
Eryptosis: Programmed Death of Nucleus-Free, Iron-Filled Blood Cells


Abstract
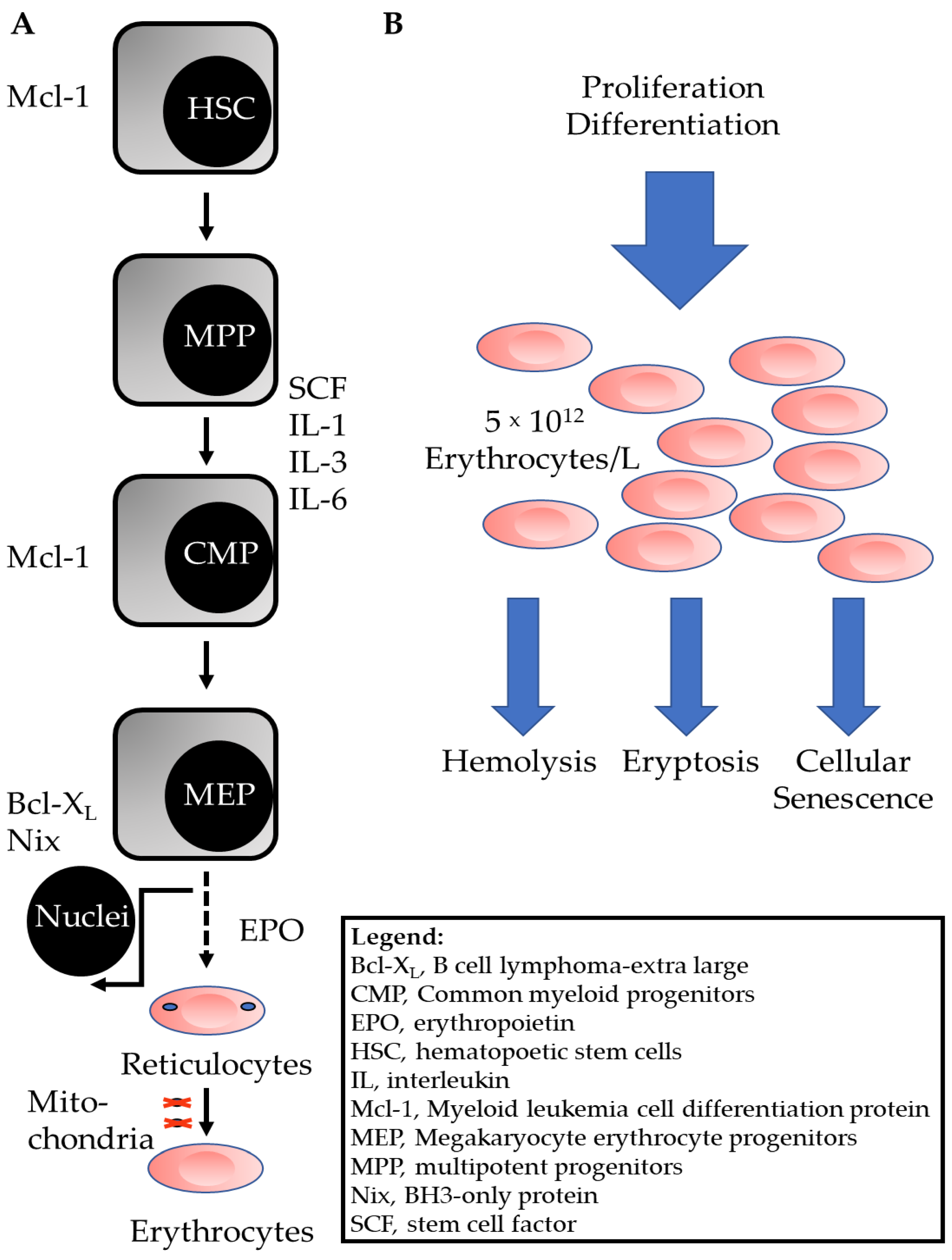
:1. Introduction
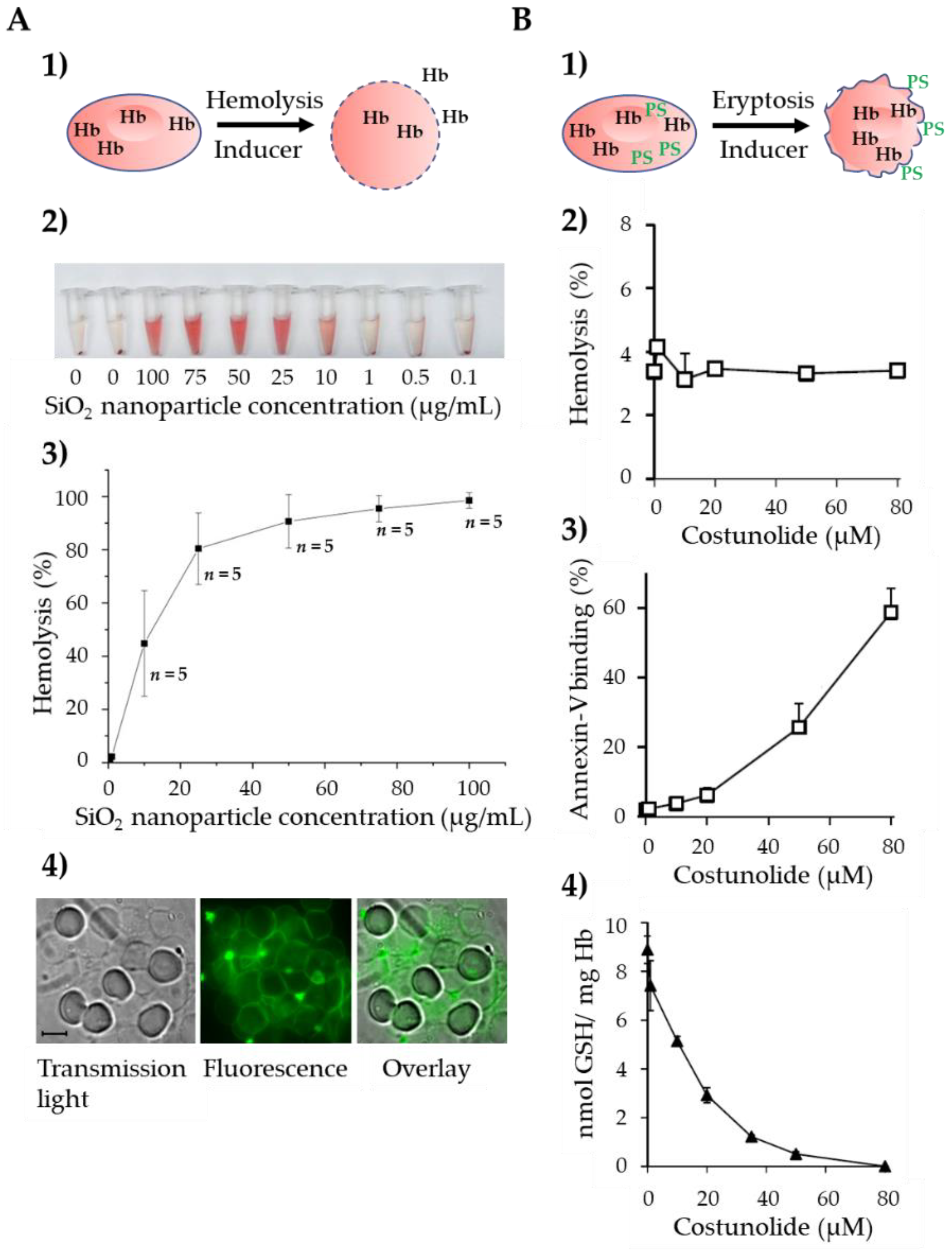
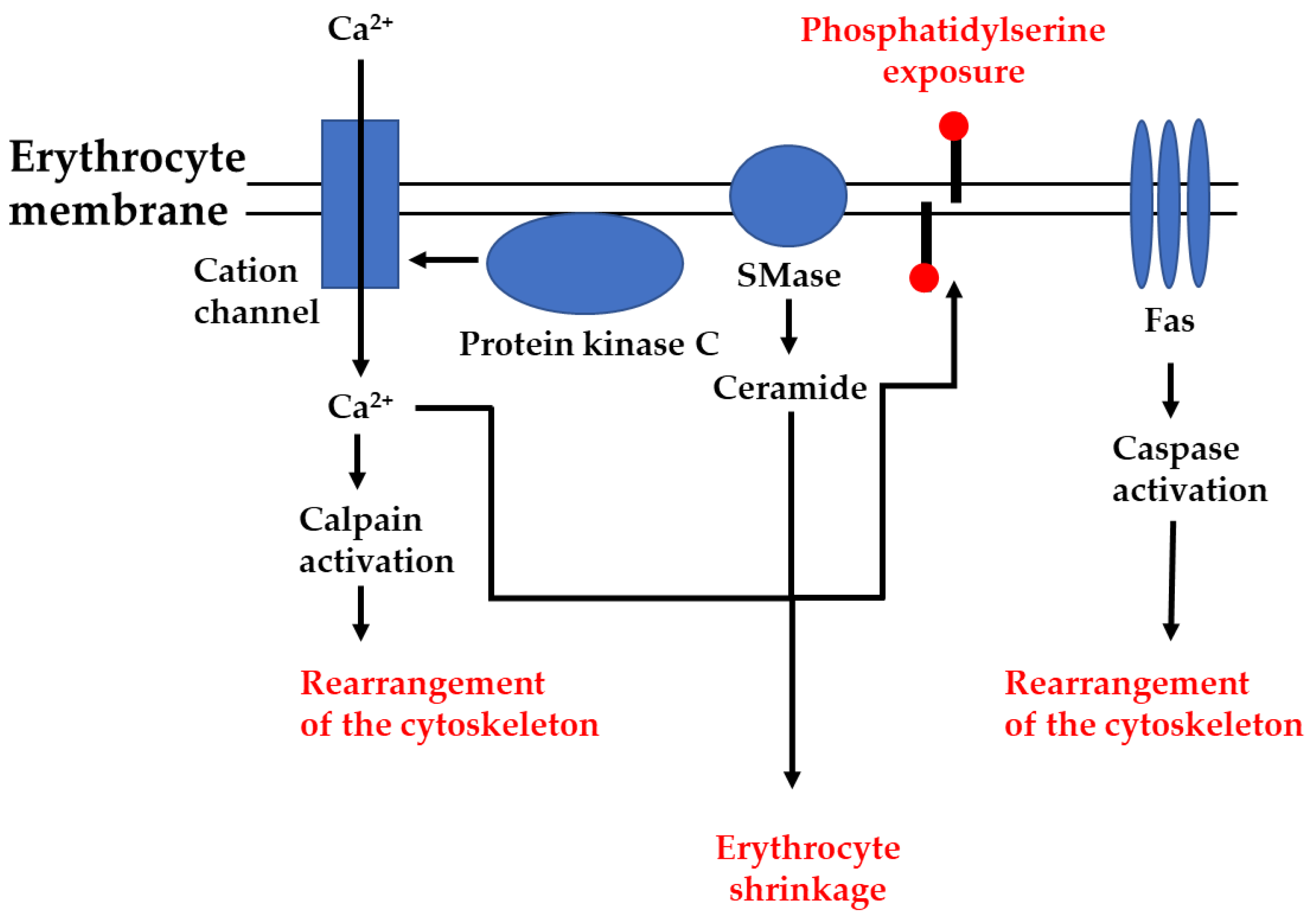
2. Pathways of Programmed Erythrocyte Death
2.1. Ca2+-Induced Eryptosis
2.2. Ceramide-Induced Eryptosis
2.3. Oxidative Stress-Induced Eryptosis
2.4. Role of Protein Kinases
2.5. Role of Proteases
2.6. Anti-Eryptotic Factors
3. Ferroptosis and Eryptosis: Similarities and Differences
4. The Role of Eryptosis in Infectious Diseases
5. The Role of Eryptosis in Hematologic Disorders and Other Diseases
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shiga, T. Oxygen transport in microcirculation. Jpn. J. Physiol. 1994, 44, 19–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, F.B. Red blood cell pH, the Bohr effect, and other oxygenation-linked phenomena in blood O2 and CO2 transport. Acta Physiol. Scand. 2004, 182, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Opferman, J.T. Life and death during hematopoietic differentiation. Curr. Opin. Immunol. 2007, 19, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Ridley, D.M.; Dawkins, F.; Perlin, E. Erythropoietin: A review. J. Natl. Med. Assoc. 1994, 86, 129–135. [Google Scholar] [PubMed]
- Yoshida, H.; Kawane, K.; Koike, M.; Mori, Y.; Uchiyama, Y.; Nagata, S. Phosphatidylserine-dependent engulfment by macrophages of nuclei from erythroid precursor cells. Nature 2005, 437, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Mankelow, T.J.; Griffiths, R.E.; Trompeter, S.; Flatt, J.F.; Cogan, N.M.; Massey, E.J.; Anstee, D.J. The ins and outs of reticulocyte maturation revisited: The role of autophagy in sickle cell disease. Autophagy 2016, 12, 590–591. [Google Scholar] [CrossRef] [Green Version]
- Callender, S.T.; Powell, E.O.; Witts, L.J. Normal red-cell survival in men and women. J. Pathol. Bacteriol. 1947, 59, 519–532. [Google Scholar] [CrossRef]
- Low, P.S.; Waugh, S.M.; Zinke, K.; Drenckhahn, D. The role of hemoglobin denaturation and band 3 clustering in red blood cell aging. Science 1985, 227, 531–533. [Google Scholar] [CrossRef]
- Lutz, H.U.; Fasler, S.; Stammler, P.; Bussolino, F.; Arese, P. Naturally occurring antiband 3 antibodies and complement in phagocytosis of oxidatively-stressed and in clearance of senescent red cells. Blood Cells 1988, 14, 175–203. [Google Scholar]
- Kay, M. Immunoregulation of cellular life span. Ann. N. Y. Acad. Sci. 2005, 1057, 85–111. [Google Scholar] [CrossRef]
- Kay, M.M.; Bosman, G.J.; Shapiro, S.S.; Bendich, A.; Bassel, P.S. Oxidation as a possible mechanism of cellular aging: Vitamin E deficiency causes premature aging and IgG binding to erythrocytes. Proc. Natl. Acad. Sci. USA 1986, 83, 2463–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoehn, R.S.; Jernigan, P.L.; Chang, A.L.; Edwards, M.J.; Pritts, T.A. Molecular mechanisms of erythrocyte aging. Biol. Chem. 2015, 396, 621–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bratosin, D.; Estaquier, J.; Petit, F.; Arnoult, D.; Quatannens, B.; Tissier, J.P.; Slomianny, C.; Sartiaux, C.; Alonso, C.; Huart, J.J.; et al. Programmed cell death in mature erythrocytes: A model for investigating death effector pathways operating in the absence of mitochondria. Cell Death Differ. 2001, 8, 1143–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, C.P.; Engels, I.H.; Rothbart, A.; Lauber, K.; Renz, A.; Schlosser, S.F.; Schulze-Osthoff, K.; Wesselborg, S. Human mature red blood cells express caspase-3 and caspase-8, but are devoid of mitochondrial regulators of apoptosis. Cell Death Differ. 2001, 8, 1197–1206. [Google Scholar] [CrossRef]
- Lang, K.S.; Duranton, C.; Poehlmann, H.; Myssina, S.; Bauer, C.; Lang, F.; Wieder, T.; Huber, S.M. Cation channels trigger apoptotic death of erythrocytes. Cell Death Differ. 2003, 10, 249–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, K.S.; Roll, B.; Myssina, S.; Schittenhelm, M.; Scheel-Walter, H.-G.; Kanz, L.; Fritz, J.; Lang, F.; Huber, S.M.; Wieder, T. Enhanced erythrocyte apoptosis in sickle cell anemia, thalassemia and glucose-6-phosphate dehydrogenase deficiency. Cell Physiol. Biochem. 2002, 12, 365–372. [Google Scholar] [CrossRef]
- Lang, K.S.; Lang, P.A.; Bauer, C.; Duranton, C.; Wieder, T.; Huber, S.M.; Lang, F. Mechanisms of suicidal erythrocyte death. Cell Physiol. Biochem. 2005, 15, 195–202. [Google Scholar] [CrossRef]
- Lang, P.A.; Kasinathan, R.S.; Brand, V.B.; Duranton, C.; Lang, C.; Koka, S.; Shumilina, E.; Kempe, D.S.; Tanneur, V.; Akel, A.; et al. Accelerated clearance of plasmodium-infected erythrocytes in sickle cell trait and annexin-A7 deficiency. Cell Physiol. Biochem. 2009, 24, 415–428. [Google Scholar] [CrossRef] [Green Version]
- Ghashghaeinia, M.; Cluitmans, J.C.A.; Akel, A.; Dreischer, P.; Toulany, M.; Köberle, M.; Skabytska, Y.; Saki, M.; Biedermann, T.; Duszenko, M.; et al. The impact of erythrocyte age on eryptosis. Br. J. Haematol. 2012, 157, 606–614. [Google Scholar] [CrossRef]
- Ghashghaeinia, M.; Koralkova, P.; Giustarini, D.; Mojzikova, R.; Fehrenbacher, B.; Dreischer, P.; Schaller, M.; Mrowietz, U.; Martínez-Ruiz, A.; Wieder, T.; et al. The specific PKC-α inhibitor chelerythrine blunts costunolide-induced eryptosis. Apoptosis 2020, 25, 674–685. [Google Scholar] [CrossRef]
- LaRocca, T.J.; Stivison, E.A.; Hod, E.A.; Spitalnik, S.L.; Cowan, P.J.; Randis, T.M.; Ratner, A.J. Human-specific bacterial pore-forming toxins induce programmed necrosis in erythrocytes. mBio 2014, 5, e01251-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, P.A.; Kempe, D.S.; Myssina, S.; Tanneur, V.; Birka, C.; Laufer, S.; Lang, F.; Wieder, T.; Huber, S.M. PGE2 in the regulation of programmed erythrocyte death. Cell Death Differ. 2005, 12, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Myssina, S.; Huber, S.M.; Birka, C.; Lang, P.A.; Lang, K.S.; Friedrich, B.; Risler, T.; Wieder, T.; Lang, F. Inhibition of erythrocyte cation channels by erythropoietin. J. Am. Soc. Nephrol. 2003, 14, 2750–2757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grygorczyk, R.; Schwarz, W.; Passow, H. Ca2+-activated K+ channels in human red cells. Comparison of single-channel currents with ion fluxes. Biophys. J. 1984, 45, 693–698. [Google Scholar] [CrossRef] [Green Version]
- Lang, P.A.; Kaiser, S.; Myssina, S.; Wieder, T.; Lang, F.; Huber, S.M. Role of Ca2+-activated K+ channels in human erythrocyte apoptosis. Am. J. Physiol. Cell Physiol. 2003, 285, C1553–C1560. [Google Scholar] [CrossRef] [Green Version]
- Koka, S.; Lang, C.; Niemoeller, O.M.; Boini, K.M.; Nicolay, J.P.; Huber, S.M.; Lang, F. Cell influence of NO synthase inhibitor L-NAME on parasitemia and survival of plasmodium berghei infected mice. Cell Physiol. Biochem. 2008, 21, 481–488. [Google Scholar] [CrossRef]
- Birulina, Y.G.; Petrova, I.V.; Rozenbaum, Y.A.; Shefer, E.A.; Smagliy, L.V.; Nosarev, A.V.; Gusakova, S.V. H2S-mediated changes in erythrocyte volume: Role of Gardos channels, Na+, K+, 2Cl− cotransport and anion exchanger. Bull. Exp. Biol. Med. 2019, 167, 508–511. [Google Scholar] [CrossRef]
- Geilen, C.C.; Wieder, T.; Orfanos, C.E. Ceramide signalling: Regulatory role in cell proliferation, differentiation and apoptosis in human epidermis. Arch. Dermatol. Res. 1997, 289, 559–566. [Google Scholar] [CrossRef]
- Trayssac, M.; Hannun, Y.A.; Obeid, L.M. Role of sphingolipids in senescence: Implication in aging and age-related diseases. J. Clin. Investig. 2018, 128, 2702–2712. [Google Scholar] [CrossRef]
- Morales, A.; Lee, H.; Goñi, F.M.; Kolesnick, R.; Fernandez-Checa, J.C. Sphingolipids and cell death. Apoptosis 2007, 12, 923–939. [Google Scholar] [CrossRef]
- Canals, D.; Salamone, S.; Hannun, Y.A. Visualizing bioactive ceramides. Chem. Phys. Lipids 2018, 216, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Lang, K.S.; Myssina, S.; Brand, V.; Sandu, C.; Lang, P.A.; Berchtold, S.; Huber, S.M.; Lang, F.; Wieder, T. Involvement of ceramide in hyperosmotic shock-induced death of erythrocytes. Cell Death Differ. 2004, 11, 231–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, P.A.; Kempe, D.S.; Tanneur, V.; Eisele, K.; Klarl, B.A.; Myssina, S.; Jendrossek, V.; Ishii, S.; Shimizu, T.; Waidmann, M.; et al. Stimulation of erythrocyte ceramide formation by platelet-activating factor. J. Cell Sci. 2005, 118, 1233–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, P.A.; Schenck, M.; Nicolay, J.P.; Becker, J.U.; Kempe, D.S.; Lupescu, A.; Koka, S.; Eisele, K.; Klarl, B.A.; Rübben, H.; et al. Liver cell death and anemia in Wilson disease involve acid sphingomyelinase and ceramide. Nat. Med. 2007, 13, 164–170. [Google Scholar] [CrossRef]
- Qadri, S.M.; Donkor, D.A.; Bhakta, V.; Eltringham-Smith, L.J.; Dwivedi, D.J.; Moore, J.C.; Pepler, L.; Ivetic, N.; Nazi, I.; Fox-Robichaud, A.E.; et al. Phosphatidylserine externalization and procoagulant activation of erythrocytes induced by pseudomonas aeruginosa virulence factor pyocyanin. J. Cell Mol. Med. 2016, 20, 710–720. [Google Scholar] [CrossRef] [Green Version]
- Allegra, M.; Restivo, I.; Fucarino, A.; Pitruzzella, A.; Vasto, S.; Livrea, M.A.; Tesoriere, L.; Attanzio, A. Proeryptotic activity of 4-hydroxynonenal: A new potential physiopathological role for lipid peroxidation products. Biomolecules 2020, 10, 770. [Google Scholar] [CrossRef]
- Ghashghaeinia, M.; Bobbala, D.; Wieder, T.; Koka, S.; Brück, J.; Fehrenbacher, B.; Röcken, M.; Schaller, M.; Lang, F.; Ghoreschi, K. Targeting glutathione by dimethylfumarate protects against experimental malaria by enhancing erythrocyte cell membrane scrambling. Am. J. Physiol. Cell. Physiol. 2010, 299, C791–C804. [Google Scholar] [CrossRef] [Green Version]
- Bissinger, R.; Bhuyan, A.A.M.; Qadri, S.M.; Lang, F. Oxidative stress, eryptosis and anemia: A pivotal mechanistic nexus in systemic diseases. FEBS J. 2019, 286, 826–854. [Google Scholar] [CrossRef] [Green Version]
- Alfhili, M.A.; Alsalmi, E.; Aljedai, A.; Alsughayyir, J.; Abudawood, M.; Basudan, A.M. Calcium-oxidative stress signaling axis and casein kinase 1α mediate eryptosis and hemolysis elicited by novel p53 agonist inauhzin. J. Chemother 2021, 19, 1–11. [Google Scholar] [CrossRef]
- Shi, X.; Wei, M.; Xu, Z.; Liu, Y.; Zhang, M.; Lv, L.; Wang, Q. Vitamin C inhibits blood-stage plasmodium parasites via oxidative stress. Front. Cell Dev. Biol. 2021, 9, 639944. [Google Scholar] [CrossRef]
- Ghashghaeinia, M.; Giustarini, D.; Koralkova, P.; Köberle, M.; Alzoubi, K.; Bissinger, R.; Hosseinzadeh, Z.; Dreischer, P.; Bernhardt, I.; Lang, F.; et al. Pharmacological targeting of glucose-6-phosphate dehydrogenase in human erythrocytes by Bay 11-7082, parthenolide and dimethyl fumarate. Sci. Rep. 2016, 6, 28754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, D.A.; Yang, L.; Low, P.S. Phorbol ester stimulates a protein kinase C-mediated agatoxin-TK-sensitive calcium permeability pathway in human red blood cells. Blood 2002, 100, 3392–3399. [Google Scholar] [CrossRef] [PubMed]
- de Jong, K.; Rettig, M.P.; Low, P.S.; Kuypers, F.A. Protein kinase C activation induces phosphatidylserine exposure on red blood cells. Biochemistry 2002, 41, 12562–12567. [Google Scholar] [CrossRef] [PubMed]
- Klarl, B.A.; Lang, P.A.; Kempe, D.S.; Niemoeller, O.M.; Akel, A.; Sobiesiak, M.; Eisele, K.; Podolski, M.; Huber, S.M.; Wieder, T.; et al. Protein kinase C mediates erythrocyte “programmed cell death” following glucose depletion. Am. J. Physiol. Cell Physiol. 2006, 290, C244–C253. [Google Scholar] [CrossRef] [Green Version]
- Adderley, J.D.; von Freyend, S.J.; Jackson, S.A.; Bird, M.J.; Burns, A.L.; Anar, B.; Metcalf, T.; Semblat, J.-P.; Billker, O.; Wilson, D.W.; et al. Analysis of erythrocyte signalling pathways during plasmodium falciparum infection identifies targets for host-directed antimalarial intervention. Nat. Commun. 2020, 11, 4015. [Google Scholar] [CrossRef]
- Adderley, J.D.; Doerig, C. Erythrocyte phospho-signalling is dynamically altered during infection with plasmodium falciparum. Microb. Cell 2020, 7, 286–288. [Google Scholar] [CrossRef]
- Lang, F.; Bissinger, R.; Abed, M.; Artunc, F. Eryptosis—The neglected cause of anemia in end stage renal disease. Kidney Blood Press. Res. 2017, 42, 749–760. [Google Scholar] [CrossRef]
- Zermati, Y.; Garrido, C.; Amsellem, S.; Fishelson, S.; Bouscary, D.; Valensi, F.; Varet, B.; Solary, E.; Hermine, O. Caspase activation is required for terminal erythroid differentiation. J. Exp. Med. 2001, 193, 247–254. [Google Scholar] [CrossRef] [Green Version]
- Dai, C.; Krantz, S.B. Interferon gamma induces upregulation and activation of caspases 1, 3, and 8 to produce apoptosis in human erythroid progenitor cells. Blood 1999, 93, 3309–3316. [Google Scholar] [CrossRef]
- Mandal, D.; Moitra, P.K.; Saha, S.; Basu, J. Caspase 3 regulates phosphatidylserine externalization and phagocytosis of oxidatively stressed erythrocytes. FEBS Lett. 2002, 513, 184–188. [Google Scholar] [CrossRef] [Green Version]
- Mandal, D.; Mazumder, A.; Das, P.; Kundu, M.; Basu, J. Fas-, caspase 8-, and caspase 3-dependent signaling regulates the activity of the aminophospholipid translocase and phosphatidylserine externalization in human erythrocytes. J. Biol. Chem. 2005, 280, 39460–39467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matarrese, P.; Straface, E.; Pietraforte, D.; Gambardella, L.; Vona, R.; Maccaglia, A.; Minetti, M.; Malorni, W. Peroxynitrite induces senescence and apoptosis of red blood cells through the activation of aspartyl and cysteinyl proteases. FASEB J. 2005, 19, 416–418. [Google Scholar] [CrossRef] [PubMed]
- Velásquez, F.C.; Maté, S.; Bakás, L.; Herlax, V. Induction of eryptosis by low concentrations of E. coli alpha-hemolysin. Biochim. Biophys. Acta 2015, 1848, 2779–2788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vota, D.M.; Crisp, R.L.; Nesse, A.B.; Vittori, D.C. Oxidative stress due to aluminum exposure induces eryptosis which is prevented by erythropoietin. J. Cell Biochem. 2012, 113, 1581–1589. [Google Scholar]
- Sun, Y.; Liu, G.; Jiang, Y.; Wang, H.; Xiao, H.; Guan, G. Erythropoietin protects erythrocytes against oxidative stress-induced eryptosis in vitro. Clin. Lab. 2018, 64, 365–369. [Google Scholar] [CrossRef]
- Bartolmäs, T.; Mayer, B.; Balola, A.H.; Salama, A. Eryptosis in autoimmune haemolytic anaemia. Eur. J. Haematol. 2018, 100, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Nicolay, J.P.; Liebig, G.; Niemoeller, O.M.; Koka, S.; Ghashghaeinia, M.; Wieder, T.; Haendeler, J.; Busse, R.; Lang, F. Inhibition of suicidal erythrocyte death by nitric oxide. Pflügers Archiv-Eur. J. Physiol. 2008, 456, 293–305. [Google Scholar] [CrossRef]
- Nader, E.; Romana, M.; Guillot, N.; Fort, R.; Stauffer, E.; Lemonne, N.; Garnier, Y.; Chambers Skinner, S.; Etienne-Julan, M.; Robert, M.; et al. Association between nitric oxide, oxidative stress, eryptosis, red blood cell microparticles, and vascular function in sickle cell anemia. Front. Immunol. 2020, 11, 551441. [Google Scholar] [CrossRef]
- Ghashghaeinia, M.; Wesseling, M.C.; Ramos, E.; Petkova-Kirova, P.; Waibel, S.; Lang, E.; Bissinger, R.; Alzoubi, K.; Edelmann, B.; Hosseinzadeh, Z.; et al. Trifluoperazine-induced suicidal erythrocyte death and S-nitrosylation inhibition, reversed by the nitric oxide donor sodium nitroprusside. Cell Physiol. Biochem. 2017, 42, 1985–1998. [Google Scholar] [CrossRef]
- Ghashghaeinia, M.; Toulany, M.; Saki, M.; Bobbala, D.; Fehrenbacher, B.; Rupec, R.; Rodemann, H.P.; Ghoreschi, K.; Röcken, M.; Schaller, M.; et al. The NFĸB pathway inhibitors Bay 11-7082 and parthenolide induce programmed cell death in anucleated erythrocytes. Cell Physiol. Biochem. 2011, 27, 45–54. [Google Scholar] [CrossRef]
- Široká, M.; Franco, C.; Guľašová, Z.; Hertelyová, Z.; Tomečková, V.; Rodella, L.F.; Rezzani, R. Nuclear factor-B and nitric oxide synthases in red blood cells: Good or bad in obesity? A preliminary study. Eur. J. Histochem. 2020, 64, 3081. [Google Scholar] [CrossRef] [PubMed]
- Ghashghaeinia, M.; Cluitmans, J.C.; Toulany, M.; Saki, M.; Köberle, M.; Lang, E.; Dreischer, P.; Biedermann, T.; Duszenko, M.; Lang, F.; et al. Age sensitivity of NFκB abundance and programmed cell death in erythrocytes induced by NFκB inhibitors. Cell Physiol. Biochem. 2013, 32, 801–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlawe, D.; Majdalani, A.; Velcicky, J.; Hessler, E.; Wieder, T.; Prokop, A.; Schmalz, H.-G. Iron-containing nucleoside analogues with pronounced apoptosis-inducing activity. Angew. Chem. Int. Ed. 2004, 43, 1731–1734. [Google Scholar] [CrossRef] [PubMed]
- Hirschhäuser, C.; Velcicky, J.; Schlawe, D.; Hessler, E.; Majdalani, A.; Neudörfl, J.-M.; Prokop, A.; Wieder, T.; Schmalz, H.-G. Nucleoside analogues with a 1,3-diene-Fe(CO)3 substructure: Stereoselective synthesis, configurational assignment, and apoptosis-inducing activity. Chemistry 2013, 19, 13017–13029. [Google Scholar] [CrossRef] [PubMed]
- Prinz, C.; Vasyutina, E.; Lohmann, G.; Schrader, A.; Romanski, S.; Hirschhäuser, C.; Mayer, P.; Frias, C.; Herling, C.D.; Hallek, M.; et al. Organometallic nucleosides induce non-classical leukemic cell death that is mitochondrial-ROS dependent and facilitated by TCL1-oncogene burden. Mol. Cancer 2015, 14, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Green, D.R.; Victor, B. The pantheon of the fallen: Why are there so many forms of cell death? Trends Cell Biol. 2012, 22, 555–556. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Henke, N.; Albrecht, P.; Bouchachia, I.; Ryazantseva, M.; Knoll, K.; Lewerenz, J.; Kaznacheyeva, E.; Maher, P.; Methner, A. The plasma membrane channel ORAI1 mediates detrimental calcium influx caused by endogenous oxidative stress. Cell Death Dis. 2013, 4, e470. [Google Scholar] [CrossRef] [Green Version]
- Battaglia, A.M.; Chirillo, R.; Aversa, I.; Sacco, A.; Costanzo, F.; Biamonte, F. Ferroptosis and cancer: Mitochondria meet the “iron maiden” cell death. Cells 2020, 9, 1505. [Google Scholar] [CrossRef] [PubMed]
- Kempe, D.S.; Lang, P.A.; Duranton, C.; Akel, A.; Lang, K.S.; Huber, S.M.; Wieder, T.; Lang, F. Enhanced programmed cell death of iron-deficient erythrocytes. FASEB J. 2006, 20, 368–370. [Google Scholar] [CrossRef] [PubMed]
- Hershko, C.; Link, G.; Cabantchik, I. Pathophysiology of iron overload. Ann. N. Y. Acad. Sci. 1998, 850, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Du Plooy, J.N.; Bester, J.; Pretorius, E. Eryptosis in haemochromatosis: Implications for rheology. Clin. Hemorheol. Microcirc. 2018, 69, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Altamura, S.; Vegi, N.M.; Hoppe, P.S.; Schroeder, T.; Aichler, M.; Walch, A.; Okreglicka, K.; Hültner, L.; Schneider, M.; Ladinig, C.; et al. Glutathione peroxidase 4 and vitamin E control reticulocyte maturation, stress erythropoiesis and iron homeostasis. Haematologica 2020, 105, 937–950. [Google Scholar] [CrossRef] [Green Version]
- Bao, Y.; Williamson, G. Phospholipid hydroperoxide peroxidase activities in erythrocytes. Biochem. Soc. Trans. 1997, 25, S557. [Google Scholar] [CrossRef] [Green Version]
- Lang, F.; Pelzl, L.; Schöls, L.; Hermann, A.; Föller, M.; Schäffer, T.E.; Stournaras, C. Neurons, erythrocytes and beyond—The diverse functions of chorein. Neurosignals 2017, 25, 117–126. [Google Scholar] [CrossRef] [Green Version]
- Calderón-Salinas, J.V.; Muñoz-Reyes, E.G.; Guerrero-Romero, J.F.; Rodríguez-Morán, M.; Bracho-Riquelme, R.L.; Carrera-Gracia, M.A.; Quintanar-Escorza, M.A. Eryptosis and oxidative damage in type 2 diabetic mellitus patients with chronic kidney disease. Mol. Cell Biochem. 2011, 357, 171–179. [Google Scholar] [CrossRef]
- Cilla, A.; López-García, G.; Collado-Díaz, V.; Blanch-Ruiz, M.A.; Garcia-Llatas, G.; Barberá, R.; Martinez-Cuesta, M.A.; Real, J.T.; Álvarez, Á.; Martínez-Hervás, S. Hypercholesterolemic patients have higher eryptosis and erythrocyte adhesion to human endothelium independently of statin therapy. Int. J. Clin. Pract. 2021, 75, e14771. [Google Scholar] [CrossRef]
- Bobbala, D.; Alesutan, I.; Föller, M.; Tschan, S.; Huber, S.M.; Lang, F. Protective effect of amiodarone in malaria. Acta Trop. 2010, 116, 39–44. [Google Scholar] [CrossRef]
- Rivas Totino, P.R.; Daniel-Ribeiro, C.T.; de Fátima Ferreira-da-Cruz, M. Refractoriness of eryptotic red blood cells to plasmodium falciparum infection: A putative host defense mechanism limiting parasitaemia. PLoS ONE 2011, 6, e26575. [Google Scholar]
- Hortle, E.; Nijagal, B.; Bauer, D.C.; Jensen, L.M.; Ahn, S.B.; Cockburn, I.A.; Lampkin, S.; Tull, D.; McConville, M.J.; McMorran, B.J.; et al. Adenosine monophosphate deaminase 3 activation shortens erythrocyte half-life and provides malaria resistance in mice. Blood 2016, 128, 1290–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulet, C.; Gaynor, T.L.; Carvalho, T.G. Eryptosis and malaria: New experimental guidelines and re-evaluation of the antimalarial potential of eryptosis inducers. Front. Cell Infect. Microbiol. 2021, 11, 630812. [Google Scholar] [CrossRef]
- Ghashghaeinia, M.; Dreischer, P.; Wieder, T.; Köberle, M. Coronavirus disease 2019 (COVID-19), human erythrocytes and the PKC-alpha/-beta inhibitor chelerythrine -possible therapeutic implication. Cell Cycle 2020, 19, 3399–3405. [Google Scholar] [CrossRef] [PubMed]
- Stalder, G.; Alberio, L. Ritonavir- and lopinavir-induced eryptosis in a SARS-CoV-2-infected patient. Blood 2020, 136, 915. [Google Scholar] [CrossRef]
- Kempe, D.S.; Akel, A.; Lang, P.A.; Hermle, T.; Biswas, R.; Muresanu, J.; Friedrich, B.; Dreischer, P.; Wolz, C.; Schumacher, U.; et al. Suicidal erythrocyte death in sepsis. J. Mol. Med. 2007, 85, 273–281. [Google Scholar] [CrossRef]
- Ibrahim, H.A.; Fouda, M.I.; Yahya, R.S.; Abousamra, N.K.; Abd Elazim, R.A. Erythrocyte phosphatidylserine exposure in β-thalassemia. Lab. Hematol. 2014, 20, 9–14. [Google Scholar] [CrossRef]
- Vomero, M.; Finucci, A.; Barbati, C.; Colasanti, T.; Ceccarelli, F.; Novelli, L.; Massaro, L.; Truglia, S.; Pensa, C.; Mauro, F.R.; et al. Increased eryptosis in patients with primary antiphospholipid syndrome (APS): A new actor in the pathogenesis of APS. Clin. Exp. Rheumatol. 2021, 39, 838–843. [Google Scholar]
- Turpin, C.; Catan, A.; Meilhac, O.; Bourdon, E.; Canonne-Hergaux, F.; Rondeau, P. Erythrocytes: Central actors in multiple scenes of atherosclerosis. Int. J. Mol. Sci. 2021, 22, 5843. [Google Scholar] [CrossRef]
- Ferreira Dias, G.; Soares Tozoni, S.; Bohnen, G.; Grobe, N.; Rodrigues, S.D.; Meireles, T.; Nakao, L.S.; Pecoits-Filho, R.; Kotanko, P.; Novais Moreno-Amaral, A. Uremia and inadequate oxygen supply induce eryptosis and intracellular hypoxia in red blood cells. Cell Physiol. Biochem. 2021, 55, 449–459. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Protein Name | Function | References |
|---|---|---|
| Cation channel | Ca2+ entry | Lang et al. [15] |
| Sphingomyelinase | Formation of ceramide | Lang et al. [32] |
| Protein kinase C | Protein phosphorylation | Klarl et al. [44] |
| Nuclear factor κB | Transcription factor 1 | Ghashghaeinia et al. [60] |
| Caspase-3 | Cysteinprotease | Mandal et al. [50] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dreischer, P.; Duszenko, M.; Stein, J.; Wieder, T. Eryptosis: Programmed Death of Nucleus-Free, Iron-Filled Blood Cells. Cells 2022, 11, 503. https://doi.org/10.3390/cells11030503
Dreischer P, Duszenko M, Stein J, Wieder T. Eryptosis: Programmed Death of Nucleus-Free, Iron-Filled Blood Cells. Cells. 2022; 11(3):503. https://doi.org/10.3390/cells11030503
Chicago/Turabian StyleDreischer, Peter, Michael Duszenko, Jasmin Stein, and Thomas Wieder. 2022. "Eryptosis: Programmed Death of Nucleus-Free, Iron-Filled Blood Cells" Cells 11, no. 3: 503. https://doi.org/10.3390/cells11030503
APA StyleDreischer, P., Duszenko, M., Stein, J., & Wieder, T. (2022). Eryptosis: Programmed Death of Nucleus-Free, Iron-Filled Blood Cells. Cells, 11(3), 503. https://doi.org/10.3390/cells11030503