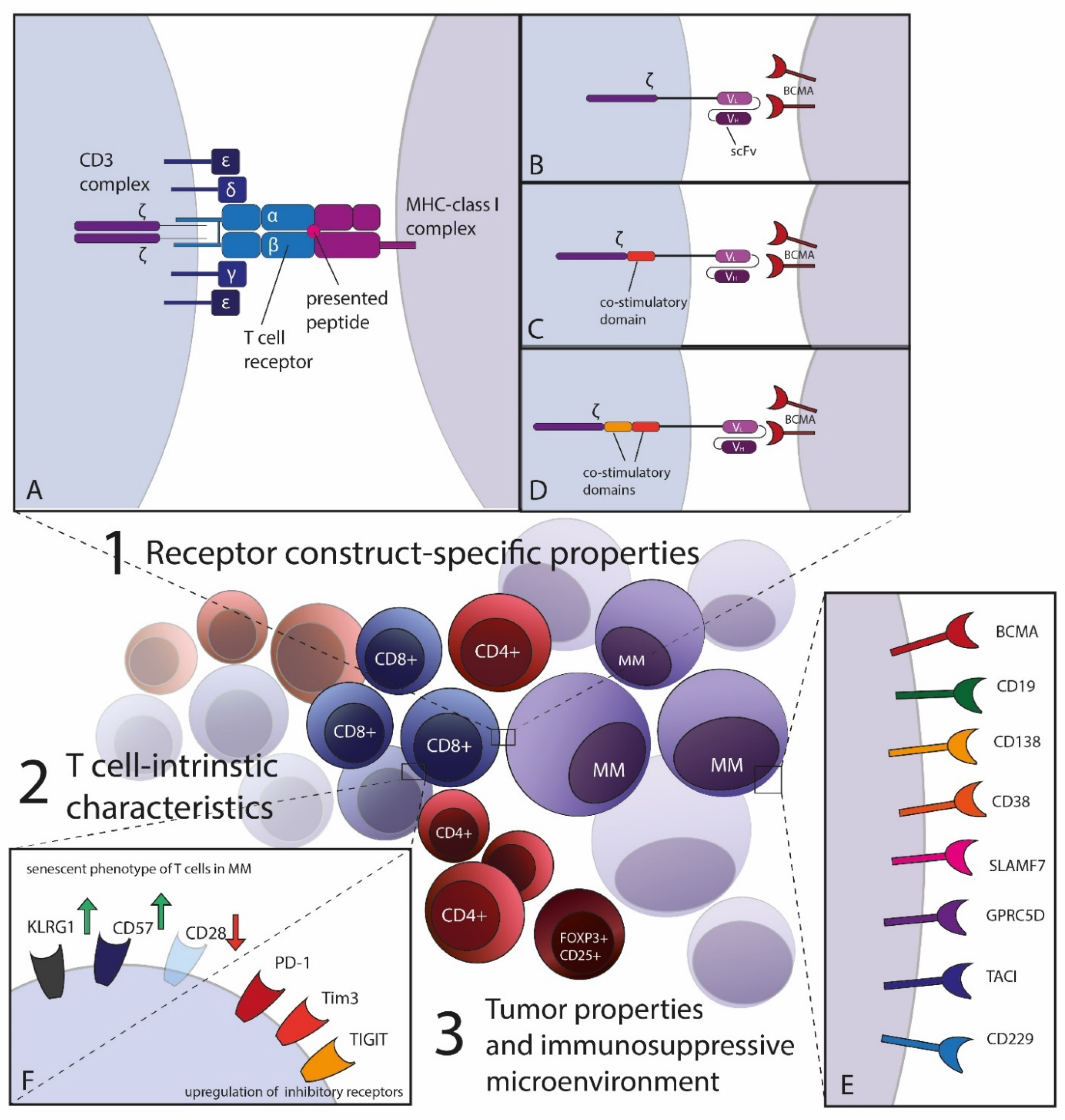{kind=link}
| Trial Number/Name | Sponsor | CAR Construct | Phase | n 1 | Origin of scFv | Co-Stimulatory Domain | Dose 2 | Conditioning Therapy 3 | ORR 4 | CRS 4 (All/ gr 3–4) | ICAN 4 (All/ gr 3–4) | Further Modifications/ Comments | References |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NCT02215967 | National Cancer Institute (NCI) | I | 30 | murine | CD28 | 0.3–9.0 × 106 | CP/ Flu | 81% | 94% | N.A. | [70] | ||
| NCT03070327 | Memorial Sloan Kettering Cancer Center | MCARH171 | I | 20 | N.A. | 4-1BB | 1 × 106– 1 × 107 | CP | N.A. | N.A. | N.A. | ±lenalidomide EGFRt (suicide gene) | |
| NCT03274219/CRB-402 | bluebird bio | bb21217 | I | 72 | murine | 4-1BB | 150, 300 or 450 × 106 | CP/ Flu | 69% | 75%/ 4.2% | 15%/ N.A. | PI3K inhibitor bb007 during ex vivo culture to enrich the drug product (DP) for memory-like T cells | [77] |
| NCT03288493 | Poseida Therapeu-tics, Inc. | P-BCMA-101 (CARTyrin) | I/II | 220 | human | 4-1BB | 0.75–15 × 106 | CP/ Flu | N.A. | N.A. | N.A. | stem cell memory T cell subset; Rimiducid (safety switch activator) can be administered as indicated | |
| NCT03338972 | Fred Hutchinson Cancer Research Center | FCARH143 | I | 28 | human | 4-1BB | 50–150 × 106; potential second dose | CP/ Flu | N.A. | N.A. | N.A. | EGFRt (suicide gene); infusion of CD8+ and CD4+ T cells in a 1:1 ratio | |
| NCT03361748/KarMMa | Celgene | bb2121/ Ide-cel | II | 149 | murine | 4-1BB | 150–450 × 106 | CP/ Flu | 73% | 84%/ 5% | 18%/ 3% | [6,7] | |
| NCT03430011/EVOLVE | Juno Therapeutics, Inc. | JCARH125 /Orva-cel | I/II | human | 4-1BB | lower: 50 or 100 × 106, higher: 300, 450 or 600 × 106 | CP/ Flu | 91% | N.A./ 2% | N.A./ 4% | 1:1 CD4/CD8 ratio preselected prior to transduction and expansion | [78] | |
| NCT03548207/CARTITUDE-1 | Janssen Research & Development, LLC | JNJ-68284528/ LCAR-B38M/ Ciltacabtagene autoleucel (Cilta-cel) | Ib/II | 126 | alpaca | 4-1BB | 0.75 × 106 | CP/ Flu | 97,9% | 95%/ 4% | 21%/ 9% | [72,73] | |
| NCT03602612 | National Cancer Institute (NCI) | FHVH33 | I | 31 | human | 4-1BB | 0.75–12 × 106 | CP/ Flu | N.A. | N.A. | N.A. | fully human heavy-chain variable domain | [79] |
| NCT03758417/CARTIFAN-1 | Nanjing Legend Biotech Co. | JNJ-68284528/ LCAR-B38M/ Ciltacabtagene autoleucel (Cilta-cel) | II | 60 | alpaca | 4-1BB | N.A. | N.A. | N.A. | N.A. | N.A. | ||
| NCT04133636/CARTITUDE-2 | Janssen Research & Development, LLC | JNJ-68284528/ LCAR-B38M/ Ciltacabtagene autoleucel (Cilta-cel) | III | 160 | alpaca | 4-1BB | 0.75 × 106 | CP/ Flu | 95% | 85%/ 10% | 20%/ 0% | +Lenalidomide/Daratumumab/ Bortezomib/ Dexamethasone | [80] |
| NCT04309981 | Sara V. Latorre | ARI0002h | I/II | 36 | humanized | 4-1BB | fractionated 3 × 106 + second infusion | CP/ Flu | 96% | 87%/ 0% | 0%/ 0% | fractionated (10%/30%/60% with at least 24 h in between) and multiple infusions; higher CD4/CD8 ratio correlated with more stringent CR | [81,82] |
| NCT05066646/FUMANBA-1 | Nanjing IASO Biotherapeutics Co., Ltd. | CT103A | I//II | 132 | human | 4-1BB | 1.0 × 106 | CP/ Flu | 94.4% | 93%/ 2.8% | 1.4%/ 0% | enrollment of patients with prior murine BCMA-CAR administrations | [83,84] |
2.4. Engineering the BCMA-CAR Construct
2.4.1. Extracellular Target-Binding Domain: scFV
2.4.2. Hinge Region/Spacer Domain and Transmembrane Domain
2.4.3. Intracellular Signaling Domain
2.4.4. Tonic Signaling
2.4.5. Reducing Toxicities
3. The TCR-Based Therapy Approach and Its Potential for Treatment of MM
3.1. General Aspects of the TCR-Peptide-MHC Interaction
3.2. Potential Targets for TCR-Based Therapy in MM
3.3. Application and Engineering Approaches for TCR-T Cells in MM
4. Therapy Resistance: Obstacles for CAR as Well as TCR T Cell-Based Therapy Approaches in MM and Engineering Strategies
4.1. Counteracting Unfavorable T Cell Intrinsic Qualities
4.2. Tumor Resistance Related to Antigen Expression
4.3. MM-Defined Suppression of T Cell Action
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gandhi, U.H.; Cornell, R.F.; Lakshman, A.; Gahvari, Z.J.; McGehee, E.; Jagosky, M.H.; Gupta, R.; Varnado, W.; Fiala, M.A.; Chhabra, S.; et al. Outcomes of patients with multiple myeloma refractory to CD38-targeted monoclonal antibody therapy. Leukemia 2019, 33, 2266–2275. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; A Dimopoulos, M.; Kastritis, E.; Terpos, E.; Nahi, H.; Goldschmidt, H.; Hillengass, J.; Leleu, X.; Beksac, M.; Alsina, M.; et al. Natural history of relapsed myeloma, refractory to immunomodulatory drugs and proteasome inhibitors: A multicenter IMWG study. Leukemia 2017, 31, 2443–2448. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and Toxicity Management of 19–28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2014, 6, 224ra25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, L.D., Jr.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef]
- Dhakal, B.; Vesole, D.H.; Hari, P. Allogeneic stem cell transplantation for multiple myeloma: Is there a future? Bone Marrow Transplant. 2016, 51, 492–500. [Google Scholar] [CrossRef]
- Noonan, K.A.; Huff, C.A.; Davis, J.; Lemas, M.V.; Fiorino, S.; Bitzan, J.; Ferguson, A.; Emerling, A.; Luznik, L.; Matsui, W.; et al. Adoptive transfer of activated marrow-infiltrating lymphocytes induces measurable antitumor immunity in the bone marrow in multiple myeloma. Sci. Transl. Med. 2015, 7, 288ra78. [Google Scholar] [CrossRef] [Green Version]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Zhao, L.; Cao, Y.J. Engineered T Cell Therapy for Cancer in the Clinic. Front. Immunol. 2019, 10, 2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Li, X.; Zhou, W.-L.; Huang, Y.; Liang, X.; Jiang, L.; Yang, X.; Sun, J.; Li, Z.; Han, W.-D.; et al. Genetically engineered T cells for cancer immunotherapy. Signal Transduct. Target. Ther. 2019, 4, 35. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.V.; et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Attal, M.; Richardson, P.G.; Rajkumar, S.V.; San-Miguel, J.; Beksac, M.; Spicka, I.; Leleu, X.; Schjesvold, F.; Moreau, P.; Dimopoulos, M.A.; et al. Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): A randomised, multicentre, open-label, phase 3 study. Lancet 2019, 394, 2096–2107. [Google Scholar] [CrossRef]
- Dimopoulos, M.; Quach, H.; Mateos, M.-V.; Landgren, O.; Leleu, X.; Siegel, D.; Weisel, K.; Yang, H.; Klippel, Z.; Zahlten-Kumeli, A.; et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): Results from a randomised, multicentre, open-label, phase 3 study. Lancet 2020, 396, 186–197. [Google Scholar] [CrossRef]
- Cohen, A.D.; Lendvai, N.; Nataraj, S.; Imai, N.; Jungbluth, A.A.; Tsakos, I.; Rahman, A.; Mei, A.H.-C.; Singh, H.; Zarychta, K.; et al. Autologous Lymphocyte Infusion Supports Tumor Antigen Vaccine–Induced Immunity in Autologous Stem Cell Transplant for Multiple Myeloma. Cancer Immunol. Res. 2019, 7, 658–669. [Google Scholar] [CrossRef]
- Rapoport, A.P.; Aqui, N.A.; Stadtmauer, E.A.; Vogl, D.T.; Xu, Y.Y.; Kalos, M.; Cai, L.; Fang, H.-B.; Weiss, B.M.; Badros, A.; et al. Combination Immunotherapy after ASCT for Multiple Myeloma Using MAGE-A3/Poly-ICLC Immunizations Followed by Adoptive Transfer of Vaccine-Primed and Costimulated Autologous T Cells. Clin. Cancer Res. 2014, 20, 1355–1365. [Google Scholar] [CrossRef] [Green Version]
- Mikkilineni, L.; Kochenderfer, J.N. CAR T cell therapies for patients with multiple myeloma. Nat. Rev. Clin. Oncol. 2020, 18, 71–84. [Google Scholar] [CrossRef]
- Hossain, N.M.; Chapuis, A.G.; Walter, R.B. T-Cell Receptor-Engineered Cells for the Treatment of Hematologic Malignancies. Curr. Hematol. Malign-Rep. 2016, 11, 311–317. [Google Scholar] [CrossRef]
- Larson, R.C.; Maus, M.V. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat. Cancer 2021, 21, 145–161. [Google Scholar] [CrossRef]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T Cell Activation. Annu. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef] [PubMed]
- Irving, B.A.; Weiss, A. The cytoplasmic domain of the T cell receptor ζ chain is sufficient to couple to receptor-associated signal transduction pathways. Cell 1991, 64, 891–901. [Google Scholar] [CrossRef]
- Letourneur, F.; Klausner, R.D. T-cell and basophil activation through the cytoplasmic tail of T-cell-receptor zeta family proteins. Proc. Natl. Acad. Sci. USA 1991, 88, 8905–8909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwu, P.; Shafer, G.E.; Treisman, J.; Schindler, D.G.; Gross, G.; Cowherd, R.; Rosenberg, S.A.; Eshhar, Z. Lysis of ovarian cancer cells by human lymphocytes redirected with a chimeric gene composed of an antibody variable region and the Fc receptor gamma chain. J. Exp. Med. 1993, 178, 361–366. [Google Scholar] [CrossRef] [Green Version]
- Hwu, P.; Yang, J.C.; Cowherd, R.; Treisman, J.; Shafer, G.E.; Eshhar, Z.; Rosenberg, S.A. In vivo antitumor activity of T cells redirected with chimeric antibody/T-cell receptor genes. Cancer Res. 1995, 55, 3369–3373. [Google Scholar]
- Brocker, T.; Karjalainen, K. Signals through T cell receptor-zeta chain alone are insufficient to prime resting T lymphocytes. J. Exp. Med. 1995, 181, 1653–1659. [Google Scholar] [CrossRef]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Rivière, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ/CD28 receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef]
- Hombach, A.; Wieczarkowiecz, A.; Marquardt, T.; Heuser, C.; Usai, L.; Pohl, C.; Seliger, B.; Abken, H. Tumor-Specific T Cell Activation by Recombinant Immunoreceptors: CD3ζ Signaling and CD28 Costimulation Are Simultaneously Required for Efficient IL-2 Secretion and Can Be Integrated Into One Combined CD28/CD3ζ Signaling Receptor Molecule. J. Immunol. 2001, 167, 6123–6131. [Google Scholar] [CrossRef] [Green Version]
- Bishop, M.R.; Maziarz, R.T.; Waller, E.K.; Jäger, U.; Westin, J.R.; McGuirk, J.P.; Fleury, I.; Holte, H.; Borchmann, P.; Del Corral, C.; et al. Tisagenlecleucel in relapsed/refractory diffuse large B-cell lymphoma patients without measurable disease at infusion. Blood Adv. 2019, 3, 2230–2236. [Google Scholar] [CrossRef] [Green Version]
- Carpenito, C.; Milone, M.C.; Hassan, R.; Simonet, J.C.; Lakhal, M.; Suhoski, M.M.; Varela-Rohena, A.; Haines, K.M.; Heitjan, D.F.; Albelda, S.M.; et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc. Natl. Acad. Sci. USA 2009, 106, 3360–3365. [Google Scholar] [CrossRef] [Green Version]
- Milone, M.C.; Fish, J.D.; Carpenito, C.; Carroll, R.G.; Binder, G.K.; Teachey, D.; Samanta, M.; Lakhal, M.; Gloss, B.; Danet-Desnoyers, G.; et al. Chimeric Receptors Containing CD137 Signal Transduction Domains Mediate Enhanced Survival of T Cells and Increased Antileukemic Efficacy In Vivo. Mol. Ther. 2009, 17, 1453–1464. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, J.; Zhang, X.; Lu, X.-A.; Xiong, M.; Zhang, J.; Zhou, X.; Qi, F.; He, T.; Ding, Y.; et al. Efficacy and Safety of CD28- or 4-1BB-Based CD19 CAR-T Cells in B Cell Acute Lymphoblastic Leukemia. Mol. Ther.-Oncolytics 2020, 18, 272–281. [Google Scholar] [CrossRef]
- Zhao, Z.; Condomines, M.; Van Der Stegen, S.J.C.; Perna, F.; Kloss, C.C.; Gunset, G.; Plotkin, J.; Sadelain, M. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 2015, 28, 415–428. [Google Scholar] [CrossRef] [Green Version]
- Guedan, S.; Posey, A.D., Jr.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3, e96976. [Google Scholar] [CrossRef] [Green Version]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; Mcgettigan, S.E.; Posey, A.D.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Boroughs, A.C.; Larson, R.C.; Marjanovic, N.D.; Gosik, K.; Castano, A.P.; Porter, C.B.; Lorrey, S.J.; Ashenberg, O.; Jerby, L.; Hofree, M.; et al. A Distinct Transcriptional Program in Human CAR T Cells Bearing the 4-1BB Signaling Domain Revealed by scRNA-Seq. Mol. Ther. 2020, 28, 2577–2592. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef] [Green Version]
- Porter, D.L.; Hwang, W.-T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef] [Green Version]
- Ying, Z.; He, T.; Wang, X.; Zheng, W.; Lin, N.; Tu, M.; Xie, Y.; Ping, L.; Zhang, C.; Liu, W.; et al. Parallel Comparison of 4-1BB or CD28 Co-stimulated CD19-Targeted CAR-T Cells for B Cell Non-Hodgkin’s Lymphoma. Mol. Ther.-Oncolytics 2019, 15, 60–68. [Google Scholar] [CrossRef] [Green Version]
- Anagnostou, T.; Riaz, I.B.; Hashmi, S.K.; Murad, M.H.; Kenderian, S.S. Anti-CD19 chimeric antigen receptor T-cell therapy in acute lymphocytic leukaemia: A systematic review and meta-analysis. Lancet Haematol. 2020, 7, e816–e826. [Google Scholar] [CrossRef]
- Wang, J.; Jensen, M.; Lin, Y.; Sui, X.; Chen, E.; Lindgren, C.G.; Till, B.; Raubitschek, A.; Forman, S.J.; Qian, X.; et al. Optimizing Adoptive Polyclonal T Cell Immunotherapy of Lymphomas, Using a Chimeric T Cell Receptor Possessing CD28 and CD137 Costimulatory Domains. Hum. Gene Ther. 2007, 18, 712–725. [Google Scholar] [CrossRef]
- Zhong, X.-S.; Matsushita, M.; Plotkin, J.; Riviere, I.; Sadelain, M. Chimeric Antigen Receptors Combining 4-1BB and CD28 Signaling Domains Augment PI3kinase/AKT/Bcl-XL Activation and CD8+ T Cell–mediated Tumor Eradication. Mol. Ther. 2010, 18, 413–420. [Google Scholar] [CrossRef]
- Karlsson, H.; Svensson, E.; Gigg, C.; Jarvius, M.; Olsson-Strömberg, U.; Savoldo, B.; Dotti, G.; Loskog, A. Evaluation of Intracellular Signaling Downstream Chimeric Antigen Receptors. PLoS ONE 2015, 10, e0144787. [Google Scholar] [CrossRef]
- Enblad, G.; Karlsson, H.; Gammelgård, G.; Wenthe, J.; Lövgren, T.; Amini, R.M.; Wikstrom, K.I.; Essand, M.; Savoldo, B.; Hallböök, H.; et al. A Phase I/IIa Trial Using CD19-Targeted Third-Generation CAR T Cells for Lymphoma and Leukemia. Clin. Cancer Res. 2018, 24, 6185–6194. [Google Scholar] [CrossRef] [Green Version]
- Till, B.G.; Jensen, M.C.; Wang, J.; Qian, X.; Gopal, A.K.; Maloney, D.G.; Lindgren, C.G.; Lin, Y.; Pagel, J.M.; Budde, L.E.; et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: Pilot clinical trial results. Blood 2012, 119, 3940–3950. [Google Scholar] [CrossRef] [Green Version]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef]
- Zhang, L.; Kerkar, S.P.; Yu, Z.; Zheng, Z.; Yang, S.; Restifo, N.P.; Rosenberg, S.A.; Morgan, R.A. Improving Adoptive T Cell Therapy by Targeting and Controlling IL-12 Expression to the Tumor Environment. Mol. Ther. 2011, 19, 751–759. [Google Scholar] [CrossRef]
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. IL-12 Release by Engineered T Cells Expressing Chimeric Antigen Receptors Can Effectively Muster an Antigen-Independent Macrophage Response on Tumor Cells That Have Shut Down Tumor Antigen Expression. Cancer Res. 2011, 71, 5697–5706. [Google Scholar] [CrossRef] [Green Version]
- Luangwattananun, P.; Junking, M.; Sujjitjoon, J.; Wutti-In, Y.; Poungvarin, N.; Thuwajit, C.; Yenchitsomanus, P.-T. Fourth-generation chimeric antigen receptor T cells targeting folate receptor alpha antigen expressed on breast cancer cells for adoptive T cell therapy. Breast Cancer Res. Treat. 2021, 186, 25–36. [Google Scholar] [CrossRef]
- Zhou, X.; Tu, S.; Wang, C.; Huang, R.; Deng, L.; Song, C.; Yue, C.; He, Y.; Yang, J.; Liang, Z.; et al. Phase I Trial of Fourth-Generation Anti-CD19 Chimeric Antigen Receptor T Cells Against Relapsed or Refractory B Cell Non-Hodgkin Lymphomas. Front. Immunol. 2020, 11, 3034. [Google Scholar] [CrossRef] [PubMed]
- Seif, M.; Einsele, H.; Löffler, J. CAR T Cells Beyond Cancer: Hope for Immunomodulatory Therapy of Infectious Diseases. Front. Immunol. 2019, 10, 2711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; Xu, Q.; Pu, C.; Zhu, K.; Lu, C.; Jiang, Y.; Xiao, L.; Han, Y.; Lu, L. Therapeutic efficacy of anti-CD19 CAR-T cells in a mouse model of systemic lupus erythematosus. Cell. Mol. Immunol. 2020, 18, 1896–1903. [Google Scholar] [CrossRef] [PubMed]
- Mougiakakos, D.; Krönke, G.; Völkl, S.; Kretschmann, S.; Aigner, M.; Kharboutli, S.; Böltz, S.; Manger, B.; Mackensen, A.; Schett, G. CD19-Targeted CAR T Cells in Refractory Systemic Lupus Erythematosus. N. Engl. J. Med. 2021, 385, 567–569. [Google Scholar] [CrossRef] [PubMed]
- Teoh, P.J.; Chng, W.J. CAR T-cell therapy in multiple myeloma: More room for improvement. Blood Cancer J. 2021, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Garfall, A.; Maus, M.; Hwang, W.-T.; Lacey, S.F.; Mahnke, Y.; Melenhorst, J.J.; Zheng, Z.; Vogl, D.T.; Cohen, A.; Weiss, B.M.; et al. Chimeric Antigen Receptor T Cells against CD19 for Multiple Myeloma. N. Engl. J. Med. 2015, 373, 1040–1047. [Google Scholar] [CrossRef]
- Prommersberger, S.; Reiser, M.; Beckmann, J.; Danhof, S.; Amberger, M.; Quade-Lyssy, P.; Einsele, H.; Hudecek, M.; Bonig, H.; Ivics, Z. CARAMBA: A first-in-human clinical trial with SLAMF7 CAR-T cells prepared by virus-free Sleeping Beauty gene transfer to treat multiple myeloma. Gene Ther. 2021, 28, 560–571. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor–transduced T cells. Blood 2012, 119, 2709–2720. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Wilson, W.H.; Janik, J.E.; Dudley, M.E.; Stetler-Stevenson, M.; Feldman, S.A.; Maric, I.; Raffeld, M.; Nathan, D.-A.N.; Lanier, B.J.; et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010, 116, 4099–4102. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Smith, E.L.; Harrington, K.; Staehr, M.; Masakayan, R.; Jones, J.; Long, T.J.; Ng, K.Y.; Ghoddusi, M.; Purdon, T.J.; Wang, X.; et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Mailankody, S.; Diamonte, C.; Fitzgerald, L.; Kane, P.; Wang, X.; Sikder, D.S.; Senechal, B.; Bermudez, V.P.; Frias, D.; Morgan, J.; et al. Phase I First-in-Class Trial of MCARH109, a G Protein Coupled Receptor Class C Group 5 Member D (GPRC5D) Tar-geted CAR T Cell Therapy in Patients with Relapsed or Refractory Multiple Myeloma. In Proceedings of the 63rd ASH Annual Meeting and Exposition, Atlanta, GA, USA, 11–14 December 2021. [Google Scholar]
- Laâbi, Y.; Gras, M.; Carbonnel, F.; Brouet, J.; Berger, R.; Larsen, C.; Tsapis, A. A new gene, BCM, on chromosome 16 is fused to the interleukin 2 gene by a t(4;16)(q26;p13) translocation in a malignant T cell lymphoma. EMBO J. 1992, 11, 3897–3904. [Google Scholar] [CrossRef]
- Laabi, Y.; Gras, M.-P.; Brouet, J.-C.; Berger, R.; Larsen, C.-J.; Tsapis, A. The BCMA gene, preferentially expressed during B lymphoid maturation, is bidirectionally transcribed. Nucleic Acids Res. 1994, 22, 1147–1154. [Google Scholar] [CrossRef]
- Madry, C.; Laabi, Y.; Callebaut, I.; Roussel, J.; Hatzoglou, A.; Le Coniat, M.; Mornon, J.P.; Berger, R.; Tsapis, A. The characterization of murine BCMA gene defines it as a new member of the tumor necrosis factor receptor superfamily. Int. Immunol. 1998, 10, 1693–1702. [Google Scholar] [CrossRef]
- Xu, S.; Lam, K.-P. B-Cell Maturation Protein, Which Binds the Tumor Necrosis Factor Family Members BAFF and APRIL, Is Dispensable for Humoral Immune Responses. Mol. Cell. Biol. 2001, 21, 4067–4074. [Google Scholar] [CrossRef] [Green Version]
- Tai, Y.-T.; Acharya, C.; An, G.; Moschetta, M.; Zhong, M.Y.; Feng, X.; Cea, M.; Cagnetta, A.; Wen, K.; Van Eenennaam, H.; et al. APRIL and BCMA promote human multiple myeloma growth and immunosuppression in the bone marrow microenvironment. Blood 2016, 127, 3225–3236. [Google Scholar] [CrossRef] [Green Version]
- Novak, A.J.; Darce, J.R.; Arendt, B.K.; Harder, B.; Henderson, K.; Kindsvogel, W.; Gross, J.A.; Greipp, P.R.; Jelinek, D.F. Expression of BCMA, TACI, and BAFF-R in multiple myeloma: A mechanism for growth and survival. Blood 2004, 103, 689–694. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, R.O.; Evbuomwan, M.O.; Pittaluga, S.; Rose, J.J.; Raffeld, M.; Yang, S.; Gress, R.E.; Hakim, F.T.; Kochenderfer, J.N. B-cell Maturation Antigen Is a Promising Target for Adoptive T-cell Therapy of Multiple Myeloma. Clin. Cancer Res. 2013, 19, 2048–2060. [Google Scholar] [CrossRef] [Green Version]
- Brudno, J.N.; Maric, I.; Hartman, S.D.; Rose, J.J.; Wang, M.; Lam, N.; Stetler-Stevenson, M.; Salem, D.; Yuan, C.; Pavletic, S.; et al. T Cells Genetically Modified to Express an Anti–B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J. Clin. Oncol. 2018, 36, 2267–2280. [Google Scholar] [CrossRef]
- Friedman, K.M.; Garrett, T.E.; Evans, J.W.; Horton, H.M.; Latimer, H.J.; Seidel, S.L.; Horvath, C.J.; Morgan, R.A. Effective Targeting of Multiple B-Cell Maturation Antigen–Expressing Hematological Malignances by Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor T Cells. Hum. Gene Ther. 2018, 29, 585–601. [Google Scholar] [CrossRef] [Green Version]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): A phase 1b/2 open-label study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef]
- Martin, T.; Usmani, S.Z.; Berdeja, J.G.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Hari, P.; Avigan, D.; Deol, A.; Htut, M.; et al. Updated Results from CARTITUDE-1: Phase 1b/2Study of Ciltacabtagene Autoleucel, a B-Cell Maturation Antigen-Directed Chimeric Antigen Receptor T Cell Therapy, in Patients With Relapsed/Refractory Multiple Myeloma. Blood 2021, 138, 549. [Google Scholar] [CrossRef]
- Mateos, M.V.; Weisel, K.; Martin, T.; Berdeja, J.G.; Jakubowiak, A.; Stewart, A.K.; Jagannath, S.; Lin, Y.; Diels, J.; Ghilotti, F.; et al. Ciltacabtagene Autoleucel for Triple-Class Exposed Multiple Myeloma: Adjusted Comparisons of CARTITUDE-1 Patient Outcomes Versus Therapies from Real-World Clinical Practice from the LocoMMo-tion Prospective Study. In Proceedings of the 63rd ASH Annual Meeting and Exposition, Atlanta, GA, USA, 11–14 December 2021. [Google Scholar]
- Brudno, J.N.; Lam, N.; Vanasse, D.; Shen, Y.-W.; Rose, J.J.; Rossi, J.; Xue, A.; Bot, A.; Scholler, N.; Mikkilineni, L.; et al. Safety and feasibility of anti-CD19 CAR T cells with fully human binding domains in patients with B-cell lymphoma. Nat. Med. 2020, 26, 270–280. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Somerville, R.P.; Lu, T.; Yang, J.C.; Sherry, R.M.; Feldman, S.A.; McIntyre, L.; Bot, A.; Rossi, J.; Lam, N.; et al. Long-Duration Complete Remissions of Diffuse Large B Cell Lymphoma after Anti-CD19 Chimeric Antigen Receptor T Cell Therapy. Mol. Ther. 2017, 25, 2245–2253. [Google Scholar] [CrossRef] [Green Version]
- Raje, N.S.; Shah, N.; Jagannath, S.; Kaufman, J.L.; Siegel, D.S.; Munshi, N.C.; Rosenblatt, J.; Lin, Y.; Jakubowiak, A.; Timm, A.; et al. Updated Clinical and Correlative Results from the Phase I CRB-402 Study of the BCMA-Targeted CAR T Cell Therapy bb21217 in Patients with Relapsed and Refractory Multiple Myeloma. Blood 2021, 138, 548. [Google Scholar] [CrossRef]
- Mailankody, S.; Jakubowiak, A.J.; Htut, M.; Costa, L.J.; Lee, K.; Ganguly, S.; Kaufman, J.L.; Siegel, D.S.D.; Bensinger, W.; Cota, M.; et al. Orvacabtagene autoleucel (orva-cel), a B-cell maturation antigen (BCMA)-directed CAR T cell therapy for patients (pts) with relapsed/refractory multiple myeloma (RRMM): Update of the phase 1/2 EVOLVE study (NCT03430011). J. Clin. Oncol. 2020, 38, 8504. [Google Scholar] [CrossRef]
- Lam, N.; Trinklein, N.D.; Buelow, B.; Patterson, G.H.; Ojha, N.; Kochenderfer, J.N. Anti-BCMA chimeric antigen receptors with fully human heavy-chain-only antigen recognition domains. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Agha, M.E.; Cohen, A.D.; Madduri, D.; Cohen, Y.C.; Delforge, M.; Hillengass, J.; Goldschmidt, H.; Weisel, K.; Raab, M.-S.; Scheid, C.; et al. CARTITUDE-2: Efficacy and safety of ciltacabtagene autoleucel (cilta-cel), a BCMA-directed CAR T-cell therapy, in patients with progressive multiple myeloma (MM) after one to three prior lines of therapy. J. Clin. Oncol. 2021, 39, 8013. [Google Scholar] [CrossRef]
- Oliver-Caldés, A.; Español-Rego, M.; Zabaleta, A.; Inogés, S.; López-Diaz de Cerio, A.; Gonzalez-Calle, V.; Cabañas, V.; Mar-tin-Antonio, B.; Pérez-Amill, L.; Reguera, J.L.; et al. Correlative Biological Studies Related to the Response, Peak and Persis-tence of ARI0002h, an Academic BCMA-Directed CAR-T Cell, with Fractionated Initial Infusion and Booster Dose for Pa-tients with Relapsed and/or Refractory Multiple Myeloma (RRMM). In Proceedings of the 63rd ASH Annual Meeting and Exposition, Atlanta, GA, USA, 11–14 December 2021. [Google Scholar]
- De Larrea, C.F.; Gonzalez-Calle, V.; Cabañas, V.; Oliver-Caldes, A.; Español-Rego, M.; Rodriguez-Otero, P.; Reguera, J.L.; Corral, L.L.; Inogés, S.; Martin-Antonio, B.; et al. Results from a Pilot Study of ARI0002h, an Academic BCMA-Directed CAR-T Cell Therapy with Fractionated Initial Infusion and Booster Dose in Patients with Relapsed and/or Refractory Multiple Myeloma. Blood 2021, 138, 2837. [Google Scholar] [CrossRef]
- Li, C.; Wang, D.; Song, Y.; Li, J.; Huang, H.; Chen, B.; Liu, J.; Hu, K.; Ren, H.; Zhang, X.; et al. A Phase 1/2 Study of a Novel Fully Human B-Cell Maturation Antigen-Specific CAR T Cells (CT103A) in Patients with Relapsed and/or Refractory Multi-ple Myeloma. In Proceedings of the 63rd ASH Annual Meeting and Exposition, Atlanta, GA, USA, 11–14 December 2021. [Google Scholar]
- Wang, D.; Wang, J.; Hu, G.; Wang, W.; Xiao, Y.; Cai, H.; Jiang, L.; Meng, L.; Yang, Y.; Zhou, X.; et al. A Phase I Study of a Novel Fully Human BCMA-Targeting CAR (CT103A) in Patients with Relapsed/Refractory Multiple Myeloma. Blood 2021. [Google Scholar] [CrossRef]
- Ghorashian, S.; Kramer, A.M.; Onuoha, S.; Wright, G.; Bartram, J.; Richardson, R.; Albon, S.J.; Casanovas-Company, J.; Castro, F.; Popova, B.; et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat. Med. 2019, 25, 1408–1414. [Google Scholar] [CrossRef]
- Chmielewski, M.; Hombach, A.; Heuser, C.; Adams, G.P.; Abken, H. T Cell Activation by Antibody-Like Immunoreceptors: Increase in Affinity of the Single-Chain Fragment Domain above Threshold Does Not Increase T Cell Activation against Antigen-Positive Target Cells but Decreases Selectivity. J. Immunol. 2004, 173, 7647–7653. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, S.; Fang, C.; Yang, S.; Olalere, D.; Pequignot, E.C.; Cogdill, A.; Li, N.; Ramones, M.; Granda, B.; et al. Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res. 2015, 75, 3596–3607. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Shevlin, E.; Vedvyas, Y.; Zaman, M.; Park, S.; Hsu, Y.-M.S.; Min, I.M.; Jin, M.M. Micromolar affinity CAR T cells to ICAM-1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Hudecek, M.; Lupo-Stanghellini, M.T.; Kosasih, P.L.; Sommermeyer, D.; Jensen, M.C.; Rader, C.; Riddell, S.R. Receptor Affinity and Extracellular Domain Modifications Affect Tumor Recognition by ROR1-Specific Chimeric Antigen Receptor T Cells. Clin. Cancer Res. 2013, 19, 3153–3164. [Google Scholar] [CrossRef] [Green Version]
- Moritz, D.; Groner, B. A spacer region between the single chain antibody- and the CD3 zeta-chain domain of chimeric T cell receptor components is required for efficient ligand binding and signaling activity. Gene Ther. 1995, 2, 539–546. [Google Scholar]
- Guest, R.D.; Hawkins, R.E.; Kirillova, N.; Cheadle, E.J.; Arnold, J.; O’Neill, A.; Irlam, J.; Chester, K.A.; Kemshead, J.T.; Shaw, D.M.; et al. The Role of Extracellular Spacer Regions in the Optimal Design of Chimeric Immune Receptors. J. Immunother. 2005, 28, 203–211. [Google Scholar] [CrossRef]
- Hombach, A.; Hombach, A.A.; Abken, H. Adoptive immunotherapy with genetically engineered T cells: Modification of the IgG1 Fc ‘spacer’ domain in the extracellular moiety of chimeric antigen receptors avoids ‘off-target’ activation and unintended initiation of an innate immune response. Gene Ther. 2010, 17, 1206–1213. [Google Scholar] [CrossRef] [Green Version]
- Da Vià, M.C.; Dietrich, O.; Truger, M.; Arampatzi, P.; Duell, J.; Heidemeier, A.; Zhou, X.; Danhof, S.; Kraus, S.; Chatterjee, M.; et al. Homozygous BCMA gene deletion in response to anti-BCMA CAR T cells in a patient with multiple myeloma. Nat. Med. 2021, 27, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Hudecek, M.; Sommermeyer, D.; Kosasih, P.L.; Silva-Benedict, A.; Liu, L.; Rader, C.; Jensen, M.C.; Riddell, S.R. The Nonsignaling Extracellular Spacer Domain of Chimeric Antigen Receptors Is Decisive for In Vivo Antitumor Activity. Cancer Immunol. Res. 2014, 3, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Haso, W.; Lee, D.W.; Shah, N.N.; Stetler-Stevenson, M.; Yuan, C.M.; Pastan, I.H.; Dimitrov, D.S.; Morgan, R.A.; Fitzgerald, D.J.; Barrett, D.M.; et al. Anti-CD22–chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood 2013, 121, 1165–1174. [Google Scholar] [CrossRef] [Green Version]
- Guedan, S.; Calderon, H.; Posey, A.D., Jr.; Maus, M.V. Engineering and Design of Chimeric Antigen Receptors. Mol. Ther. Methods Clin. Dev. 2019, 12, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Alabanza, L.; Pegues, M.; Geldres, C.; Shi, V.; Wiltzius, J.J.; Sievers, S.A.; Yang, S.; Kochenderfer, J.N. Function of Novel Anti-CD19 Chimeric Antigen Receptors with Human Variable Regions Is Affected by Hinge and Transmembrane Domains. Mol. Ther. 2017, 25, 2452–2465. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, K.; Tsunei, A.; Kusabuka, H.; Ogaki, E.; Tachibana, M.; Okada, N. Hinge and Transmembrane Domains of Chimeric Antigen Receptor Regulate Receptor Expression and Signaling Threshold. Cells 2020, 9, 1182. [Google Scholar] [CrossRef]
- Jensen, M.; Tan, G.; Forman, S.; Wu, A.; Raubitschek, A. CD20 is a molecular target for scFvFc:zeta receptor redirected T cells: Implications for cellular immunotherapy of CD20+ malignancy. Biol. Blood Marrow Transplant. 1998, 4, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Kochenderfer, J.N.; Feldman, S.A.; Zhao, Y.; Xu, H.; Black, M.A.; Morgan, R.A.; Wilson, W.H.; Rosenberg, S.A. Construction and Preclinical Evaluation of an Anti-CD19 Chimeric Antigen Receptor. J. Immunother. 2009, 32, 689–702. [Google Scholar] [CrossRef] [Green Version]
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.-H.; Geiger, T.L.; Campana, D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004, 18, 676–684. [Google Scholar] [CrossRef] [Green Version]
- Salter, A.I.; Ivey, R.G.; Kennedy, J.J.; Voillet, V.; Rajan, A.; Alderman, E.J.; Voytovich, U.J.; Lin, C.; Sommermeyer, D.; Liu, L.; et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Salter, A.I.; Rajan, A.; Kennedy, J.J.; Ivey, R.G.; Shelby, S.A.; Leung, I.; Templeton, M.L.; Muhunthan, V.; Voillet, V.; Sommermeyer, D.; et al. Comparative analysis of TCR and CAR signaling informs CAR designs with superior antigen sensitivity and in vivo function. Sci. Signal. 2021, 14, eabe2606. [Google Scholar] [CrossRef]
- Feucht, J.; Sun, J.; Eyquem, J.; Ho, Y.-J.; Zhao, Z.; Leibold, J.; Dobrin, A.; Cabriolu, A.; Hamieh, M.; Sadelain, M. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med. 2019, 25, 82–88. [Google Scholar] [CrossRef]
- Majzner, R.G.; Rietberg, S.P.; Sotillo, E.; Dong, R.; Vachharajani, V.T.; Labanieh, L.; Myklebust, J.H.; Kadapakkam, M.; Weber, E.W.; Tousley, A.M.; et al. Tuning the Antigen Density Requirement for CAR T-cell Activity. Cancer Discov. 2020, 10, 702–723. [Google Scholar] [CrossRef] [Green Version]
- Duan, D.; Wang, K.; Wei, C.; Feng, D.; Liu, Y.; He, Q.; Xu, X.; Wang, C.; Zhao, S.; Lv, L.; et al. The BCMA-Targeted Fourth-Generation CAR-T Cells Secreting IL-7 and CCL19 for Therapy of Refractory/Recurrent Multiple Myeloma. Front. Immunol. 2021, 12, 561. [Google Scholar] [CrossRef]
- Ajina, A.; Maher, J. Strategies to Address Chimeric Antigen Receptor Tonic Signaling. Mol. Cancer Ther. 2018, 17, 1795–1815. [Google Scholar] [CrossRef] [Green Version]
- Frigault, M.J.; Lee, J.; Basil, M.C.; Carpenito, C.; Motohashi, S.; Scholler, J.; Kawalekar, O.U.; Guedan, S.; McGettigan, S.E.; Posey, A.D., Jr.; et al. Identification of Chimeric Antigen Receptors That Mediate Constitutive or Inducible Proliferation of T Cells. Cancer Immunol. Res. 2015, 3, 356–367. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Marquez, P.; Calleja-Cervantes, M.E.; Serrano, G.; Palacios-Berraquero, M.L.; Alignani, D.; Martin-Uriz, P.S.; Vilas-Zornoza, A.; Ceballos, C.; Martin-Mallo, A.; Rodríguez-Diaz, S.; et al. CAR Density Influences Antitumoral Efficacy of BCMA CAR-T Cells and Correlates with Clinical Outcome. Blood 2021, 138, 735. [Google Scholar] [CrossRef]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Stenger, D.; Stief, T.A.; Kaeuferle, T.; Willier, S.; Rataj, F.; Schober, K.; Vick, B.; Lotfi, R.; Wagner, B.; Grünewald, T.G.P.; et al. Endogenous TCR promotes in vivo persistence of CD19-CAR-T cells compared to a CRISPR/Cas9-mediated TCR knockout CAR. Blood 2020, 136, 1407–1418. [Google Scholar] [CrossRef]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Stefanová, I.; Dorfman, J.; Germain, R.N. Self-recognition promotes the foreign antigen sensitivity of naive T lymphocytes. Nature 2002, 420, 429–434. [Google Scholar] [CrossRef] [Green Version]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR–T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.A.; Shi, V.; Maric, I.; Wang, M.; Stroncek, D.F.; Rose, J.J.; Brudno, J.N.; Stetler-Stevenson, M.; Feldman, S.A.; Hansen, B.G.; et al. T cells expressing an anti–B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood 2016, 128, 1688–1700. [Google Scholar] [CrossRef]
- Brudno, J.N.; Kochenderfer, J.N. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. 2019, 34, 45–55. [Google Scholar] [CrossRef]
- Santomasso, B.D.; Park, J.H.; Salloum, D.; Riviere, I.; Flynn, J.; Mead, E.; Halton, E.; Wang, X.; Senechal, B.; Purdon, T.; et al. Clinical and Biological Correlates of Neurotoxicity Associated with CAR T-cell Therapy in Patients with B-cell Acute Lymphoblastic Leukemia. Cancer Discov. 2018, 8, 958–971. [Google Scholar] [CrossRef] [Green Version]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Hudecek, M.; Pender, B.; Robinson, E.; Hawkins, R.; Chaney, C.; Cherian, S.; Chen, X.; et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8 + and CD4 + CD19-specific chimeric antigen receptor–modified T cells. Sci. Transl. Med. 2016, 8, 355ra116. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.W.; Gardner, R.A.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.C.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef] [Green Version]
- Teachey, D.T.; Lacey, S.F.; Shaw, P.A.; Melenhorst, J.J.; Maude, S.L.; Jeffrey, F.; Pequignot, E.; Gonzalez, V.E.; Chen, F.; Finklestein, J.; et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016, 6, 664–679. [Google Scholar] [CrossRef] [Green Version]
- Amatya, C.; Pegues, M.A.; Lam, N.; Vanasse, D.; Geldres, C.; Choi, S.; Hewitt, S.M.; Feldman, S.A.; Kochenderfer, J.N. Development of CAR T Cells Expressing a Suicide Gene Plus a Chimeric Antigen Receptor Targeting Signaling Lymphocytic-Activation Molecule F7. Mol. Ther. 2020, 29, 702–717. [Google Scholar] [CrossRef]
- Jan, M.; Scarfò, I.; Larson, R.C.; Walker, A.; Schmidts, A.; Guirguis, A.A.; Gasser, J.A.; Słabicki, M.; Bouffard, A.A.; Castano, A.P.; et al. Reversible ON- and OFF-switch chimeric antigen receptors controlled by lenalidomide. Sci. Transl. Med. 2021, 13, eabb6295. [Google Scholar] [CrossRef]
- Drent, E.; Poels, R.; Mulders, M.J.; Van De Donk, N.W.C.J.; Themeli, M.; Lokhorst, H.M.; Mutis, T. Feasibility of controlling CD38-CAR T cell activity with a Tet-on inducible CAR design. PLoS ONE 2018, 13, e0197349. [Google Scholar] [CrossRef]
- Davis, M.M.; Boniface, J.J.; Reich, Z.; Lyons, D.; Hampl, J.; Arden, B.; Chien, Y. Ligand recognition by alpha beta T cell receptors. Annu. Rev. Immunol. 1998, 16, 523–544. [Google Scholar] [CrossRef]
- Chan, A.; Mak, T.W. Genomic organization of the T cell receptor. Cancer Detect. Prev. 1989, 14, 261–267. [Google Scholar] [PubMed]
- Nikolich-Žugich, J.; Slifka, M.K.; Messaoudi, I. The many important facets of T-cell repertoire diversity. Nat. Rev. Immunol. 2004, 4, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.K.; Jameson, S.C.; Hogquist, K.A. Positive and Negative Selection of T Cells. Annu. Rev. Immunol. 2003, 21, 139–176. [Google Scholar] [CrossRef]
- Lawlor, D.A.; Zemmour, J.; Ennis, P.D.; Parham, P. Evolution of Class-I MHC Genes and Proteins: From Natural Selection to Thymic Selection. Annu. Rev. Immunol. 1990, 8, 23–63. [Google Scholar] [CrossRef]
- Sasmal, D.K.; Feng, W.; Roy, S.; Leung, P.; He, Y.; Cai, C.; Cao, G.; Lian, H.; Qin, J.; Hui, E.; et al. TCR–pMHC bond conformation controls TCR ligand discrimination. Cell. Mol. Immunol. 2019, 17, 203–217. [Google Scholar] [CrossRef] [Green Version]
- Osman, N.; Lucas, S.; Cantrell, D. The role of tyrosine phosphorylation in the interaction of cellular tyrosine kinases with the T cell receptor ζ chain tyrosine-based activation motif. Eur. J. Immunol. 1995, 25, 2863–2869. [Google Scholar] [CrossRef]
- Huang, J.; Brameshuber, M.; Zeng, X.; Xie, J.; Li, Q.-J.; Chien, Y.-H.; Valitutti, S.; Davis, M.M. A Single Peptide-Major Histocompatibility Complex Ligand Triggers Digital Cytokine Secretion in CD4+ T Cells. Immunity 2013, 39, 846–857. [Google Scholar] [CrossRef] [Green Version]
- Valitutti, S.; Müller, S.; Cella, M.; Padovan, E.; Lanzavecchia, A. Serial triggering of many T-cell receptors by a few peptide–MHC complexes. Nature 1995, 375, 148–151. [Google Scholar] [CrossRef]
- Purbhoo, M.A.; Irvine, D.J.; Huppa, J.B.; Davis, M.M. T cell killing does not require the formation of a stable mature immunological synapse. Nat. Immunol. 2004, 5, 524–530. [Google Scholar] [CrossRef]
- Campillo-Davo, D.; Flumens, D.; Lion, E. The Quest for the Best: How TCR Affinity, Avidity, and Functional Avidity Affect TCR-Engineered T-Cell Antitumor Responses. Cells 2020, 9, 1720. [Google Scholar] [CrossRef]
- Allard, M.; Hebeisen, M.; Rufer, N. Assessing T Cell Receptor Affinity and Avidity Against Tumor Antigens. Oncoimmunology 2017, 665–679. [Google Scholar] [CrossRef]
- Alexander-Miller, M.A.; Leggatt, G.; Berzofsky, J.A. Selective expansion of high- or low-avidity cytotoxic T lymphocytes and efficacy for adoptive immunotherapy. Proc. Natl. Acad. Sci. USA 1996, 93, 4102–4107. [Google Scholar] [CrossRef] [Green Version]
- Almeida, J.R.; Price, D.A.; Papagno, L.; Arkoub, Z.A.; Sauce, D.; Bornstein, E.; Asher, T.E.; Samri, A.; Schnuriger, A.; Theodorou, I.; et al. Superior control of HIV-1 replication by CD8+ T cells is reflected by their avidity, polyfunctionality, and clonal turnover. J. Exp. Med. 2007, 204, 2473–2485. [Google Scholar] [CrossRef] [Green Version]
- Berger, C.T.; Frahm, N.; Price, D.A.; Mothe, B.; Ghebremichael, M.; Hartman, K.L.; Henry, L.M.; Brenchley, J.M.; Ruff, L.E.; Venturi, V.; et al. High-Functional-Avidity Cytotoxic T Lymphocyte Responses to HLA-B-Restricted Gag-Derived Epitopes Associated with Relative HIV Control. J. Virol. 2011, 85, 9334–9345. [Google Scholar] [CrossRef] [Green Version]
- Neveu, B.; Debeaupuis, E.; Echasserieau, K.; Le Moullac-Vaidye, B.; Gassin, M.; Jegou, L.; Decalf, J.; Albert, M.; Ferry, N.; Gournay, J.; et al. Selection of high-avidity CD8 T cells correlates with control of hepatitis C virus infection. Hepatology 2008, 48, 713–722. [Google Scholar] [CrossRef]
- Zeh, H.J., 3rd; Perry-Lalley, D.; Dudley, M.E.; Rosenberg, S.A.; Yang, J.C. High avidity CTLs for two self-antigens demon-strate superior in vitro and in vivo antitumor efficacy. J. Immunol. 1999, 162, 989–994. [Google Scholar]
- Hebeisen, M.; Allard, M.; Gannon, P.O.; Schmidt, J.; Speiser, D.; Rufer, N. Identifying Individual T Cell Receptors of Optimal Avidity for Tumor Antigens. Front. Immunol. 2015, 6, 582. [Google Scholar] [CrossRef] [Green Version]
- Lövgren, T.; Baumgaertner, P.; Wieckowski, S.; Devêvre, E.; Guillaume, P.; Luescher, I.; Rufer, N.; Speiser, D.E. Enhanced cytotoxicity and decreased CD8 dependence of human cancer-specific cytotoxic T lymphocytes after vaccination with low peptide dose. Cancer Immunol. Immunother. 2011, 61, 817–826. [Google Scholar] [CrossRef] [Green Version]
- Aranda, F.; Llopiz, D.; Diaz-Valdes, N.; Riezu-Boj, J.-I.; Bezunartea, J.; Ruiz, M.; Martinez, M.; Durantez, M.; Mansilla, C.; Prieto, J.; et al. Adjuvant Combination and Antigen Targeting as a Strategy to Induce Polyfunctional and High-Avidity T-Cell Responses against Poorly Immunogenic Tumors. Cancer Res. 2011, 71, 3214–3224. [Google Scholar] [CrossRef] [Green Version]
- Derby, M.A.; Alexander-Miller, M.A.; Tse, R.; Berzofsky, J.A. High-Avidity CTL Exploit Two Complementary Mechanisms to Provide Better Protection Against Viral Infection Than Low-Avidity CTL. J. Immunol. 2001, 166, 1690–1697. [Google Scholar] [CrossRef] [Green Version]
- Savage, P.A.; Boniface, J.J.; Davis, M.M. A Kinetic Basis For T Cell Receptor Repertoire Selection during an Immune Response. Immunity 1999, 10, 485–492. [Google Scholar] [CrossRef] [Green Version]
- Price, D.A.; Brenchley, J.M.; Ruff, L.E.; Betts, M.R.; Hill, B.J.; Roederer, M.; Koup, R.A.; Migueles, S.A.; Gostick, E.; Wooldridge, L.; et al. Avidity for antigen shapes clonal dominance in CD8+ T cell populations specific for persistent DNA viruses. J. Exp. Med. 2005, 202, 1349–1361. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, J.J.; Huang, J.; Zhu, C.; Evavold, B. High prevalence of low affinity peptide–MHC II tetramer–negative effectors during polyclonal CD4+ T cell responses. J. Exp. Med. 2011, 208, 81–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zehn, D.; Lee, S.Y.; Bevan, M.J. Complete but curtailed T-cell response to very low-affinity antigen. Nature 2009, 458, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Sim, M.J.W.; Lu, J.; Spencer, M.; Hopkins, F.; Tran, E.; Rosenberg, S.A.; Long, E.O.; Sun, P.D. High-affinity oligoclonal TCRs define effective adoptive T cell therapy targeting mutant KRAS-G12D. Proc. Natl. Acad. Sci. USA 2020, 117, 12826–12835. [Google Scholar] [CrossRef]
- Oliveira, G.; Stromhaug, K.; Klaeger, S.; Kula, T.; Frederick, D.T.; Le, P.M.; Forman, J.; Huang, T.; Li, S.; Zhang, W.; et al. Phenotype, specificity and avidity of antitumour CD8+ T cells in melanoma. Nature 2021, 596, 119–125. [Google Scholar] [CrossRef]
- Presotto, D.; Erdes, E.; Duong, M.N.; Allard, M.; Regamey, P.-O.; Quadroni, M.; Doucey, M.-A.; Rufer, N.; Hebeisen, M. Fine-Tuning of Optimal TCR Signaling in Tumor-Redirected CD8 T Cells by Distinct TCR Affinity-Mediated Mechanisms. Front. Immunol. 2017, 8, 1564. [Google Scholar] [CrossRef] [Green Version]
- Bräunlein, E.; Lupoli, G.; Füchsl, F.; Abualrous, E.T.; Krätzig, N.d.A.; Gosmann, D.; Wietbrock, L.; Lange, S.; Engleitner, T.; Lan, H.; et al. Functional analysis of peripheral and intratumoral neoantigen-specific TCRs identified in a patient with melanoma. J. Immunother. Cancer 2021, 9, e002754. [Google Scholar] [CrossRef]
- Bassani-Sternberg, M.; Bräunlein, E.; Klar, R.; Engleitner, T.; Sinitcyn, P.; Audehm, S.; Straub, M.; Weber, J.; Slotta-Huspenina, J.; Specht, K.; et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun. 2016, 7, 13404. [Google Scholar] [CrossRef] [Green Version]
- Schmid, D.A.; Irving, M.B.; Posevitz, V.; Hebeisen, M.; Posevitz-Fejfar, A.; Sarria, J.-C.F.; Gomez-Eerland, R.; Thome, M.; Schumacher, T.; Romero, P.; et al. Evidence for a TCR Affinity Threshold Delimiting Maximal CD8 T Cell Function. J. Immunol. 2010, 184, 4936–4946. [Google Scholar] [CrossRef]
- Slansky, J.E.; Jordan, K.R. The Goldilocks Model for TCR—Too Much Attraction Might Not Be Best for Vaccine Design. PLoS Biol. 2010, 8, e1000482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Ippolito, E.; Schober, K.; Nauerth, M.; Busch, D.H. T cell engineering for adoptive T cell therapy: Safety and receptor avidity. Cancer Immunol. Immunother. 2019, 68, 1701–1712. [Google Scholar] [CrossRef] [PubMed]
- Robey, E.; Allison, J.P. T-cell activation: Integration of signals from the antigen receptor and costimulatory molecules. Immunol. Today 1995, 16, 306–310. [Google Scholar] [CrossRef]
- Wu, H.; Witzl, A.; Ueno, H. Assessment of TCR signal strength of antigen-specific memory CD8+ T cells in human blood. Blood Adv. 2019, 3, 2153–2163. [Google Scholar] [CrossRef]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242, Erratum in Nat. Rev. Immunol. 2013, 13, 542. [Google Scholar] [CrossRef]
- Blum, J.S.; Wearsch, P.A.; Cresswell, P. Pathways of Antigen Processing. Annu. Rev. Immunol. 2013, 31, 443–473. [Google Scholar] [CrossRef] [Green Version]
- Cresswell, P. Antigen processing and presentation. Immunol. Rev. 2005, 207, 5–7. [Google Scholar] [CrossRef]
- Laumont, C.M.; Vincent, K.; Hesnard, L.; Audemard, É.; Bonneil, É.; Laverdure, J.-P.; Gendron, P.; Courcelles, M.; Hardy, M.-P.; Côté, C.; et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Wei, Z.; Zhang, L.; Yang, Z.; Liu, Q. Systematically Characterizing A-to-I RNA Editing Neoantigens in Cancer. Front. Oncol. 2020, 10, 2753. [Google Scholar] [CrossRef]
- Leen, A.M.; Christin, A.; Myers, G.D.; Liu, H.; Cruz, C.R.; Hanley, P.J.; Kennedy-Nasser, A.A.; Leung, K.S.; Gee, A.P.; Krance, R.A.; et al. Cytotoxic T lymphocyte therapy with donor T cells prevents and treats adenovirus and Epstein-Barr virus infections after haploidentical and matched unrelated stem cell transplantation. Blood 2009, 114, 4283–4292. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.K.; Blyth, E.; Clancy, L.; Simms, R.; Burgess, J.; Brown, R.; Deo, S.; Micklethwaite, K.; Gottlieb, D.J. Addition of varicella zoster virus–specific T cells to cytomegalovirus, Epstein-Barr virus and adenovirus tri-specific T cells as adoptive immunotherapy in patients undergoing allogeneic hematopoietic stem cell transplantation. Cytotherapy 2015, 17, 1406–1420. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, A.; Gerdemann, U.; Katari, U.L.; Tzannou, I.; Liu, H.; Martinez, C.; Leung, K.; Carrum, G.; Gee, A.P.; Vera, J.F.; et al. Activity of Broad-Spectrum T Cells as Treatment for AdV, EBV, CMV, BKV, and HHV6 Infections after HSCT. Sci. Transl. Med. 2014, 6, 242ra83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perruccio, K.; Tosti, A.; Burchielli, E.; Topini, F.; Ruggeri, L.; Carotti, A.; Capanni, M.; Urbani, E.; Mancusi, A.; Aversa, F.; et al. Transferring functional immune responses to pathogens after haploidentical hematopoietic transplantation. Blood 2005, 106, 4397–4406. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, O.; Caballero, O.L.; Stevenson, B.J.; Chen, Y.-T.; Cohen, T.; Chua, R.; Maher, C.A.; Panji, S.; Schaefer, U.; Kruger, A.; et al. Genome-wide analysis of cancer/testis gene expression. Proc. Natl. Acad. Sci. USA 2008, 105, 20422–20427. [Google Scholar] [CrossRef] [Green Version]
- Andrade, V.C.; Vettore, A.L.; Felix, R.S.; Almeida, M.S.; Carvalho, F.; Oliveira, J.S.; Chauffaille, M.L.; Andriolo, A.; Caballe-ro, O.L.; Zago, M.A.; et al. Prognostic impact of cancer/testis antigen expression in advanced stage multiple myeloma patients. Cancer Immun. 2008, 8, 2. [Google Scholar] [CrossRef]
- Tarte, K.; De Vos, J.; Thykjaer, T.; Zhan, F.; Fiol, G.; Costes, V.; Rème, T.; Legouffe, E.; Rossi, J.-F.; Shaughnessy, J.J.; et al. Generation of polyclonal plasmablasts from peripheral blood B cells: A normal counterpart of malignant plasmablasts. Blood 2002, 100, 1113–1122. [Google Scholar] [CrossRef]
- Van Duin, M.; Broyl, A.; de Knegt, Y.; Goldschmidt, H.; Richardson, P.G.; Hop, W.C.J.; van der Holt, B.; Joseph-Pietras, D.; Mulligan, G.; Neuwirth, R.; et al. Cancer testis antigens in newly diagnosed and relapse multiple myeloma: Prognostic markers and potential targets for immunotherapy. Haematologica 2011, 96, 1662–1669. [Google Scholar] [CrossRef] [Green Version]
- Atanackovic, D.; Arfsten, J.; Cao, Y.; Gnjatic, S.; Schnieders, F.; Bartels, K.; Schilling, G.; Faltz, C.; Wolschke, C.; Dierlamm, J.; et al. Cancer-testis antigens are commonly expressed in multiple myeloma and induce systemic immunity following allogeneic stem cell transplantation. Blood 2006, 109, 1103–1112. [Google Scholar] [CrossRef] [Green Version]
- Condomines, M.; Hose, D.; Raynaud, P.; Hundemer, M.; De Vos, J.; Baudard, M.; Moehler, T.; Pantesco, V.; Moos, M.; Schved, J.-F.; et al. Cancer/Testis Genes in Multiple Myeloma: Expression Patterns and Prognosis Value Determined by Microarray Analysis. J. Immunol. 2007, 178, 3307–3315. [Google Scholar] [CrossRef] [Green Version]
- Ilyas, S.; Yang, J.C. Landscape of Tumor Antigens in T Cell Immunotherapy. J. Immunol. 2015, 195, 5117–5122. [Google Scholar] [CrossRef] [Green Version]
- Aleksic, M.; Liddy, N.; Molloy, P.; Pumphrey, N.; Vuidepot, A.; Chang, K.-M.; Jakobsen, B.K. Different affinity windows for virus and cancer-specific T-cell receptors: Implications for therapeutic strategies. Eur. J. Immunol. 2012, 42, 3174–3179. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.P.; Gerry, A.B.; Brewer, J.E.; Melchiori, L.; Bridgeman, J.S.; Bennett, A.D.; Pumphrey, N.J.; Jakobsen, B.K.; Price, D.A.; Ladell, K.; et al. T cell receptor binding affinity governs the functional profile of cancer-specific CD8+ T cells. Clin. Exp. Immunol. 2015, 180, 255–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, S.A.; Yannelli, J.R.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of Patients with Metastatic Melanoma with Autologous Tumor-Infiltrating Lymphocytes and Interleukin 2. JNCI J. Natl. Cancer Inst. 1994, 86, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.C.; Postow, M.A.; Orlowski, R.J.; Mick, R.; Bengsch, B.; Manne, S.; Xu, W.; Harmon, S.; Giles, J.R.; Wenz, B.; et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017, 545, 60–65. [Google Scholar] [CrossRef] [Green Version]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [Green Version]
- Robbins, P.F.; Lu, Y.C.; El-Gamil, M.; Li, Y.F.; Gross, C.; Gartner, J.; Lin, J.C.; Teer, J.K.; Cliften, P.; Tycksen, E.; et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med. 2013, 19, 747–752. [Google Scholar] [CrossRef]
- Schwitalle, Y.; Kloor, M.; Eiermann, S.; Linnebacher, M.; Kienle, P.; Knaebel, H.P.; Tariverdian, M.; Benner, A.; von Knebel Doeberitz, M. Immune Response Against Frameshift-Induced Neopeptides in HNPCC Patients and Healthy HNPCC Mutation Carriers. Gastroenterology 2008, 134, 988–997. [Google Scholar] [CrossRef]
- Yang, W.; Lee, K.-W.; Srivastava, R.M.; Kuo, F.; Krishna, C.; Chowell, D.; Makarov, V.; Hoen, D.; Dalin, M.G.; Wexler, L.; et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat. Med. 2019, 25, 767–775. [Google Scholar] [CrossRef]
- Dalet, A.; Robbins, P.F.; Stroobant, V.; Vigneron, N.; Li, Y.F.; El-Gamil, M.; Hanada, K.-I.; Yang, J.C.; Rosenberg, S.A.; Eynde, B.J.V.D. An antigenic peptide produced by reverse splicing and double asparagine deamidation. Proc. Natl. Acad. Sci. USA 2011, 108, E323–E331. [Google Scholar] [CrossRef] [Green Version]
- Kumai, T.; Ishibashi, K.; Oikawa, K.; Matsuda, Y.; Aoki, N.; Kimura, S.; Hayashi, S.; Kitada, M.; Harabuchi, Y.; Celis, E.; et al. Induction of tumor-reactive T helper responses by a posttranslational modified epitope from tumor protein p53. Cancer Immunol. Immunother. 2014, 63, 469–478. [Google Scholar] [CrossRef]
- Yarchoan, M.; Johnson, B.A., III; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.J.; Gartner, J.J.; Horovitz-Fried, M.; Shamalov, K.; Trebska-McGowan, K.; Bliskovsky, V.V.; Parkhurst, M.R.; Ankri, C.; Prickett, T.D.; Crystal, J.S.; et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J. Clin. Investig. 2015, 125, 3981–3991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobisse, S.; Genolet, R.; Roberti, A.; Tanyi, J.L.; Racle, J.; Stevenson, B.J.; Iseli, C.; Michel, A.; Le Bitoux, M.-A.; Guillaume, P.; et al. Sensitive and frequent identification of high avidity neo-epitope specific CD8 + T cells in immunotherapy-naive ovarian cancer. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kim, S.; Hundal, J.; Herndon, J.M.; Li, S.; Petti, A.A.; Soysal, S.D.; Li, L.; McLellan, M.D.; Hoog, J.; et al. Breast Cancer Neoantigens Can Induce CD8+ T-Cell Responses and Antitumor Immunity. Cancer Immunol. Res. 2017, 5, 516–523. [Google Scholar] [CrossRef] [Green Version]
- Tran, E.; Ahmadzadeh, M.; Lu, Y.-C.; Gros, A.; Turcotte, S.; Robbins, P.F.; Gartner, J.J.; Zheng, Z.; Li, Y.F.; Ray, S.; et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015, 350, 1387–1390. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Perumal, D.; Imai, N.; Laganà, A.; Finnigan, J.; Melnekoff, D.T.; Leshchenko, V.V.; Solovyov, A.; Madduri, D.; Chari, A.; Cho, H.J.; et al. Mutation-derived Neoantigen-specific T-cell Responses in Multiple Myeloma. Clin. Cancer Res. 2019, 26, 450–464. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.; Asmann, Y.; Cattaneo, L.; Braggio, E.; Keats, J.; Auclair, D.; Lonial, S.; The MMRF CoMMpass Network; Russell, S.J.; Stewart, A.K. High somatic mutation and neoantigen burden are correlated with decreased progression-free survival in multiple myeloma. Blood Cancer J. 2017, 7, e612. [Google Scholar] [CrossRef] [Green Version]
- Andreatta, M.; Nielsen, M. Gapped sequence alignment using artificial neural networks: Application to the MHC class I system. Bioinformatics 2015, 32, 511–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, M.; Lundegaard, C.; Worning, P.; Lauemøller, S.L.; Lamberth, K.; Buus, S.; Brunak, S.; Lund, O. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003, 12, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Samur, M.K.; Samur, A.A.; Fulciniti, M.; Szalat, R.; Han, T.; Shammas, M.; Richardson, P.; Magrangeas, F.; Minvielle, S.; Corre, J.; et al. Genome-Wide Somatic Alterations in Multiple Myeloma Reveal a Superior Outcome Group. J. Clin. Oncol. 2020, 38, 3107–3118. [Google Scholar] [CrossRef] [PubMed]
- Pasca, S.; Tomuleasa, C.; Teodorescu, P.; Ghiaur, G.; Dima, D.; Moisoiu, V.; Berce, C.; Stefan, C.; Ciechanover, A.; Einsele, H. KRAS/NRAS/BRAF Mutations as Potential Targets in Multiple Myeloma. Front. Oncol. 2019, 9, 1137. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread Genetic Heterogeneity in Multiple Myeloma: Implications for Targeted Therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Wolfl, M.; Kuball, J.; Ho, W.Y.; Nguyen, H.; Manley, T.J.; Bleakley, M.; Greenberg, P.D. Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities. Blood 2007, 110, 201–210. [Google Scholar] [CrossRef]
- Yossef, R.; Tran, E.; Deniger, D.C.; Gros, A.; Pasetto, A.; Parkhurst, M.R.; Gartner, J.J.; Prickett, T.D.; Cafri, G.; Robbins, P.F.; et al. Enhanced detection of neoantigen-reactive T cells targeting unique and shared oncogenes for personalized cancer immunotherapy. JCI Insight 2018, 3, e122467. [Google Scholar] [CrossRef] [Green Version]
- Inozume, T.; Hanada, K.-I.; Wang, Q.J.; Ahmadzadeh, M.; Wunderlich, J.R.; Rosenberg, S.A.; Yang, J.C. Selection of CD8+PD-1+ Lymphocytes in Fresh Human Melanomas Enriches for Tumor-reactive T Cells. J. Immunother. 2010, 33, 956–964. [Google Scholar] [CrossRef] [Green Version]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen–specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef]
- Ali, M.; Foldvari, Z.; Giannakopoulou, E.; Böschen, M.-L.; Strønen, E.; Yang, W.; Toebes, M.; Schubert, B.; Kohlbacher, O.; Schumacher, T.; et al. Induction of neoantigen-reactive T cells from healthy donors. Nat. Protoc. 2019, 14, 1926–1943. [Google Scholar] [CrossRef]
- Hadrup, S.R.; Bakker, A.; Shu, C.J.; Andersen, R.S.; Van Veluw, J.; Hombrink, P.; Castermans, E.; Straten, P.T.; Blank, C.; Haanen, J.B.; et al. Parallel detection of antigen-specific T-cell responses by multidimensional encoding of MHC multimers. Nat. Methods 2009, 6, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-C.; Zheng, Z.; Lowery, F.J.; Gartner, J.J.; Prickett, T.D.; Robbins, P.F.; Rosenberg, S.A. Direct identification of neoantigen-specific TCRs from tumor specimens by high-throughput single-cell sequencing. J. Immunother. Cancer 2021, 9, e002595. [Google Scholar] [CrossRef] [PubMed]
- Paria, B.C.; Levin, N.; Lowery, F.J.; Pasetto, A.; Deniger, D.C.; Parkhurst, M.R.; Yossef, R.; Kim, S.P.; Florentin, M.; Ngo, L.T.; et al. Rapid Identification and Evaluation of Neoantigen-reactive T-Cell Receptors From Single Cells. J. Immunother. 2020, 44, 1–8. [Google Scholar] [CrossRef]
- Arnaud, M.; Duchamp, M.; Bobisse, S.; Renaud, P.; Coukos, G.; Harari, A. Biotechnologies to tackle the challenge of neoantigen identification. Curr. Opin. Biotechnol. 2020, 65, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Stadtmauer, E.A.; Faitg, T.H.; Lowther, D.E.; Badros, A.Z.; Chagin, K.; Dengel, K.; Iyengar, M.; Melchiori, L.; Navenot, J.-M.; Norry, E.; et al. Long-term safety and activity of NY-ESO-1 SPEAR T cells after autologous stem cell transplant for myeloma. Blood Adv. 2019, 3, 2022–2034. [Google Scholar] [CrossRef] [PubMed]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, A.P.; Stadtmauer, E.A.; Binder-Scholl, G.K.; Goloubeva, O.; Vogl, D.T.; Lacey, S.F.; Badros, A.Z.; Garfall, A.; Weiss, B.M.; Finklestein, J.; et al. NY-ESO-1–specific TCR–engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med. 2015, 21, 914–921. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Moysey, R.; Molloy, P.E.; Vuidepot, A.-L.; Mahon, T.; Baston, E.; Dunn, S.; Liddy, N.; Jacob, J.; Jakobsen, B.K.; et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat. Biotechnol. 2005, 23, 349–354. [Google Scholar] [CrossRef]
- Robbins, P.F.; Li, Y.F.; El-Gamil, M.; Zhao, Y.; Wargo, J.A.; Zheng, Z.; Xu, H.; Morgan, R.A.; Feldman, S.A.; Johnson, L.A.; et al. Single and Dual Amino Acid Substitutions in TCR CDRs Can Enhance Antigen-Specific T Cell Functions. J. Immunol. 2008, 180, 6116–6131. [Google Scholar] [CrossRef] [Green Version]
- Holler, P.D.; Holman, P.O.; Shusta, E.V.; O’Herrin, S.; Wittrup, K.D.; Kranz, D.M. In vitro evolution of a T cell receptor with high affinity for peptide/MHC. Proc. Natl. Acad. Sci. USA 2000, 97, 5387–5392. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.; Xue, S.-A.; Bangham, C.R.M.; Jakobsen, B.K.; Morris, E.C.; Stauss, H.J. Human T cells expressing affinity-matured TCR display accelerated responses but fail to recognize low density of MHC-peptide antigen. Blood 2011, 118, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Y.; Cheng, C.; Cheng, A.; Zhang, X.; Li, N.; Xia, C.; Wei, X.; Liu, X.; Wang, H. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res. 2016, 27, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Zhang, X.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget 2017, 8, 17002–17011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, E.K.; Ranganathan, R.; Eruslanov, E.; Kim, S.; Newick, K.; O’Brien, S.; Lo, A.; Liu, X.; Zhao, Y.; Albelda, S.M. Blockade of Programmed Death 1 Augments the Ability of Human T Cells Engineered to Target NY-ESO-1 to Control Tumor Growth after Adoptive Transfer. Clin. Cancer Res. 2015, 22, 436–447. [Google Scholar] [CrossRef] [Green Version]
- Odorizzi, P.M.; Pauken, K.E.; Paley, M.A.; Sharpe, A.H.; Wherry, E.J. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J. Exp. Med. 2015, 212, 1125–1137. [Google Scholar] [CrossRef]
- Wartewig, T.; Kurgyis, Z.; Keppler, S.J.; Pechloff, K.; Hameister, E.; Öllinger, R.; Maresch, R.; Buch, T.; Steiger, K.; Winter, C.; et al. PD-1 is a haploinsufficient suppressor of T cell lymphomagenesis. Nature 2017, 552, 121–125. [Google Scholar] [CrossRef] [Green Version]
- Jahn, L.; Hombrink, P.; Hagedoorn, R.S.; Kester, M.G.D.; van der Steen, D.M.; Rodriguez, T.; Pentcheva-Hoang, T.; de Ru, A.H.; Schoonakker, M.P.; Meeuwsen, M.H.; et al. TCR-based therapy for multiple myeloma and other B-cell malignancies targeting intracellular transcription factor BOB1. Blood 2017, 129, 1284–1295. [Google Scholar] [CrossRef] [Green Version]
- Meeuwsen, M.H.; Wouters, A.K.; Jahn, L.; Hagedoorn, R.S.; Kester, M.G.; Remst, D.F.; Morton, L.T.; van der Steen, D.M.; Kweekel, C.; de Ru, A.H.; et al. A broad and systematic approach to identify B cell malignancy-targeting TCRs for multi-antigen-based T cell therapy. Mol. Ther. 2021. [Google Scholar] [CrossRef]
- Gattinoni, L.; Klebanoff, C.A.; Restifo, N.P. Paths to stemness: Building the ultimate antitumour T cell. Nat. Rev. Cancer 2012, 12, 671–684. [Google Scholar] [CrossRef]
- Gattinoni, L.; Klebanoff, C.A.; Palmer, D.C.; Wrzesinski, C.; Kerstann, K.W.; Yu, Z.; Finkelstein, S.E.; Theoret, M.R.; Rosenberg, S.A.; Restifo, N.P. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J. Clin. Investig. 2005, 115, 1616–1626. [Google Scholar] [CrossRef]
- Sadelain, M.; Rivière, M.S.I.; Riddell, S. Therapeutic T cell engineering. Nature 2017, 545, 423–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busch, D.H.; Fräßle, S.P.; Sommermeyer, D.; Buchholz, V.R.; Riddell, S.R. Role of memory T cell subsets for adoptive immunotherapy. Semin. Immunol. 2016, 28, 28–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinrichs, C.S.; Borman, Z.A.; Cassard, L.; Gattinoni, L.; Spolski, R.; Yu, Z.; Sanchez-Perez, L.; Muranski, P.; Kern, S.J.; Logun, C.; et al. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc. Natl. Acad. Sci. USA 2009, 106, 17469–17474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, C.T.; Hassan, M.; Morris, A.B.; Jeffery, J.; Lee, K.; Jagirdar, N.; Staton, A.D.; Raikar, S.S.; Spencer, H.T.; Sulchek, T.; et al. Improving T-cell expansion and function for adoptive T-cell therapy using ex vivo treatment with PI3Kδ inhibitors and VIP antagonists. Blood Adv. 2018, 2, 210–223. [Google Scholar] [CrossRef] [Green Version]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- Sommer, C.; Boldajipour, B.; Kuo, T.C.; Bentley, T.; Sutton, J.; Chen, A.; Geng, T.; Dong, H.; Galetto, R.; Valton, J.; et al. Preclinical Evaluation of Allogeneic CAR T Cells Targeting BCMA for the Treatment of Multiple Myeloma. Mol. Ther. 2019, 27, 1126–1138. [Google Scholar] [CrossRef]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Gros, A.; Robbins, P.F.; Yao, X.; Li, Y.F.; Turcotte, S.; Tran, E.; Wunderlich, J.R.; Mixon, A.; Farid, S.; Dudley, M.E.; et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J. Clin. Investig. 2014, 124, 2246–2259. [Google Scholar] [CrossRef]
- Baitsch, L.; Baumgaertner, P.; Devêvre, E.; Raghav, S.K.; Legat, A.; Barba, L.; Wieckowski, S.; Bouzourene, H.; Deplancke, B.; Romero, P.; et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J. Clin. Investig. 2011, 121, 2350–2360. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosser, R.; Cherkassky, L.; Chintala, N.; Adusumilli, P.S. Combination Immunotherapy with CAR T Cells and Checkpoint Blockade for the Treatment of Solid Tumors. Cancer Cell 2019, 36, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Dichwalkar, T.; Chang, J.Y.H.; Cossette, B.; Garafola, D.; Zhang, A.Q.; Fichter, M.; Wang, C.; Liang, S.; Silva, M.; et al. Enhanced CAR–T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science 2019, 365, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Reinhard, K.; Rengstl, B.; Oehm, P.; Michel, K.; Billmeier, A.; Hayduk, N.; Klein, O.; Kuna, K.; Ouchan, Y.; Wöll, S.; et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science 2020, 367, 446–453. [Google Scholar] [CrossRef]
- Hedrick, S.M. Thymus Lineage Commitment: A Single Switch. Immunity 2008, 28, 297–299. [Google Scholar] [CrossRef] [Green Version]
- Zander, R.; Schauder, D.; Xin, G.; Nguyen, C.; Wu, X.; Zajac, A.; Cui, W. CD4+ T Cell Help Is Required for the Formation of a Cytolytic CD8+ T Cell Subset that Protects against Chronic Infection and Cancer. Immunity 2019, 51, 1028–1042.e4. [Google Scholar] [CrossRef]
- Kennedy, R.; Celis, E. Multiple roles for CD4+T cells in anti-tumor immune responses. Immunol. Rev. 2008, 222, 129–144. [Google Scholar] [CrossRef]
- Pardoll, D.M.; Topalian, S.L. The role of CD4+ T cell responses in antitumor immunity. Curr. Opin. Immunol. 1998, 10, 588–594. [Google Scholar] [CrossRef]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2015, 30, 492–500. [Google Scholar] [CrossRef] [Green Version]
- Samur, M.K.; Fulciniti, M.; Aktas Samur, A.; Bazarbachi, A.H.; Tai, Y.-T.; Prabhala, R.; Alonso, A.; Sperling, A.S.; Campbell, T.; Petrocca, F.; et al. Biallelic loss of BCMA as a resistance mechanism to CAR T cell therapy in a patient with multiple myeloma. Nat. Commun. 2021, 12, 868. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Bluhm, J.; Kieback, E.; Marino, S.F.; Oden, F.; Westermann, J.; Chmielewski, M.; Abken, H.; Uckert, W.; Höpken, U.E.; Rehm, A. CAR T Cells with Enhanced Sensitivity to B Cell Maturation Antigen for the Targeting of B Cell Non-Hodgkin’s Lymphoma and Multiple Myeloma. Mol. Ther. 2018, 26, 1906–1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dogan, A.; Siegel, D.; Tran, N.; Fu, A.; Fowler, J.; Belani, R.; Landgren, O. B-cell maturation antigen expression across hematologic cancers: A systematic literature review. Blood Cancer J. 2020, 10, 73. [Google Scholar] [CrossRef] [PubMed]
- Laurent, S.A.; Hoffmann, F.S.; Kuhn, P.-H.; Cheng, Q.; Chu, Y.; Schmidt-Supprian, M.; Hauck, S.; Schuh, E.; Krumbholz, M.; Rübsamen, H.; et al. γ-secretase directly sheds the survival receptor BCMA from plasma cells. Nat. Commun. 2015, 6, 7333. [Google Scholar] [CrossRef]
- Pont, M.J.; Hill, T.; Cole, G.O.; Abbott, J.J.; Kelliher, J.; Salter, A.I.; Hudecek, M.; Comstock, M.L.; Rajan, A.; Patel, B.K.R.; et al. γ-Secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood 2019, 134, 1585–1597. [Google Scholar] [CrossRef]
- Cowan, A.J.; Pont, M.J.; Sather, B.; Turtle, C.J.; Till, B.; Libby, E.; Coffey, D.G.; Tuazon, S.A.; Wood, B.L.; Gooley, T.A.; et al. Safety and Efficacy of Fully Human BCMA CAR T Cells in Combination with a Gamma Secretase Inhibitor to Increase BCMA Surface Expression in Patients with Relapsed or Refractory Multiple Myeloma. In Proceedings of the 63rd ASH An-nual Meeting and Exposition, Atlanta, GA, USA, 11–14 December 2021. [Google Scholar]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.-P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259–1271.e11. [Google Scholar] [CrossRef] [Green Version]
- Anagnostou, V.; Smith, K.N.; Forde, P.M.; Niknafs, N.; Bhattacharya, R.; White, J.; Zhang, T.; Adleff, V.; Phallen, J.; Wali, N.; et al. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non–Small Cell Lung Cancer. Cancer Discov. 2016, 7, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Chen, L.-J.; Yang, S.-S.; Sun, Y.; Wu, W.; Liu, Y.-F.; Zhuang, Y.; Zhang, W.; Weng, X.-Q.; Wu, J.; et al. Exploratory trial of a biepitopic CAR T-targeting B cell maturation antigen in relapsed/refractory multiple myeloma. Proc. Natl. Acad. Sci. USA 2019, 116, 9543–9551. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.-H.; Liu, J.; Wang, B.-Y.; Chen, Y.-X.; Cao, X.-M.; Yang, Y.; Zhang, Y.-L.; Wang, F.-X.; Zhang, P.-Y.; Lei, B.; et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J. Hematol. Oncol. 2018, 11, 141. [Google Scholar] [CrossRef]
- Liu, S.; Deng, B.; Lin, Y.; Yin, Z.; Pan, J.; Wu, T.; Gao, Z.; Song, Y.; Zhao, Y.; Tong, C. Sequential CD19- and CD22-CART Cell Therapies for Relapsed B-Cell Acute Lymphoblastic Leukemia after Allogeneic Hematopoietic Stem Cell Transplantation. Blood 2018, 132, 2126. [Google Scholar] [CrossRef]
- Hackl, H.; Charoentong, P.; Finotello, F.; Trajanoski, Z. Computational genomics tools for dissecting tumour–immune cell interactions. Nat. Rev. Genet. 2016, 17, 441–458. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, V.P.; Łuksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Sendabaoglu, Y.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Łuksza, M.; Riaz, N.; Makarov, V.; Balachandran, V.P.; Hellmann, M.D.; Solovyov, A.; Rizvi, N.A.; Merghoub, T.; Levine, A.J.; Chan, T.A.; et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature 2017, 551, 517–520. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Neoantigen quality, not quantity. Sci. Transl. Med. 2019, 11, eaax7918. [Google Scholar] [CrossRef]
- Richman, L.; Vonderheide, R.H.; Rech, A.J. Neoantigen Dissimilarity to the Self-Proteome Predicts Immunogenicity and Response to Immune Checkpoint Blockade. Cell Syst. 2019, 9, 375–382.e4. [Google Scholar] [CrossRef]
- Linette, G.P.; Becker-Hapak, M.; Skidmore, Z.L.; Baroja, M.L.; Xu, C.; Hundal, J.; Spencer, D.H.; Fu, W.; Cummins, C.; Robnett, M.; et al. Immunological ignorance is an enabling feature of the oligo-clonal T cell response to melanoma neoantigens. Proc. Natl. Acad. Sci. USA 2019, 116, 23662–23670. [Google Scholar] [CrossRef]
- Lam, H.; McNeil, L.K.; Starobinets, H.; DeVault, V.L.; Cohen, R.B.; Twardowski, P.; Johnson, M.L.; Gillison, M.L.; Stein, M.N.; Vaishampayan, U.N.; et al. An Empirical Antigen Selection Method Identifies Neoantigens That Either Elicit Broad Antitumor T-cell Responses or Drive Tumor Growth. Cancer Discov. 2021, 11, 696–713. [Google Scholar] [CrossRef]
- Janicki, C.N.; Jenkinson, S.R.; Williams, N.A.; Morgan, D.J. Loss of CTL Function among High-Avidity Tumor-Specific CD8+ T Cells following Tumor Infiltration. Cancer Res. 2008, 68, 2993–3000. [Google Scholar] [CrossRef] [Green Version]
- Hellstrom, K.E.; Hellstrom, I. From the Hellstrom paradox toward cancer cure. Prog. Mol. Biol. Transl. Sci. 2019, 164, 1–24. [Google Scholar] [CrossRef]
- Leone, P.; Di Lernia, G.; Solimando, A.G.; Cicco, S.; Saltarella, I.; Lamanuzzi, A.; Ria, R.; Frassanito, M.A.; Ponzoni, M.; Ditonno, P.; et al. Bone marrow endothelial cells sustain a tumor-specific CD8+ T cell subset with suppressive function in myeloma patients. OncoImmunology 2018, 8, e1486949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Haart, S.J.; Van De Donk, N.W.; Minnema, M.C.; Huang, J.H.; Aarts-Riemens, T.; Bovenschen, N.; Yuan, H.; Groen, R.; McMillin, D.W.; Jakubikova, J.; et al. Accessory Cells of the Microenvironment Protect Multiple Myeloma from T-Cell Cytotoxicity through Cell Adhesion-Mediated Immune Resistance. Clin. Cancer Res. 2013, 19, 5591–5601. [Google Scholar] [CrossRef] [Green Version]
- Leone, P.; Solimando, A.G.; Malerba, E.; Fasano, R.; Buonavoglia, A.; Pappagallo, F.; De Re, V.; Argentiero, A.; Silvestris, N.; Vacca, A.; et al. Actors on the Scene: Immune Cells in the Myeloma Niche. Front. Oncol. 2020, 10, 599098. [Google Scholar] [CrossRef] [PubMed]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Joshua, D.E.; Vuckovic, S.; Favaloro, J.; Lau, K.H.A.; Yang, S.; Bryant, C.E.; Gibson, J.; Ho, P.J. Treg and Oligoclonal Expansion of Terminal Effector CD8+ T Cell as Key Players in Multiple Myeloma. Front. Immunol. 2021, 12, 145. [Google Scholar] [CrossRef]
- Raja, K.R.M.; Rihova, L.; Zahradova, L.; Klincova, M.; Penka, M.; Hajek, R. Increased T Regulatory Cells Are Associated with Adverse Clinical Features and Predict Progression in Multiple Myeloma. PLoS ONE 2012, 7, e47077. [Google Scholar] [CrossRef]
- Alrasheed, N.; Lee, L.; Ghorani, E.; Henry, J.Y.; Conde, L.; Chin, M.; Galas-Filipowicz, D.; Furness, A.J.; Chavda, S.J.; Richards, H.; et al. Marrow-Infiltrating Regulatory T Cells Correlate with the Presence of Dysfunctional CD4+PD-1+ Cells and Inferior Survival in Patients with Newly Diagnosed Multiple Myeloma. Clin. Cancer Res. 2020, 26, 3443–3454. [Google Scholar] [CrossRef] [Green Version]
- Favaloro, J.; Brown, R.; Aklilu, E.; Yang, S.; Suen, H.; Hart, D.; Fromm, P.; Gibson, J.; Khoo, L.; Ho, P.J.; et al. Myeloma skews regulatory T and pro-inflammatory T helper 17 cell balance in favor of a suppressive state. Leuk. Lymphoma 2013, 55, 1090–1098. [Google Scholar] [CrossRef]
- Prabhala, R.H.; Neri, P.; Bae, J.E.; Tassone, P.; Shammas, M.A.; Allam, C.K.; Daley, J.F.; Chauhan, D.; Blanchard, E.; Thatte, H.S.; et al. Dysfunctional T regulatory cells in multiple myeloma. Blood 2006, 107, 301–304. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Ganeshan, P.; Hakim, M.; Verma, R.; Sharma, A.; Kumar, L. Significantly reduced regulatory T cell population in patients with untreated multiple myeloma. Leuk. Res. 2011, 35, 874–878. [Google Scholar] [CrossRef]
- Feng, X.; Zhang, L.; Acharya, C.; An, G.; Wen, K.; Qiu, L.; Munshi, N.C.; Tai, Y.-T.; Anderson, K.C. Targeting CD38 Suppresses Induction and Function of T Regulatory Cells to Mitigate Immunosuppression in Multiple Myeloma. Clin. Cancer Res. 2017, 23, 4290–4300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; Van De Donk, N.W.C.J.; Weiss, B.M.; et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.C.; Hayman, E.; Pegram, H.J.; Santos, E.; Heller, G.; Sadelain, M.; Brentjens, R.J. In vivo Inhibition of Human CD19-Targeted Effector T Cells by Natural T Regulatory Cells in a Xenotransplant Murine Model of B Cell Malignancy. Cancer Res. 2011, 71, 2871–2881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brentjens, R.J.; Rivière, I.; Park, J.H.; Davila, M.L.; Wang, X.; Stefanski, J.; Taylor, C.; Yeh, R.; Bartido, S.; Borquez-Ojeda, O.; et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 2011, 118, 4817–4828. [Google Scholar] [CrossRef]
- Ahmadzadeh, M.; Rosenberg, S.A. IL-2 administration increases CD4+CD25hi Foxp3+ regulatory T cells in cancer patients. Blood 2006, 107, 2409–2414. [Google Scholar] [CrossRef] [Green Version]
- Shameli, A.; Yamanouchi, J.; Tsai, S.; Yang, Y.; Clemente-Casares, X.; Moore, A.; Serra, P.; Santamaria, P. IL-2 promotes the function of memory-like autoregulatory CD8+T cells but suppresses their development via FoxP3+Treg cells. Eur. J. Immunol. 2012, 43, 394–403. [Google Scholar] [CrossRef]
- Perna, S.K.; Pagliara, D.; Mahendravada, A.; Liu, H.; Brenner, M.K.; Savoldo, B.; Dotti, G. Interleukin-7 Mediates Selective Expansion of Tumor-redirected Cytotoxic T Lymphocytes (CTLs) without Enhancement of Regulatory T-cell Inhibition. Clin. Cancer Res. 2013, 20, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Golumba-Nagy, V.; Kuehle, J.; Hombach, A.A.; Abken, H. CD28-ζ CAR T Cells Resist TGF-β Repression through IL-2 Signaling, Which Can Be Mimicked by an Engineered IL-7 Autocrine Loop. Mol. Ther. 2018, 26, 2218–2230. [Google Scholar] [CrossRef] [Green Version]
- Kofler, D.M.; Chmielewski, M.; Rappl, G.; Hombach, A.; Riet, T.; Schmidt, A.; A Hombach, A.; Wendtner, C.-M.; Abken, H. CD28 Costimulation Impairs the Efficacy of a Redirected T-cell Antitumor Attack in the Presence of Regulatory T cells Which Can Be Overcome by Preventing Lck Activation. Mol. Ther. 2011, 19, 760–767. [Google Scholar] [CrossRef]
- Koehler, H.; Kofler, D.; Hombach, A.; Abken, H. CD28 Costimulation Overcomes Transforming Growth Factor-β–Mediated Repression of Proliferation of Redirected Human CD4+ and CD8+ T Cells in an Antitumor Cell Attack. Cancer Res. 2007, 67, 2265–2273. [Google Scholar] [CrossRef] [Green Version]
- Tang, N.; Cheng, C.; Zhang, X.; Qiao, M.; Li, N.; Mu, W.; Wei, X.-F.; Han, W.; Wang, H. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight 2020, 5, e133977. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, M.; Raje, N. Anti-BCMA CAR T-cell therapy in multiple myeloma: Can we do better? Leukemia 2020, 34, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfannenstiel, L.W.; Diaz-Montero, C.M.; Tian, Y.F.; Scharpf, J.; Ko, J.S.; Gastman, B.R.; Diaz-Montero, M. Immune-Checkpoint Blockade Opposes CD8+ T-cell Suppression in Human and Murine Cancer. Cancer Immunol. Res. 2019, 7, 510–525. [Google Scholar] [CrossRef] [Green Version]
- Hallett, W.H.; Jing, W.; Drobyski, W.R.; Johnson, B.D. Immunosuppressive Effects of Multiple Myeloma Are Overcome by PD-L1 Blockade. Biol. Blood Marrow Transplant. 2011, 17, 1133–1145. [Google Scholar] [CrossRef] [Green Version]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J. Clin. Oncol. 2016, 34, 2698–2704. [Google Scholar] [CrossRef] [Green Version]
- Mateos, M.-V.; Blacklock, H.; Schjesvold, F.; Oriol, A.; Simpson, D.; George, A.; Goldschmidt, H.; LaRocca, A.; Chanan-Khan, A.; Sherbenou, D.; et al. Pembrolizumab plus pomalidomide and dexamethasone for patients with relapsed or refractory multiple myeloma (KEYNOTE-183): A randomised, open-label, phase 3 trial. Lancet Haematol. 2019, 6, e459–e469. [Google Scholar] [CrossRef]
- Suen, H.; Brown, R.; Yang, S.; Weatherburn, C.; Ho, P.J.; Woodland, N.; Nassif, N.; Barbaro, P.; Bryant, C.; Hart, D.; et al. Multiple myeloma causes clonal T-cell immunosenescence: Identification of potential novel targets for promoting tumour immunity and implications for checkpoint blockade. Leukemia 2016, 30, 1716–1724. [Google Scholar] [CrossRef]
- Zelle-Rieser, C.; Thangavadivel, S.; Biedermann, R.; Brunner, A.; Stoitzner, P.; Willenbacher, E.; Greil, R.; Jöhrer, K. T cells in multiple myeloma display features of exhaustion and senescence at the tumor site. J. Hematol. Oncol. 2016, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Quach, H.; Ritchie, D.; Stewart, A.K.; Neeson, P.; Harrison, S.; Smyth, M.; Prince, H.M. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia 2009, 24, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Walter, M.; Urak, R.; Weng, L.; Huynh, C.; Lim, L.; Wong, C.W.; Chang, W.-C.; Thomas, S.; Sanchez, J.F.; et al. Lenalidomide Enhances the Function of CS1 Chimeric Antigen Receptor–Redirected T Cells Against Multiple Myeloma. Clin. Cancer Res. 2017, 24, 106–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van De Donk, N.W.; Usmani, S.Z. CD38 Antibodies in Multiple Myeloma: Mechanisms of Action and Modes of Resistance. Front. Immunol. 2018, 9, 2134. [Google Scholar] [CrossRef] [PubMed]
- Alabanza, L.; Vu, B.; Wu, D.; Zhu, Z.; Dropulic, B.; Schneider, D. A Fully-Human Armored BCMA CAR Boosts Function of CD4+ CAR-T Cells and Resists TGF-β Suppression in Pre-Clinical Models of Multiple Myeloma. Blood 2020, 136, 37–38. [Google Scholar] [CrossRef]
- Kuhn, N.F.; Purdon, T.J.; van Leeuwen, D.G.; Lopez, A.V.; Curran, K.J.; Daniyan, A.F.; Brentjens, R.J. CD40 Ligand-Modified Chimeric Antigen Receptor T Cells Enhance Antitumor Function by Eliciting an Endogenous Antitumor Response. Cancer Cell 2019, 35, 473–488.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; Van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef] [PubMed]

| Target | Other Names | Physiological Single Cell-Type Enrichment (Proteinatlas.Org) | Identified Ligands (Uniprot) | Involvement in Biological Process (Uniprot) | Car-Based Clinical Trials in Mm (Selection from Clinicaltrials.Gov) |
|---|---|---|---|---|---|
| CD19 | (naïve and memory) B cells | co-receptor for B cell receptor, B cell activation, proliferation, differentiation and antibody-production | NCT04194931, NCT03706547, NCT04603872, NCT02794246 | ||
| CD38 | ciliated cells, erythroid cells, granulocytes, Kupffer cells, T cells, NK cells | NAD, NADP | production of second messengers cyclic ADP-ribose and nicotinate-adenine dinucleotide phosphate, cADPr hydrolase activity | NCT03464916, NCT03767751, NCT03778346 | |
| CD138 | Syndecan 1, SDC1 | hepatocytes, urothelial cells, cholangiocytes, memory B cells | linking of cytoskeleton and interstitial matrix, regulation of exosome biogenesis | NCT03672318, NCT03778346 | |
| BCMA (B cell maturation antigen) | TNF receptor superfamily member 17, TNFRSF13a, CD269 | melanocytes, erythroid cells, (naïve and memory) B cells, plasmacytoid DCs) | TNFSF13B/BLyS/ BAFF and TNFSF13/APRIL | B cell survival, regulation of humoral immunity, activation of NF-kappa-B and JNK | see Table 2 |
| Integrin β7 | B cells, granulocytes, T cells | Magnesium, Metal-binding | cell adhesion, lymphocyte migration and homing to gut tissue | NCT03778346 | |
| SLAMF7/SLAM-family member 7 | CS1, CD319 | Monocytes | immune cell activation, connection of innate and adaptive immunity | NCT04499339, NCT03958656, NCT03778346 | |
| GPRC5D (G-protein-coupled Receptor Class C Group 5 Member D) | early spermatids, melanocytes, late spermatids, B-cells | not yet determined in detail | NCT04555551, NCT05016778 | ||
| Immunoglobulin light chain | B cells | NCT00881920 | |||
| CD229 | Lymphocyte antigen 9 (LY 9) | melanocytes, B cells, T cells, erythroid cells, plasmacytoid DCs | member of the SLAM-family, activation and differentiation of a variety of immune cells | ||
| TACI (Transmembrane activator and CAML interactor) | TNF receptor superfamily member 13B | TNFSF13/APRIL and TNFSF13B/ TALL1/BAFF/BLYS | stimulation of B and T cell function, Calcineurin-dependent NFAT-activation, NFkB and AP-1 |
| Gene | Expression in BM-Samples from MM Patients Across Different Studies [in %] | ||||
|---|---|---|---|---|---|
| Andrade et al., 2008 [169] (n = 39) | Van Duin et al., 2011 [171] | Atanackovic et al., 2007 [172] (n = 55) | Condomines et al., 2007 [173] (n = 64) | ||
| newly diagnosed (n = 320) | relapsed (n = 264) | ||||
| BAGE1 | 32 | 14.5 | |||
| CTAGE5 | 95.6 | 48.5 | |||
| CTNNA2 | 60.6 | 26.5 | |||
| FAMI133A | 86.3 | 79.2 | |||
| GAGE (family) | 36 | 17 | |||
| GAGE8 | 15 | 61.4 | |||
| GAGEA | 16.6 | 71.2 | |||
| JARID1B | 82.5 | 33.7 | |||
| LAGE-1 | 49 | ||||
| MAGE A3/6 | 46 | 37.8/45 | 47.3/49.2 | 54.5 | 33/31 |
| MAGE A9 | 10.9 | 5.7 | |||
| MAGE B1 | 5.3 | 3.8 | 0.9 | ||
| MAGEA1 | 31 | 21.9 | 42 | 3.7 | |
| MAGEA12 | 20.5 | 15.3 | 33.7 | 25 | |
| MAGEA2 | 41 | 9.4 | 8.3 | 2.0 | |
| MAGEA4 | 3.1 | 5.7 | 0.2 | ||
| MAGEB2 | 47.2 | 27.7 | |||
| MAGEB4 | 5.3 | 1.1 | |||
| MAGEC1/CT7 | 77 | 71.3 | 60.6 | 66 | |
| MAGEC2 | 29.1 | 9.5 | 56.4 | 13 | |
| NY-ESO-1 | 36 | 7.3 | |||
| PAGE2 | 5.9 | 2.3 | |||
| PBK | 94.1 | 86.4 | |||
| PRAME | 23 | 31.9 | 37.9 | ||
| SPA17 | 38.1 | 9.1 | |||
| SPAG9 | 100 | 99.6 | |||
| SPANXC | 5 | 3 | 0.1 | ||
| SSX1 | 28 | 30.3 | 29.5 | 34.5 | 20 |
| SSX2 | 6.6 | 6.4 | 16.4 | 0.6 | |
| SSX3 | 2.5 | 5.7 | 0.4 | ||
| SSX5 | 20.0 | ||||
| TEX14 | 7.2 | 3 | |||
| TSPY1 | 10.6 | 13.6 | |||
| ZNF165 | 83.1 | 13.6 | |||
| Trial Number/Name | Sponsor | TCR-Specificity | HLA-Restriction | Phase | n | Diagnosis | Dose 1 | Conditioning Therapy 2 | Further Modifications | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| NCT01892293 | Adapt-immune | NY-ESO-1c259 | HLA-A*0201 | I/IIa | 6 | relapsed or progressive MM | 1 × ≥ 0.1–1 × 1010; in case of progression: second dose of up to 5 × 1010 | |||
| NCT01352286 | Glaxo SmithKline | NY-ESO-1c259 (high affinity) | HLA-A*0201 | I/IIa | 25 | relapsed or refractory MM (at least one prior therapy line) | >0.1–1 × 1010 | affinity maturated TCR/ specific peptide enhanced affinity receptor (SPEAR) T cells | [209] | |
| NCT03399448 | University of Pennsyl-vania | NY-ESO-1c259 (high affinity) | HLA-A*0201 | I/IIa | 3 | refractory metastatic sarcoma, relapsed or refractory MM (at least three prior therapy regimen) | 1 × 108 | CP, FLU | electroporated with CRISPR guide RNA to disrupt expression of endogenous TCRα, TCRβ and PD-1 (NYCE T Cells) | [210] |
| NCT02457650 | Shenzhen Second People’s Hospital | NY-ESO-1 | HLA-A*0201 | I | 36 | various entities, amongst them MM | N.A. | CP, FLU |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Füchsl, F.; Krackhardt, A.M. Adoptive Cellular Therapy for Multiple Myeloma Using CAR- and TCR-Transgenic T Cells: Response and Resistance. Cells 2022, 11, 410. https://doi.org/10.3390/cells11030410
Füchsl F, Krackhardt AM. Adoptive Cellular Therapy for Multiple Myeloma Using CAR- and TCR-Transgenic T Cells: Response and Resistance. Cells. 2022; 11(3):410. https://doi.org/10.3390/cells11030410
Chicago/Turabian StyleFüchsl, Franziska, and Angela M. Krackhardt. 2022. "Adoptive Cellular Therapy for Multiple Myeloma Using CAR- and TCR-Transgenic T Cells: Response and Resistance" Cells 11, no. 3: 410. https://doi.org/10.3390/cells11030410
APA StyleFüchsl, F., & Krackhardt, A. M. (2022). Adoptive Cellular Therapy for Multiple Myeloma Using CAR- and TCR-Transgenic T Cells: Response and Resistance. Cells, 11(3), 410. https://doi.org/10.3390/cells11030410