
Mechanisms of Choice in X-Chromosome Inactivation


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. X-Chromosome Inactivation: A Special Case of Dosage Compensation between the Sexes
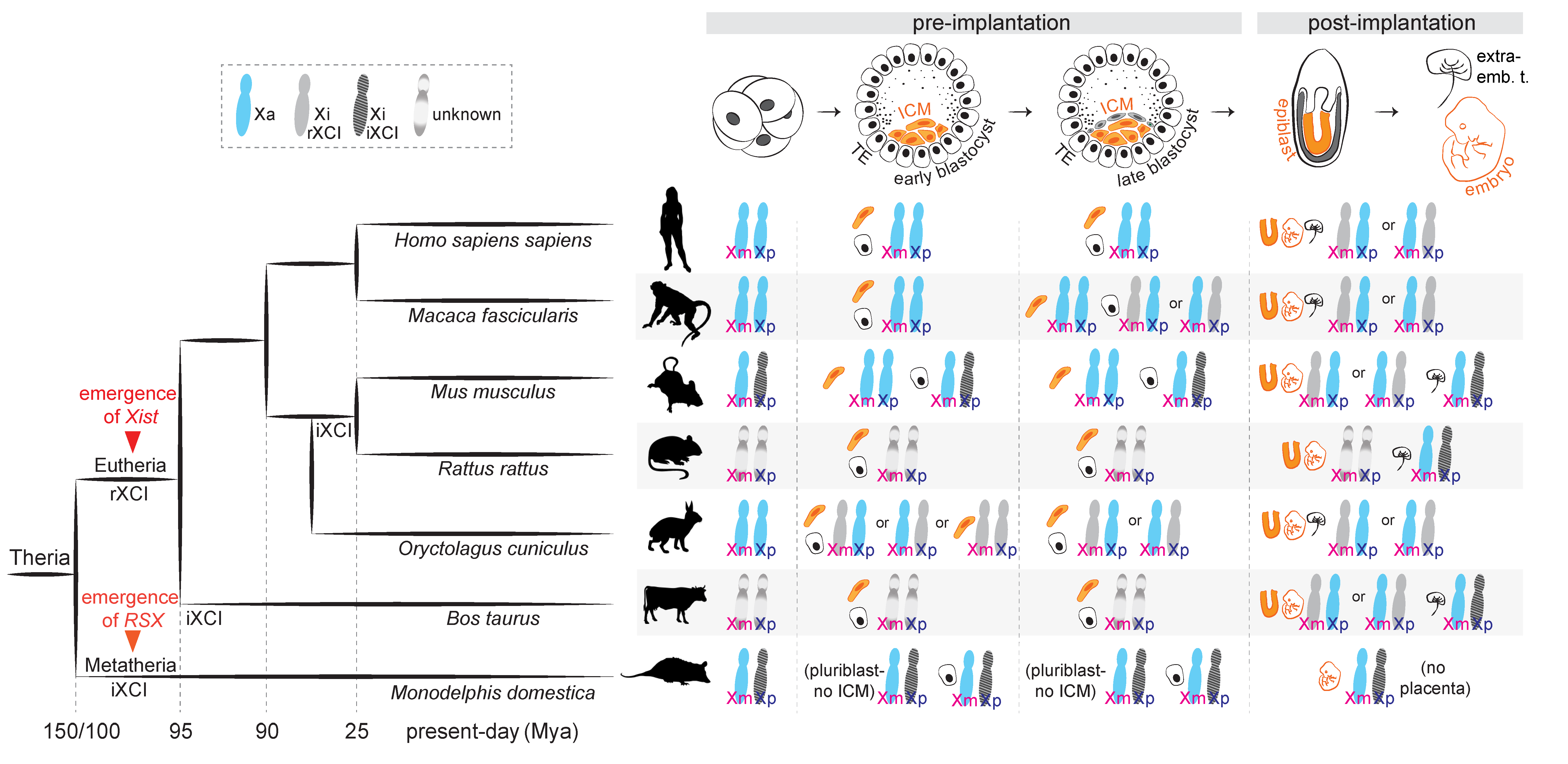
2. Types of XCI Choice across Mammals: Predetermined or Rolling Dice
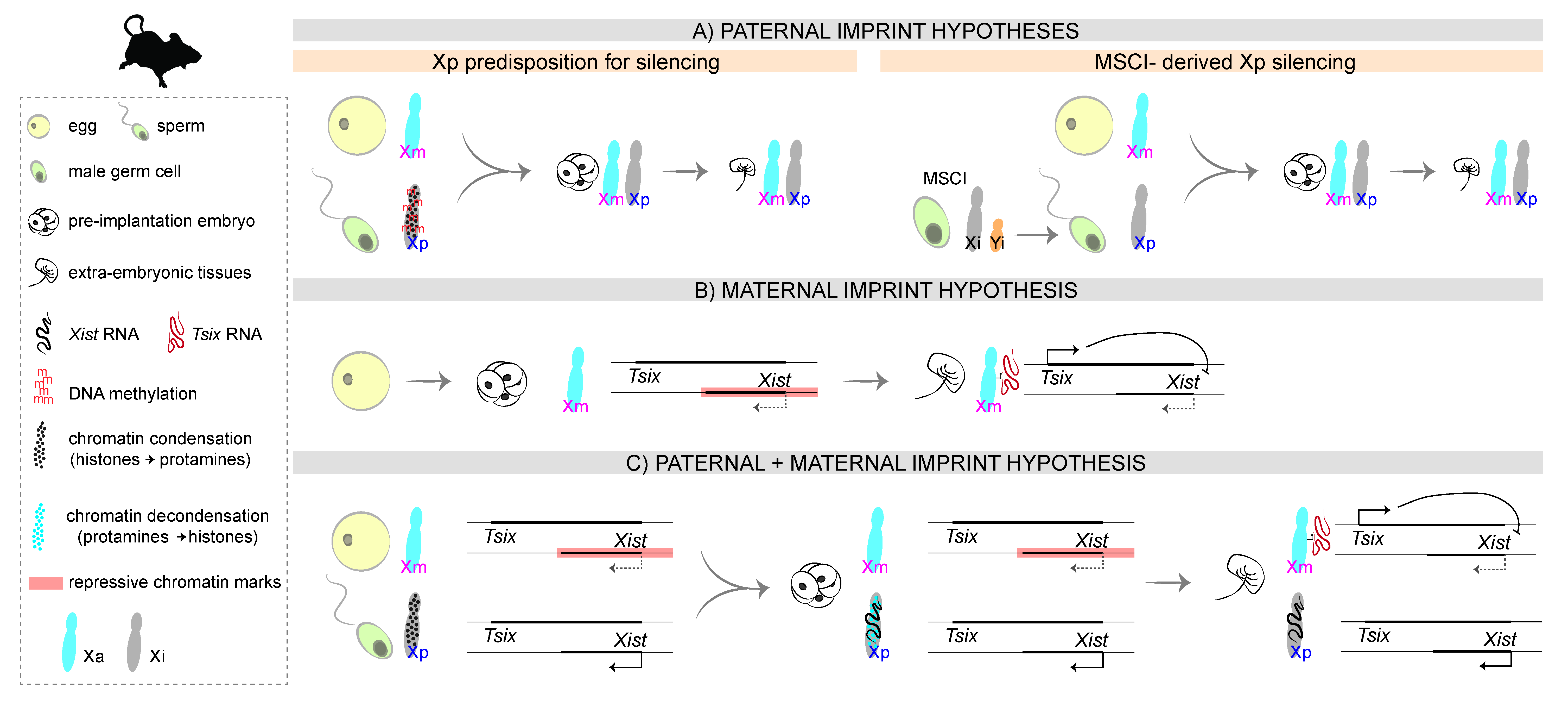
3. Mechanisms of iXCI: Choosing to Inactivate the Xp
3.1. The First Proposals: A Paternal Imprint
3.2. The Unexpected Outcome: A Non-Canonical Maternal Imprint
3.3. Evolutionary Considerations about iXCI
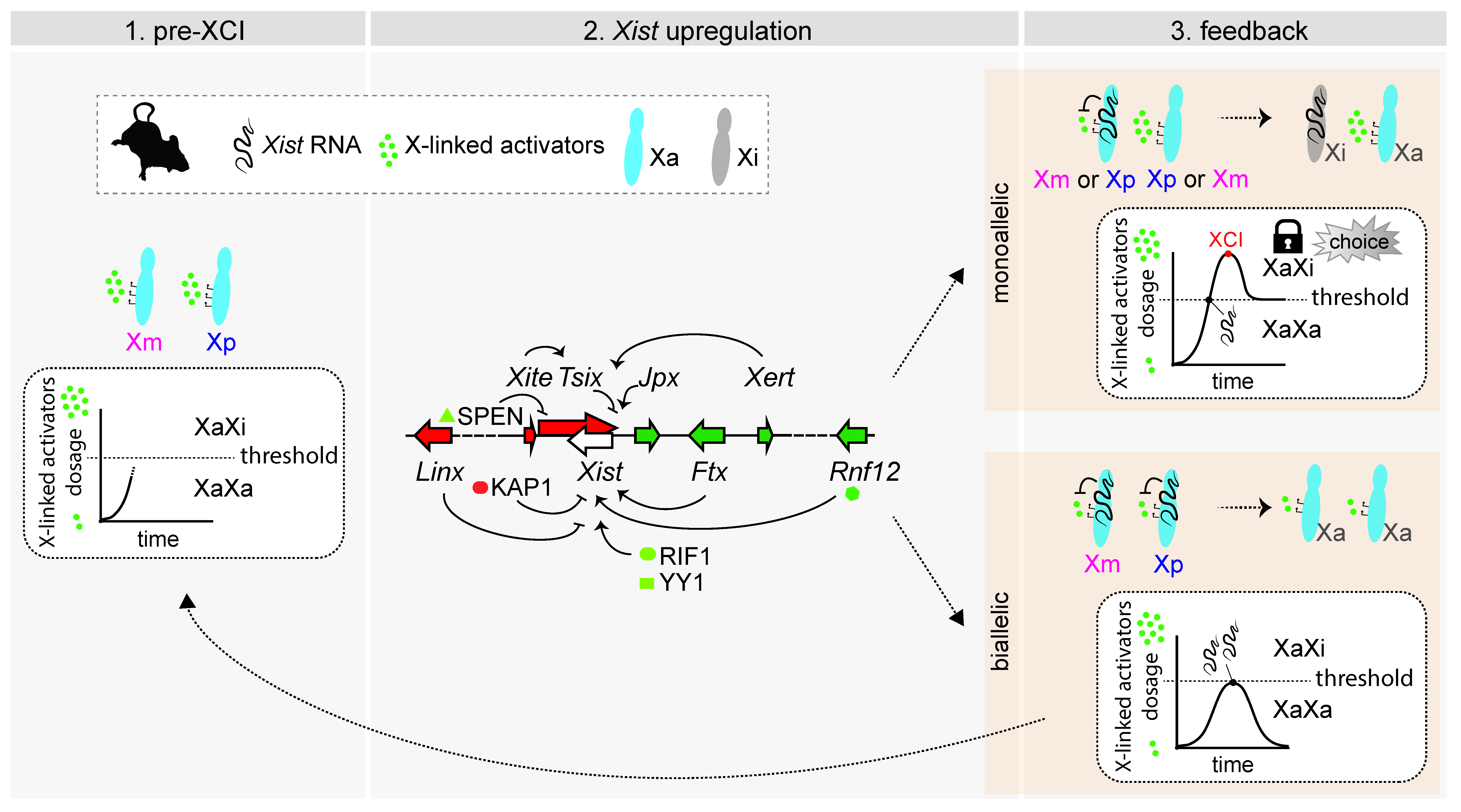
4. Mechanisms of rXCI in Mouse: A Race for Inactivation
4.1. Influencing Choice by Influencing Xist Upregulation
4.2. Influencing Choice by Preventing Xist Upregulation from the Second Chromosome
5. Choice in Human rXCI: Biallelic Dampening or Direct Monoallelic Inactivation?
6. Preferences in Choice: Random XCI Patterns Are Often Skewed
7. XCI Choice: The Second Most Important Moment in the Lives of XX Mammals?
Funding
Acknowledgments
Conflicts of Interest
References
- Disteche, C.M. Dosage Compensation of the Sex Chromosomes. Annu. Rev. Genet. 2012, 46, 537–560. [Google Scholar] [CrossRef] [PubMed]
- Lyon, M.F. Gene Action in the X-chromosome of the Mouse (Mus musculus L.). Nature 1961, 190, 372–373. [Google Scholar] [CrossRef] [PubMed]
- Whitworth, D.J.; Pask, A.J. The X factor: X chromosome dosage compensation in the evolutionarily divergent monotremes and marsupials. Semin. Cell Dev. Biol. 2016, 56, 117–121. [Google Scholar] [CrossRef]
- Sharman, G.B. Late DNA Replication in the Paternally Derived X Chromosome of Female Kangaroos. Nature 1971, 230, 231–232. [Google Scholar] [CrossRef] [PubMed]
- Evans, H.J.; Ford, C.E.; Lyon, M.F.; Gray, J.; Evans, C.E.F.H.J. DNA Replication and Genetic Expression in Female Mice with Morphologically Distinguishable X Chromosomes. Nature 1965, 206, 900–903. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, I.; Otte, A.P.; Allis, C.D.; Reinberg, D.; Heard, E. Epigenetic Dynamics of Imprinted X Inactivation During Early Mouse Development. Science 2004, 303, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Takagi, N.; Sasaki, M. Preferential inactivation of the paternally derived X chromosome in the extraembryonic membranes of the mouse. Nature 1975, 256, 640–642. [Google Scholar] [CrossRef] [PubMed]
- Harper, M.I.; Fosten, M.; Monk, M. Preferential paternal X inactivation in extraembryonic tissues of early mouse embryos. Development 1982, 67, 127–135. [Google Scholar] [CrossRef]
- Huynh, K.D.; Lee, J.T. Inheritance of a pre-inactivated paternal X chromosome in early mouse embryos. Nature 2003, 426, 857–862. [Google Scholar] [CrossRef]
- Latham, K.E. X chromosome imprinting and inactivation in the early mammalian embryo. Trends Genet. 1996, 12, 134–138. [Google Scholar] [CrossRef]
- Mak, W.; Nesterova, T.B.; de Napoles, M.; Appanah, R.; Yamanaka, S.; Otte, A.P.; Brockdorff, N. Reactivation of the Paternal X Chromosome in Early Mouse Embryos. Science 2004, 303, 666–669. [Google Scholar] [CrossRef]
- West, J.D.; Frels, W.I.; Chapman, V.M.; Papaioannou, V. Preferential expression of the maternally derived X chromosome in the mouse yolk sac. Cell 1977, 12, 873–882. [Google Scholar] [CrossRef]
- Wake, N.; Takagi, N.; Sasaki, M. Non-random inactivation of X chromosome in the rat yolk sac. Nature 1976, 262, 580–581. [Google Scholar] [CrossRef]
- Dindot, S.V.; Kent, K.C.; Evers, B.; Loskutoff, N.; Womack, J.; Piedrahita, J.A. Conservation of genomic imprinting at the XIST, IGF2, and GTL2 loci in the bovine. Mamm. Genome 2004, 15, 966–974. [Google Scholar] [CrossRef]
- Xue, F.; Tian, X.C.; Du, F.; Kubota, C.; Taneja, M.; Dinnyes, A.; Dai, Y.; Levine, H.; Pereira, L.V.; Yang, X. Aberrant patterns of X chromosome inactivation in bovine clones. Nat. Genet. 2002, 31, 216–220. [Google Scholar] [CrossRef]
- Goto, T.; Wright, E.; Monk, M. Paternal X-chromosome inactivation in human trophoblastic cells. Mol. Hum. Reprod. 1997, 3, 77–80. [Google Scholar] [CrossRef]
- Harrison, K. X-chromosome inactivation in the human cytotrophoblast. Cytogenet. Genome Res. 1989, 52, 37–41. [Google Scholar] [CrossRef]
- De Mello, J.C.M.; De Araújo, É.S.S.; Stabellini, R.; Fraga, A.M.; De Souza, J.E.S.; Sumita, D.R.; Camargo, A.A.; Pereira, L.V. Random X Inactivation and Extensive Mosaicism in Human Placenta Revealed by Analysis of Allele-Specific Gene Expression along the X Chromosome. PLoS ONE 2010, 5, e10947. [Google Scholar] [CrossRef]
- Hamada, H.; Okae, H.; Toh, H.; Chiba, H.; Hiura, H.; Shirane, K.; Sato, T.; Suyama, M.; Yaegashi, N.; Sasaki, H.; et al. Allele-Specific Methylome and Transcriptome Analysis Reveals Widespread Imprinting in the Human Placenta. Am. J. Hum. Genet. 2016, 99, 1045–1058. [Google Scholar] [CrossRef]
- Okamoto, I.; Patrat, C.; Thépot, D.; Peynot, N.; Fauque, P.; Daniel, N.; Diabangouaya, P.; Wolf, J.-P.; Renard, J.-P.; Duranthon, V.; et al. Eutherian mammals use diverse strategies to initiate X-chromosome inactivation during development. Nature 2011, 472, 370–374. [Google Scholar] [CrossRef]
- Zou, H.; Yu, D.; Du, X.; Wang, J.; Chen, L.; Wang, Y.; Xu, H.; Zhao, Y.; Zhao, S.; Pang, Y.; et al. No imprinted XIST expression in pigs: Biallelic XIST expression in early embryos and random X inactivation in placentas. Cell. Mol. Life. Sci. 2019, 76, 4525–4538. [Google Scholar] [CrossRef]
- Ramos-Ibeas, P.; Sang, F.; Zhu, Q.; Tang, W.W.C.; Withey, S.; Klisch, D.; Wood, L.; Loose, M.; Surani, M.A.; Alberio, R. Pluripotency and X chromosome dynamics revealed in pig pre-gastrulating embryos by single cell analysis. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef]
- Okamoto, I.; Nakamura, T.; Sasaki, K.; Yabuta, Y.; Iwatani, C.; Tsuchiya, H.; Nakamura, S.-I.; Ema, M.; Yamamoto, T.; Saitou, M. The X chromosome dosage compensation program during the development of cynomolgus monkeys. Science 2021, 374, eabd8887. [Google Scholar] [CrossRef]
- Dossin, F.; Heard, E. The Molecular and Nuclear Dynamics of X-Chromosome Inactivation. Cold Spring Harb. Perspect. Biol. 2021, 22, a040196. [Google Scholar] [CrossRef]
- Penny, G.D.; Kay, G.F.; Sheardown, S.A.; Rastan, S.; Brockdorff, N. Requirement for Xist in X chromosome inactivation. Nature 1996, 379, 131–137. [Google Scholar] [CrossRef]
- Borensztein, M.; Syx, L.; Ancelin, K.; Diabangouaya, P.; Picard, C.; Liu, T.; Liang, J.-B.; Vassilev, I.; Galupa, R.; Servant, N.; et al. Xist-dependent imprinted X inactivation and the early developmental consequences of its failure. Nat. Struct. Mol. Biol. 2017, 24, 226–233. [Google Scholar] [CrossRef]
- Namekawa, S.H.; Payer, B.; Huynh, K.D.; Jaenisch, R.; Lee, J.T. Two-Step Imprinted X Inactivation: Repeat versus Genic Silencing in the Mouse. Mol. Cell. Biol. 2010, 30, 3187–3205. [Google Scholar] [CrossRef]
- Grant, J.; Mahadevaiah, S.K.; Khil, P.; Sangrithi, M.; Royo, H.; Duckworth, J.; McCarrey, J.R.; VandeBerg, J.L.; Renfree, M.; Taylor, W.; et al. Rsx is a metatherian RNA with Xist-like properties in X-chromosome inactivation. Nature 2012, 487, 254–258. [Google Scholar] [CrossRef]
- Singh, D.; Sun, D.; King, A.G.; Alquezar-Planas, D.E.; Johnson, R.N.; Alvarez-Ponce, D.; Yi, S.V. Koala methylomes reveal divergent and conserved DNA methylation signatures of X chromosome regulation. Proc. R. Soc. B Boil. Sci. 2021, 288, 20202244. [Google Scholar] [CrossRef]
- Wang, X.; Douglas, K.C.; VandeBerg, J.L.; Clark, A.G.; Samollow, P.B. Chromosome-wide profiling of X-chromosome inactivation and epigenetic states in fetal brain and placenta of the opossum, Monodelphis domestica. Genome Res. 2013, 24, 70–83. [Google Scholar] [CrossRef]
- Kay, G.F.; Barton, S.C.; Surani, A.; Rastan, S. Imprinting and X chromosome counting mechanisms determine Xist expression in early mouse development. Cell 1994, 77, 639–650. [Google Scholar] [CrossRef]
- Mullins, L.J.; Veres, G.; Caskey, C.T.; Chapman, V. Differential methylation of the ornithine carbamoyl transferase gene on active and inactive mouse X chromosomes. Mol. Cell. Biol. 1987, 7, 3916–3922. [Google Scholar] [CrossRef] [PubMed]
- Sanford, J.P.; Clark, H.J.; Chapman, V.M.; Rossant, J. Differences in DNA methylation during oogenesis and spermatogenesis and their persistence during early embryogenesis in the mouse. Genes Dev. 1987, 1, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Monk, M.; Boubelik, M.; Lehnert, S. Temporal and regional changes in DNA methylation in the embryonic, extraembryonic and germ cell lineages during mouse embryo development. Development 1987, 99, 371–382. [Google Scholar] [CrossRef]
- Krumlauf, R.; Chapman, V.M.; Hammer, R.E.; Brinster, R.; Tilghman, S.M. Differential expression of α-fetoprotein genes on the inactive X chromosome in extraembryonic and somatic tissues of a transgenic mouse line. Nature 1986, 319, 224–226. [Google Scholar] [CrossRef]
- Grant, S.G.; Chapman, V.M. Mechanisms of X-chromosome Regulation. Annu. Rev. Genet. 1988, 22, 199–233. [Google Scholar] [CrossRef]
- Lifschytz, E.; Lindsley, D.L. The Role of X-Chromosome Inactivation during Spermatogenesis. Proc. Natl. Acad. Sci. USA 1972, 69, 182–186. [Google Scholar] [CrossRef]
- Cloutier, J.M.; Turner, J.M. Meiotic sex chromosome inactivation. Curr. Biol. 2010, 20, R962–R963. [Google Scholar] [CrossRef]
- Cooper, D.W. Directed Genetic Change Model for X Chromosome Inactivation in Eutherian Mammals. Nature 1971, 230, 292–294. [Google Scholar] [CrossRef]
- Kalantry, S.; Purushothaman, S.; Bowen, R.B.; Starmer, J.; Magnuson, T. Evidence of Xist RNA-independent initiation of mouse imprinted X-chromosome inactivation. Nature 2009, 460, 647–651. [Google Scholar] [CrossRef]
- Okamoto, I.; Arnaud, D.; Le Baccon, P.; Otte, A.P.; Disteche, C.M.; Avner, P.; Heard, E. Evidence for de novo imprinted X-chromosome inactivation independent of meiotic inactivation in mice. Nature 2005, 438, 369–373. [Google Scholar] [CrossRef]
- Patrat, C.; Okamoto, I.; Diabangouaya, P.; Vialon, V.; Le Baccon, P.; Chow, J.; Heard, E. Dynamic changes in paternal X-chromosome activity during imprinted X-chromosome inactivation in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 5198–5203. [Google Scholar] [CrossRef]
- Duret, L.; Chureau, C.; Samain, S.; Weissenbach, J.; Avner, P. The Xist RNA Gene Evolved in Eutherians by Pseudogenization of a Protein-Coding Gene. Science 2006, 312, 1653–1655. [Google Scholar] [CrossRef]
- Hore, T.A.; Koina, A.; Wakefield, M.J.; Graves, J.A.M. The region homologous to the X-chromosome inactivation centre has been disrupted in marsupial and monotreme mammals. Chromosom. Res. 2007, 15, 147–161. [Google Scholar] [CrossRef]
- Davidow, L.S.; Breen, M.; Duke, S.E.; Samollow, P.B.; McCarrey, J.R.; Lee, J.T. The search for a marsupial XIC reveals a break with vertebrate synteny. Chromosom. Res. 2007, 15, 137–146. [Google Scholar] [CrossRef]
- Shevchenko, A.I.; Zakharova, I.S.; Elisaphenko, E.A.; Kolesnikov, N.N.; Whitehead, S.; Bird, C.; Ross, M.; Weidman, J.R.; Jirtle, R.L.; Karamysheva, T.V.; et al. Genes flanking Xist in mouse and human are separated on the X chromosome in American marsupials. Chromosom. Res. 2007, 15, 127–136. [Google Scholar] [CrossRef]
- Mahadevaiah, S.K.; Royo, H.; VandeBerg, J.L.; McCarrey, J.R.; Mackay, S.; Turner, J.M. Key Features of the X Inactivation Process Are Conserved between Marsupials and Eutherians. Curr. Biol. 2009, 19, 1478–1484. [Google Scholar] [CrossRef]
- Lyon, M.F.; Rastan, S. Parental source of chromosome imprinting and its relevance for X chromosome inactivation. Differentiation 1984, 26, 63–67. [Google Scholar] [CrossRef]
- Tada, T.; Obata, Y.; Tada, M.; Goto, Y.; Nakatsuji, N.; Tan, S.; Kono, T.; Takagi, N. Imprint switching for non-random X-chromosome inactivation during mouse oocyte growth. Development 2000, 127, 3101–3105. [Google Scholar] [CrossRef]
- Goto, Y.; Takagi, N. Tetraploid embryos rescue embryonic lethality caused by an additional maternally inherited X chromosome in the mouse. Development 1998, 125, 3353–3363. [Google Scholar] [CrossRef]
- Tada, T.; Takagi, N.; Adler, I.-D. Parental imprinting on the mouse X chromosome: Effects on the early development of X0, XXY and XXX embryos. Genet. Res. 1993, 62, 139–148. [Google Scholar] [CrossRef]
- Shao, C.; Takagi, N. An extra maternally derived X chromosome is deleterious to early mouse development. Development 1990, 110, 969–975. [Google Scholar] [CrossRef]
- Kaufman, M.H.; Guc-Cubrilo, M.; Lyon, M.F.; Kaufman, M.G.-C.M.H. X chromosome inactivation in diploid parthenogenetic mouse embryos. Nature 1978, 271, 547–549. [Google Scholar] [CrossRef] [PubMed]
- Rastan, S.; Kaufman, M.H.; Handyside, A.H.; Lyon, M.F. X-chromosome inactivation in extra-embryonic membranes of diploid parthenogenetic mouse embryos demonstrated by differential staining. Nature 1980, 288, 172–173. [Google Scholar] [CrossRef]
- Oikawa, M.; Inoue, K.; Shiura, H.; Matoba, S.; Kamimura, S.; Hirose, M.; Mekada, K.; Yoshiki, A.; Tanaka, S.; Abe, K.; et al. Understanding the X chromosome inactivation cycle in mice. Epigenetics 2014, 9, 204–211. [Google Scholar] [CrossRef]
- Hanna, C.W.; Kelsey, G. Features and mechanisms of canonical and noncanonical genomic imprinting. Genes Dev. 2021, 35, 821–834. [Google Scholar] [CrossRef]
- Chiba, H.; Hirasawa, R.; Kaneda, M.; Amakawa, Y.; Li, E.; Sado, T.; Sasaki, H. De novoDNA methylation independent establishment of maternal imprint on X chromosome in mouse oocytes. Genes 2008, 46, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Jiang, L.; Lu, F.; Suzuki, T.; Zhang, Y. Maternal H3K27me3 controls DNA methylation-independent imprinting. Nature 2017, 547, 419–424. [Google Scholar] [CrossRef]
- Inoue, A.; Jiang, L.; Lu, F.; Zhang, Y. Genomic imprinting of Xist by maternal H3K27me3. Genes Dev. 2017, 31, 1927–1932. [Google Scholar] [CrossRef]
- Collombet, S.; Ranisavljevic, N.; Nagano, T.; Varnai, C.; Shisode, T.; Leung, W.; Piolot, T.; Galupa, R.; Borensztein, M.; Servant, N.; et al. Parental-to-Embryo Switch of Chromosome Organization in Early Embryogenesis. Obstet. Gynecol. Surv. 2020, 75, 414–415. [Google Scholar] [CrossRef]
- Fukuda, A.; Tomikawa, J.; Miura, T.; Hata, K.; Nakabayashi, K.; Eggan, K.; Akutsu, H.; Umezawa, A. The role of maternal-specific H3K9me3 modification in establishing imprinted X-chromosome inactivation and embryogenesis in mice. Nat. Commun. 2014, 5, 5464. [Google Scholar] [CrossRef]
- Furlan, G.; Hernandez, N.G.; Huret, C.; Galupa, R.; van Bemmel, J.G.; Romito, A.; Heard, E.; Morey, C.; Rougeulle, C. The Ftx Noncoding Locus Controls X Chromosome Inactivation Independently of Its RNA Products. Mol. Cell 2018, 70, 462–472.e8. [Google Scholar] [CrossRef]
- Tian, D.; Sun, S.; Lee, J.T. The Long Noncoding RNA, Jpx, Is a Molecular Switch for X Chromosome Inactivation. Cell 2010, 143, 390–403. [Google Scholar] [CrossRef]
- Lee, J.T.; Lu, N. Targeted Mutagenesis of Tsix Leads to Nonrandom X Inactivation. Cell 1999, 99, 47–57. [Google Scholar] [CrossRef]
- Lee, J.T. Disruption of Imprinted X Inactivation by Parent-of-Origin Effects at Tsix. Cell 2000, 103, 17–27. [Google Scholar] [CrossRef]
- Sado, T.; Wang, Z.; Sasaki, H.; Li, E. Regulation of imprinted X-chromosome inactivation in mice by Tsix. Development 2001, 128, 1275–1286. [Google Scholar] [CrossRef]
- Soma, M.; Fujihara, Y.; Okabe, M.; Ishino, F.; Kobayashi, S. Ftx is dispensable for imprinted X-chromosome inactivation in preimplantation mouse embryos. Sci. Rep. 2014, 4, 5181. [Google Scholar] [CrossRef] [PubMed]
- Mahadevaiah, S.K.; Sangrithi, M.N.; Hirota, T.; Turner, J.M.A. A single-cell transcriptome atlas of marsupial embryogenesis and X inactivation. Nature 2020, 586, 612–617. [Google Scholar] [CrossRef]
- McLay, D.W.; Clarke, H.J. Remodelling the paternal chromatin at fertilization in mammals. Reproduction 2003, 125, 625. [Google Scholar] [CrossRef]
- Lee, J.T.; Bartolomei, M.S. X-Inactivation, Imprinting, and Long Noncoding RNAs in Health and Disease. Cell 2013, 152, 1308–1323. [Google Scholar] [CrossRef] [PubMed]
- Namekawa, S.H.; VandeBerg, J.L.; McCarrey, J.R.; Lee, J.T. Sex chromosome silencing in the marsupial male germ line. Proc. Natl. Acad. Sci. USA 2007, 104, 9730–9735. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.W.; Chandra, H.S. Inactivation System of the Mammalian X Chromosome. Proc. Natl. Acad. Sci. USA 1973, 70, 195–199. [Google Scholar] [CrossRef]
- Berg, I.M.V.D.; Galjaard, R.J.; Laven, J.S.E.; van Doorninck, J.H. XCI in preimplantation mouse and human embryos: First there is remodelling…. Qual. Life Res. 2011, 130, 203–215. [Google Scholar] [CrossRef]
- Sado, T. What makes the maternal X chromosome resistant to undergoing imprinted X inactivation? Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160365. [Google Scholar] [CrossRef]
- Sado, T.; Sakaguchi, T. Species-specific differences in X chromosome inactivation in mammals. Reproduction 2013, 146, R131–R139. [Google Scholar] [CrossRef]
- Mutzel, V.; Okamoto, I.; Dunkel, I.; Saitou, M.; Giorgetti, L.; Heard, E.; Schulz, E.G. A symmetric toggle switch explains the onset of random X inactivation in different mammals. Nat. Struct. Mol. Biol. 2019, 26, 350–360. [Google Scholar] [CrossRef]
- Marahrens, Y.; Panning, B.; Dausman, J.; Strauss, W.; Jaenisch, R. Xist-deficient mice are defective in dosage compensation but not spermatogenesis. Genes Dev. 1997, 11, 156–166. [Google Scholar] [CrossRef]
- Petropoulos, S.; Edsgärd, D.; Reinius, B.; Deng, Q.; Panula, S.P.; Codeluppi, S.; Reyes, A.P.; Linnarsson, S.; Sandberg, R.; Lanner, F. Single-Cell RNA-Seq Reveals Lineage and X Chromosome Dynamics in Human Preimplantation Embryos. Cell 2016, 165, 1012–1026. [Google Scholar] [CrossRef]
- Vallot, C.; Patrat, C.; Collier, A.; Huret, C.; Casanova, M.; Ali, T.M.L.; Tosolini, M.; Frydman, N.A.; Heard, E.; Rugg-Gunn, P.; et al. XACT Noncoding RNA Competes with XIST in the Control of X Chromosome Activity during Human Early Development. Cell Stem Cell 2017, 20, 102–111. [Google Scholar] [CrossRef]
- Mutzel, V.; Schulz, E.G. Dosage Sensing, Threshold Responses, and Epigenetic Memory: A Systems Biology Perspective on Random X-Chromosome Inactivation. BioEssays 2020, 42, e1900163. [Google Scholar] [CrossRef]
- Enervald, E.; Powell, L.M.; Boteva, L.; Foti, R.; Ruiz, N.B.; Kibar, G.; Piszczek, A.; Cavaleri, F.; Vingron, M.; Cerase, A.; et al. RIF1 and KAP1 differentially regulate the choice of inactive versus active X chromosomes. EMBO J. 2021, 40, e105862. [Google Scholar] [CrossRef] [PubMed]
- Makhlouf, M.; Ouimette, J.-F.; Oldfield, A.; Navarro, P.; Neuillet, D.; Rougeulle, C. A prominent and conserved role for YY1 in Xist transcriptional activation. Nat. Commun. 2014, 5, 4878. [Google Scholar] [CrossRef]
- Sun, B.; Deaton, A.; Lee, J.T. A Transient Heterochromatic State in Xist Preempts X Inactivation Choice without RNA Stabilization. Mol. Cell 2006, 21, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Sado, T.; Hoki, Y.; Sasaki, H. Tsix Silences Xist through Modification of Chromatin Structure. Dev. Cell 2005, 9, 159–165. [Google Scholar] [CrossRef]
- Navarro, P.; Page, D.R.; Avner, P.; Rougeulle, C. Tsix-mediated epigenetic switch of a CTCF-flanked region of the Xist promoter determines the Xist transcription program. Genes Dev. 2006, 20, 2787–2792. [Google Scholar] [CrossRef]
- Debrand, E.; Chureau, C.; Arnaud, D.; Avner, P.; Heard, E. Functional Analysis of the DXPas34 Locus, a 3′ Regulator of Xist Expression. Mol. Cell. Biol. 1999, 19, 8513–8525. [Google Scholar] [CrossRef]
- Yin, H.; Wei, C.; Lee, J.T. Revisiting the consequences of deleting the X inactivation center. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Loda, A.; Brandsma, J.H.; Vassilev, I.; Servant, N.; Loos, F.; Amirnasr, A.; Splinter, E.; Barillot, E.; Poot, R.A.; Heard, E.; et al. Genetic and epigenetic features direct differential efficiency of Xist-mediated silencing at X-chromosomal and autosomal locations. Nat. Commun. 2017, 8, 690. [Google Scholar] [CrossRef]
- Sado, T.; Hoki, Y.; Sasaki, H. Tsix defective in splicing is competent to establish Xist silencing. Development 2006, 133, 4925–4931. [Google Scholar] [CrossRef]
- Shibata, S.; Lee, J.T. Tsix Transcription- versus RNA-Based Mechanisms in Xist Repression and Epigenetic Choice. Curr. Biol. 2004, 14, 1747–1754. [Google Scholar] [CrossRef]
- Luikenhuis, S.; Wutz, A.; Jaenisch, R. Antisense Transcription through the Xist Locus Mediates Tsix Function in Embryonic Stem Cells. Mol. Cell. Biol. 2001, 21, 8512–8520. [Google Scholar] [CrossRef] [PubMed]
- Navarro, P.; Pichard, S.; Ciaudo, C.; Avner, P.; Rougeulle, C. Tsix transcription across the Xist gene alters chromatin conformation without affecting Xist transcription: Implications for X-chromosome inactivation. Genes Dev. 2005, 19, 1474–1484. [Google Scholar] [CrossRef] [PubMed]
- Ohhata, T.; Hoki, Y.; Sasaki, H.; Sado, T. Crucial role of antisense transcription across the Xist promoter in Tsix-mediated Xist chromatin modification. Development 2008, 135, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Stavropoulos, N.; Lu, N.; Lee, J.T. A functional role for Tsix transcription in blocking Xist RNA accumulation but not in X-chromosome choice. Proc. Natl. Acad. Sci. USA 2001, 98, 10232–10237. [Google Scholar] [CrossRef]
- Migeon, B.R.; Lee, C.H.; Chowdhury, A.K.; Carpenter, H. Species Differences in TSIX/Tsix Reveal the Roles of These Genes in X-Chromosome Inactivation. Am. J. Hum. Genet. 2002, 71, 286–293. [Google Scholar] [CrossRef]
- Migeon, B.R.; Chowdhury, A.K.; Dunston, J.A.; McIntosh, I. Identification of TSIX, Encoding an RNA Antisense to Human XIST, Reveals Differences from its Murine Counterpart: Implications for X Inactivation. Am. J. Hum. Genet. 2001, 69, 951–960. [Google Scholar] [CrossRef]
- Galupa, R.; Heard, E. X-Chromosome Inactivation: A Crossroads Between Chromosome Architecture and Gene Regulation. Annu. Rev. Genet. 2018, 52, 535–566. [Google Scholar] [CrossRef]
- Ogawa, Y.; Lee, J.T. Xite, X-Inactivation Intergenic Transcription Elements that Regulate the Probability of Choice. Mol. Cell 2003, 11, 731–743. [Google Scholar] [CrossRef]
- Galupa, R.; Nora, E.P.; Worsley-Hunt, R.; Picard, C.; Gard, C.; van Bemmel, J.G.; Servant, N.; Zhan, Y.; El Marjou, F.; Johanneau, C.; et al. A Conserved Noncoding Locus Regulates Random Monoallelic Xist Expression across a Topological Boundary. Mol. Cell 2020, 77, 352–367.e8. [Google Scholar] [CrossRef]
- Tena, J.J.; Santos-Pereira, J.M. Topologically Associating Domains and Regulatory Landscapes in Development, Evolution and Disease. Front. Cell Dev. Biol. 2021, 9, 1817. [Google Scholar] [CrossRef]
- Nora, E.; Lajoie, B.R.; Schulz, E.G.; Giorgetti, L.; Okamoto, I.; Servant, N.; Piolot, T.; Van Berkum, N.L.; Meisig, J.; Sedat, J.; et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 2012, 485, 381–385. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef]
- Van Bemmel, J.G.; Galupa, R.; Gard, C.; Servant, N.; Picard, C.; Davies, J.; Szempruch, A.J.; Zhan, Y.; Żylicz, J.J.; Nora, E.P.; et al. The bipartite TAD organization of the X-inactivation center ensures opposing developmental regulation of Tsix and Xist. Nat. Genet. 2019, 51, 1024–1034. [Google Scholar] [CrossRef]
- Gjaltema, R.A.; Schwämmle, T.; Kautz, P.; Robson, M.; Schöpflin, R.; Lustig, L.R.; Brandenburg, L.; Dunkel, I.; Vechiatto, C.; Ntini, E.; et al. Distal and proximal cis-regulatory elements sense X chromosome dosage and developmental state at the Xist locus. Mol. Cell 2021, 82, 190–208.e17. [Google Scholar] [CrossRef]
- Rosspopoff, O.; Huret, C.; Collier, A.J.; Casanova, M.; Rugg-Gunn, P.J.; Ouimette, J.-F.; Rougeulle, C. Mechanistic diversifica-tion of XIST regulatory network in mammals. BioRxiv 2019, 689430. [Google Scholar]
- Sun, S.; Del Rosario, B.C.; Szanto, A.; Ogawa, Y.; Jeon, Y.; Lee, J.T. Jpx RNA Activates Xist by Evicting CTCF. Cell 2013, 153, 1537–1551. [Google Scholar] [CrossRef]
- Nesterova, T.B.; Johnston, C.M.; Appanah, R.; Newall, A.E.; Godwin, J.; Alexiou, M.; Brockdorff, N. Skewing X chromosome choice by modulating sense transcription across theXistlocus. Genes Dev. 2003, 17, 2177–2190. [Google Scholar] [CrossRef]
- Sarkar, M.K.; Gayen, S.; Kumar, S.; Maclary, E.; Buttigieg, E.; Hinten, M.; Kumari, A.; Harris, C.; Sado, T.; Kalantry, S. An Xist-activating antisense RNA required for X-chromosome inactivation. Nat. Commun. 2015, 6, 8564. [Google Scholar] [CrossRef]
- Dossin, F.; Pinheiro, I.; Zylicz, J.; Roensch, J.; Collombet, S.; Le Saux, A.; Chelmicki, T.; Attia, M.; Kapoor, V.; Zhan, Y.; et al. SPEN integrates transcriptional and epigenetic control of X-inactivation. Nature 2020, 578, 455–460. [Google Scholar] [CrossRef]
- Robert-Finestra, T.; Tan, B.F.; Mira-Bontenbal, H.; Timmers, E.; Gontan, C.; Merzouk, S.; Giaimo, B.D.; Dossin, F.; van Ijcken, W.F.J.; Martens, J.W.M.; et al. SPEN is required for Xist upregulation during initiation of X chromosome inactivation. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Schulz, E.G.; Nora, E.P.; Heard, E. Rnf12—A Jack of All Trades in X Inactivation? PLoS Genet. 2011, 7, e1002002. [Google Scholar] [CrossRef]
- Monkhorst, K.; Jonkers, I.; Rentmeester, E.; Grosveld, F.; Gribnau, J. X Inactivation Counting and Choice Is a Stochastic Process: Evidence for Involvement of an X-Linked Activator. Cell 2008, 132, 410–421. [Google Scholar] [CrossRef]
- Jonkers, I.; Barakat, T.S.; Achame, E.M.; Monkhorst, K.; Kenter, A.; Rentmeester, E.; Grosveld, F.; Grootegoed, J.A.; Gribnau, J. RNF12 Is an X-Encoded Dose-Dependent Activator of X Chromosome Inactivation. Cell 2009, 139, 999–1011. [Google Scholar] [CrossRef]
- Gontan, C.; Achame, E.M.; Demmers, J.; Barakat, S.; Rentmeester, E.; van Ijcken, W.; Grootegoed, J.A.; Gribnau, J. RNF12 initiates X-chromosome inactivation by targeting REX1 for degradation. Nature 2012, 485, 386–390. [Google Scholar] [CrossRef]
- Barakat, T.S.; Gunhanlar, N.; Pardo, C.G.; Achame, E.M.; Ghazvini, M.; Boers, R.; Kenter, A.; Rentmeester, E.; Grootegoed, J.A.; Gribnau, J. RNF12 Activates Xist and Is Essential for X Chromosome Inactivation. PLoS Genet. 2011, 7, e1002001. [Google Scholar] [CrossRef]
- Shin, J.; Bossenz, M.; Chung, Y.; Ma, H.; Byron, M.; Taniguchi-Ishigaki, N.; Zhu, X.; Jiao, B.; Hall, L.L.; Green, M.R.; et al. Maternal Rnf12/RLIM is required for imprinted X-chromosome inactivation in mice. Nature 2010, 467, 977–981. [Google Scholar] [CrossRef]
- Shin, J.; Wallingford, M.C.; Gallant, J.; Marcho, C.; Jiao, B.; Byron, M.; Bossenz, M.; Lawrence, J.B.; Jones, S.N.; Mager, J.; et al. RLIM is dispensable for X-chromosome inactivation in the mouse embryonic epiblast. Nature 2014, 511, 86–89. [Google Scholar] [CrossRef]
- Hayashi, S.; Lewis, P.; Pevny, L.; McMahon, A.P. Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Mech. Dev. 2002, 119, S97–S101. [Google Scholar] [CrossRef]
- Zhou, Q.; Wang, T.; Leng, L.; Zheng, W.; Huang, J.; Fang, F.; Yang, L.; Chen, F.; Lin, G.; Wang, W.J.; et al. Single-cell RNA-seq reveals distinct dynamic behavior of sex chromosomes during early human embryogenesis. Mol. Reprod. Dev. 2019, 86, 871–882. [Google Scholar] [CrossRef]
- Heard, E.; Rougeulle, C. Digging into X chromosome inactivation. Science 2021, 374, 942–943. [Google Scholar] [CrossRef]
- Mekhoubad, S.; Bock, C.; de Boer, A.S.; Kiskinis, E.; Meissner, A.; Eggan, K. Erosion of Dosage Compensation Impacts Human iPSC Disease Modeling. Cell Stem Cell 2012, 10, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Sahakyan, A.; Kim, R.; Chronis, C.; Sabri, S.; Bonora, G.; Theunissen, T.; Kuoy, E.; Langerman, J.; Clark, A.T.; Jaenisch, R.; et al. Human Naive Pluripotent Stem Cells Model X Chromosome Dampening and X Inactivation. Cell Stem Cell 2017, 20, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Saiba, R.; Arava, M.; Gayen, S. Dosage compensation in human pre-implantation embryos: X-chromosome inactivation or dampening? EMBO Rep. 2018, 19, e46294. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Rv, P.; Gayen, S. Dampened X-chromosomes in human pluripotent stem cells: Dampening or erasure of X-upregulation? Chromosoma 2019, 129, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Spaziano, A.; Cantone, I. X-chromosome reactivation: A concise review. Biochem. Soc. Trans. 2021, 49, 2797–2805. [Google Scholar] [CrossRef]
- Mandal, S.; Chandel, D.; Kaur, H.; Majumdar, S.; Arava, M.; Gayen, S. Single-Cell Analysis Reveals Partial Reactivation of X Chromosome instead of Chromosome-wide Dampening in Naive Human Pluripotent Stem Cells. Stem Cell Rep. 2020, 14, 745–754. [Google Scholar] [CrossRef]
- Casanova, M.; Moscatelli, M.; Chauvière, L.É.; Huret, C.; Samson, J.; Ali, T.M.L.; Rosspopoff, O.; Rougeulle, C. A primate-specific retroviral enhancer wires the XACT lncRNA into the core pluripotency network in humans. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Brown, C.; Robinson, W. The causes and consequences of random and non-random X chromosome inactivation in humans. Clin. Genet. 2000, 58, 353–363. [Google Scholar] [CrossRef]
- Belmont, J.W. Genetic control of X inactivation and processes leading to X-inactivation skewing. Am. J. Hum. Genet. 1996, 58, 1101–1108. [Google Scholar]
- Puck, J.M.; Willard, H.F. X Inactivation in Females with X-Linked Disease. N. Engl. J. Med. 1998, 338, 325–328. [Google Scholar] [CrossRef]
- Ørstavik, K.H. X chromosome inactivation in clinical practice. Qual. Life Res. 2009, 126, 363–373. [Google Scholar] [CrossRef]
- Wu, H.; Luo, J.; Yu, H.; Rattner, A.; Mo, A.; Wang, Y.; Smallwood, P.M.; Erlanger, B.; Wheelan, S.J.; Nathans, J. Cellular Resolution Maps of X Chromosome Inactivation: Implications for Neural Development, Function, and Disease. Neuron 2014, 81, 103–119. [Google Scholar] [CrossRef]
- Gale, R.E.; Wheadon, H.; Linch, D.C. X-chromosome inactivation patterns using HPRT and PGK polymorphisms in haematologically normal and post-chemotherapy females. Br. J. Haematol. 1991, 79, 193–197. [Google Scholar] [CrossRef]
- Fey, M.F.; Peter, H.J.; Hinds, H.L.; Zimmermann, A.; Liechti-Gallati, S.; Gerber, H.; Studer, H.; Tobler, A. Clonal analysis of human tumors with M27 beta, a highly informative polymorphic X chromosomal probe. J. Clin. Investig. 1992, 89, 1438–1444. [Google Scholar] [CrossRef]
- Amos-Landgraf, J.; Cottle, A.; Plenge, R.M.; Friez, M.; Schwartz, C.E.; Longshore, J.; Willard, H.F. X Chromosome–Inactivation Patterns of 1,005 Phenotypically Unaffected Females. Am. J. Hum. Genet. 2006, 79, 493–499. [Google Scholar] [CrossRef]
- Migeon, B.R. Why females are mosaics, x-chromosome inactivation, and sex differences in disease. Gend. Med. 2007, 4, 97–105. [Google Scholar] [CrossRef]
- Bolduc, V.; Chagnon, P.; Provost, S.; Dubé, M.-P.; Belisle, C.; Gingras, M.; Mollica, L.; Busque, L. No evidence that skewing of X chromosome inactivation patterns is transmitted to offspring in humans. J. Clin. Investig. 2008, 118, 333–341. [Google Scholar] [CrossRef]
- Shvetsova, E.; BIOS Consortium; Sofronova, A.; Monajemi, R.; Gagalova, K.; Draisma, H.; White, S.J.; Santen, G.W.E.; Lopes, S.M.C.D.S.; Heijmans, B.T.; et al. Skewed X-inactivation is common in the general female population. Eur. J. Hum. Genet. 2019, 27, 455–465. [Google Scholar] [CrossRef]
- Cattanach, B.M.; Papworth, D. Controlling elements in the mouse: V. Linkage tests with X-linked genes. Genet. Res. 1981, 38, 57–70. [Google Scholar] [CrossRef]
- Calaway, J.D.; Lenarcic, A.B.; Didion, J.P.; Wang, J.R.; Searle, J.B.; McMillan, L.; Valdar, W.; De Villena, F.P.-M. Genetic Architecture of Skewed X Inactivation in the Laboratory Mouse. PLoS Genet. 2013, 9, e1003853. [Google Scholar] [CrossRef]
- Chadwick, L.H.; Pertz, L.M.; Broman, K.; Bartolomei, M.S.; Willard, H.F. Genetic Control of X Chromosome Inactivation in Mice: Definition of the Xce Candidate Interval. Genetics 2006, 173, 2103–2110. [Google Scholar] [CrossRef] [PubMed]
- Simmler, M.-C.; Cattanach, B.M.; Rasberry, C.; Rougeulle, C.; Avner, P. Mapping the murine Xce locus with (CA)n repeats. Mamm. Genome 1993, 4, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsen, J.L.; Krapp, C.; Willard, H.F.; Bartolomei, M.S. Nonrandom X Chromosome Inactivation Is Influenced by Multiple Regions on the Murine X Chromosome. Genetics 2012, 192, 1095–1107. [Google Scholar] [CrossRef] [PubMed]
- Galupa, R.; Heard, E. X-chromosome inactivation: New insights into cis and trans regulation. Curr. Opin. Genet. Dev. 2015, 31, 57–66. [Google Scholar] [CrossRef]
- Sun, K.Y.; Oreper, D.; Schoenrock, S.A.; McMullan, R.; Giusti-Rodríguez, P.; Zhabotynsky, V.; Miller, D.R.; Tarantino, L.M.; de Villena, F.P.-M.; Valdar, W. Bayesian modeling of skewed X inactivation in genetically diverse mice identifies a novel Xce allele associated with copy number changes. BioRxiv 2021, 153, 1537–1551. [Google Scholar] [CrossRef]
- Peeters, S.B.; Yang, C.; Brown, C.J. Have humans lost control: The elusive X-controlling element. Semin. Cell Dev. Biol. 2016, 56, 71–77. [Google Scholar] [CrossRef]
- Morey, C.; Avner, P. Genetics and epigenetics of the X chromosome. Ann. N. Y. Acad. Sci. USA 2010, 1214, E18–E33. [Google Scholar] [CrossRef]
- Veyver, I.B.V.D. Skewed X Inactivation in X-Linked Disorders. Semin. Reprod. Med. 2001, 19, 183–192. [Google Scholar] [CrossRef]
- Schmidt, M.; Du Sart, D.; Kalitsis, P.; Fraser, N.; Leversha, M.; Voullaire, L.; Foster, D.; Davies, J.; Hills, L.; Petrovic, V.; et al. X chromosome inactivation in fibroblasts of mentally retarded female carriers of the fragile site Xq27.3: Application of the probe M27β to evaluate X inactivation status. Am. J. Med Genet. 1991, 38, 411–415. [Google Scholar] [CrossRef]
- Ham, A.L.; Kumar, A.; Deeter, R.; Schanen, N.C. Does Genotype Predict Phenotype in Rett Syndrome? J. Child Neurol. 2005, 20, 768–778. [Google Scholar] [CrossRef]
- Carrette, L.; Wang, C.-Y.; Wei, C.; Press, W.; Ma, W.; Kelleher, R.J.; Lee, J.T. A mixed modality approach towards Xi reactivation for Rett syndrome and other X-linked disorders. Proc. Natl. Acad. Sci. USA 2018, 115, E668–E675. [Google Scholar] [CrossRef]
- McMahon, A.; Monk, M. X-chromosome activity in female mouse embryos heterozygous forPgk-1and Searle’s translocation, T(X; 16) 16H. Genet. Res. 1983, 41, 69–83. [Google Scholar] [CrossRef]
- Viggiano, E.; Ergoli, M.; Picillo, E.; Politano, L. Determining the role of skewed X-chromosome inactivation in developing muscle symptoms in carriers of Duchenne muscular dystrophy. Qual. Life Res. 2016, 135, 685–698. [Google Scholar] [CrossRef]
- Mattei, M.-G.; Mattei, J.F.; Ayme, S.; Giraud, F. X-Autosome translocations: Cytogenetic characteristics and their consequences. Qual. Life Res. 1982, 61, 295–309. [Google Scholar] [CrossRef]
- Mattei, M.-G.; Mattei, J.F.; Vidal, I.; Giraud, F. Structural anomalies of the X chromosome and inactivation center. Qual. Life Res. 1981, 56, 401–408. [Google Scholar] [CrossRef]
- Smith, J.C. Lewis Wolpert (1929-2021). Development 2021, 148, dev199618. [Google Scholar] [CrossRef]
- Bainbridge, D. The X in Sex: How the X Chromosome Controls Our Lives; Harvard University Press: Cambridge, MA, USA, 2003. [Google Scholar]
- Gomez, M.R.; Engel, A.G.; Dewald, G.; Peterson, H.A. Failure of inactivation of Duchenne dystrophy X-chromosome in one of female identical twins. Neurology 1977, 27, 537. [Google Scholar] [CrossRef]
- Richards, C.S.; Watkins, S.C.; Hoffman, E.P.; Schneider, N.R.; Milsark, I.W.; Katz, K.S.; Cook, J.D.; Kunkel, L.M.; Cortada, J.M. Skewed X inactivation in a female MZ twin results in Duchenne muscular dystrophy. Am. J. Hum. Genet. 1990, 46, 672–681. [Google Scholar]
- Lupski, J.R.; Garcia, C.A.; Zoghbi, H.Y.; Hoffman, E.; Fenwick, R.G. Discordance of muscular dystrophy in monozygotic female twins: Evidence supporting asymmetric splitting of the inner cell mass in a manifesting carrier of Duchenne dystrophy. Am. J. Med Genet. 1991, 40, 354–364. [Google Scholar] [CrossRef]
- Talon, I.; Janiszewski, A.; Chappell, J.; Vanheer, L.; Pasque, V. Recent Advances in Understanding the Reversal of Gene Silencing During X Chromosome Reactivation. Front. Cell Dev. Biol. 2019, 7, 169. [Google Scholar] [CrossRef]
- Chaligné, R.; Heard, E. X-chromosome inactivation in development and cancer. FEBS Lett. 2014, 588, 2514–2522. [Google Scholar] [CrossRef]
- Panda, A.; Zylicz, J.J.; Pasque, V. New Insights Into X-Chromosome Reactivation during Reprogramming to Pluripotency. Cells 2020, 9, 2706. [Google Scholar] [CrossRef] [PubMed]
- Katz, D.M.; Bird, A.; Coenraads, M.; Gray, S.J.; Menon, D.U.; Philpot, B.D.; Tarquinio, D.C. Rett Syndrome: Crossing the Threshold to Clinical Translation. Trends Neurosci. 2016, 39, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Przanowski, P.; Wasko, U.; Zheng, Z.; Yu, J.; Sherman, R.; Zhu, L.J.; McConnell, M.J.; Tushir-Singh, J.; Green, M.R.; Bhatnagar, S. Pharmacological reactivation of inactive X-linked Mecp2 in cerebral cortical neurons of living mice. Proc. Natl. Acad. Sci. USA 2018, 115, 7991–7996. [Google Scholar] [CrossRef]
- Lee, H.-M.; Kuijer, M.B.; Blanes, N.R.; Clark, E.P.; Aita, M.; Arjona, L.G.; Kokot, A.; Sciaky, N.; Simon, J.M.; Bhatnagar, S.; et al. A small-molecule screen reveals novel modulators of MeCP2 and X-chromosome inactivation maintenance. J. Neurodev. Disord. 2020, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furlan, G.; Galupa, R. Mechanisms of Choice in X-Chromosome Inactivation. Cells 2022, 11, 535. https://doi.org/10.3390/cells11030535
Furlan G, Galupa R. Mechanisms of Choice in X-Chromosome Inactivation. Cells. 2022; 11(3):535. https://doi.org/10.3390/cells11030535
Chicago/Turabian StyleFurlan, Giulia, and Rafael Galupa. 2022. "Mechanisms of Choice in X-Chromosome Inactivation" Cells 11, no. 3: 535. https://doi.org/10.3390/cells11030535
APA StyleFurlan, G., & Galupa, R. (2022). Mechanisms of Choice in X-Chromosome Inactivation. Cells, 11(3), 535. https://doi.org/10.3390/cells11030535