
Interferon-γ Preferentially Promotes Necroptosis of Lung Epithelial Cells by Upregulating MLKL


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
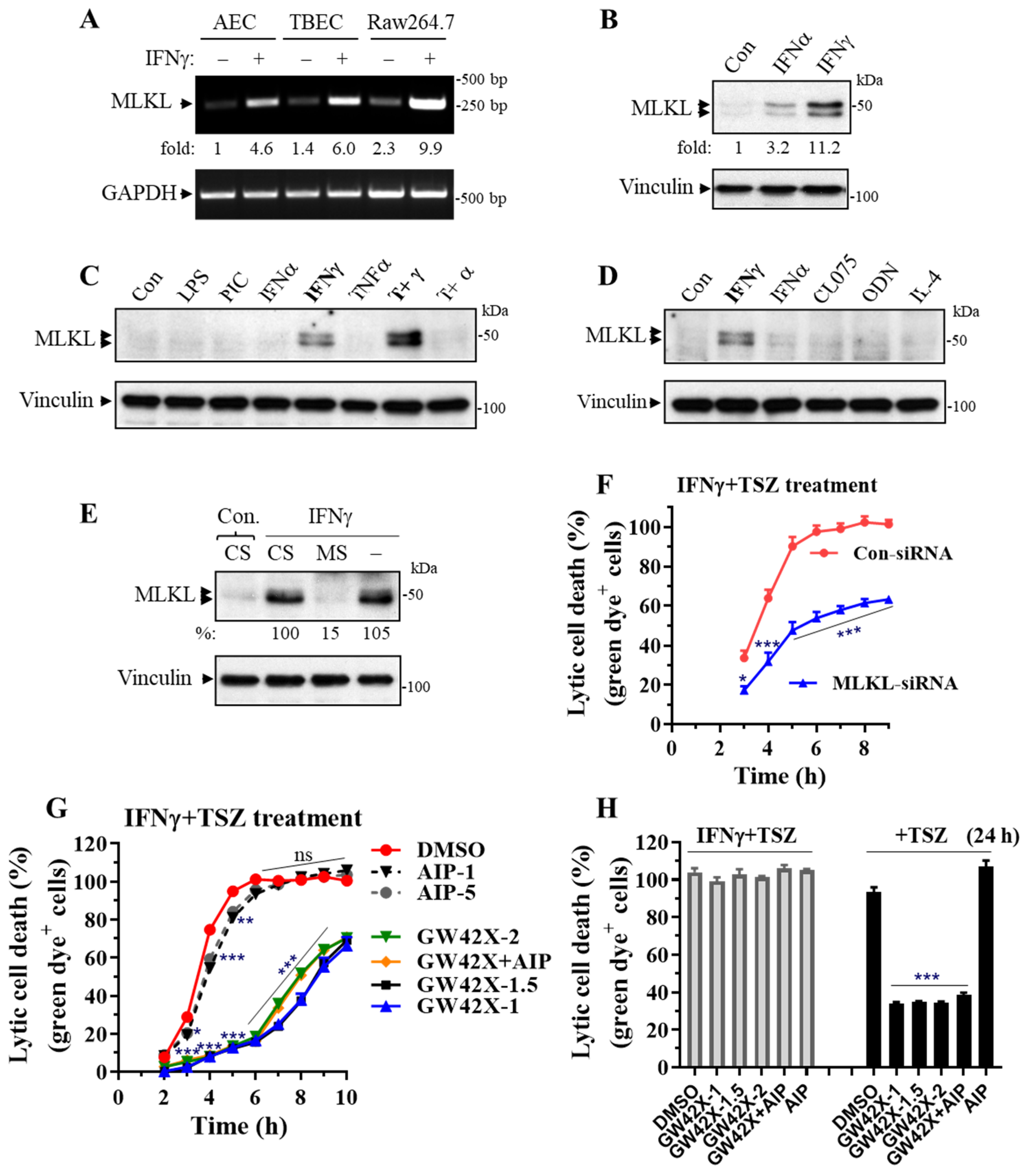
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies and Reagents
2.2. Cell Cultures
2.3. Cell Viability, Lactate Dehydrogenase Release and Real Time Necroptosis Assays
2.4. DNA and High-Mobility Group Protein B1 Release Assays
2.5. Real Time Measurement of ATP Release
2.6. Western Blot Analysis
2.7. RNA Isolation and Reverse Transcription (RT)-PCR
2.8. SiRNAs and siRNA Transfection
2.9. Statistical Analysis
3. Results
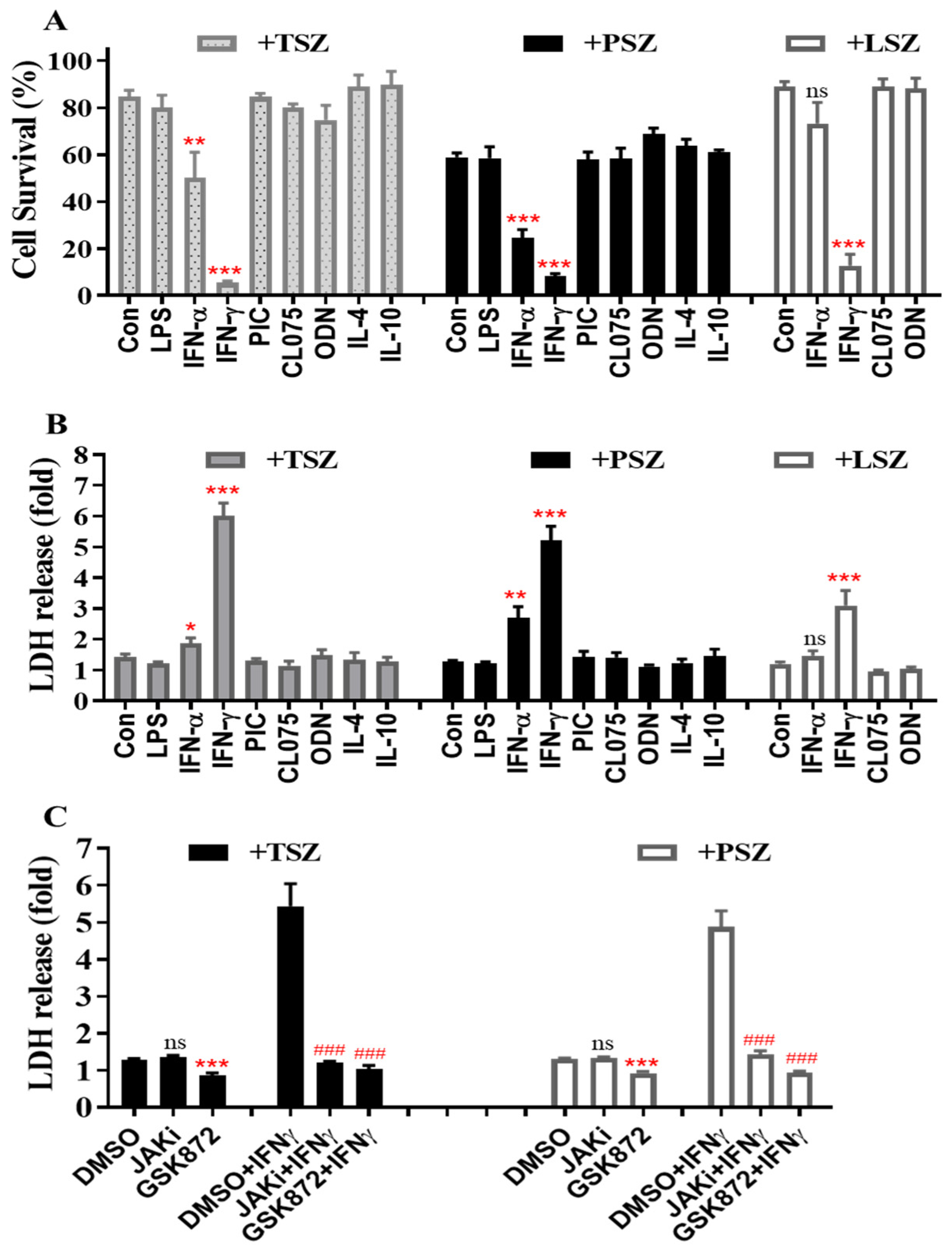
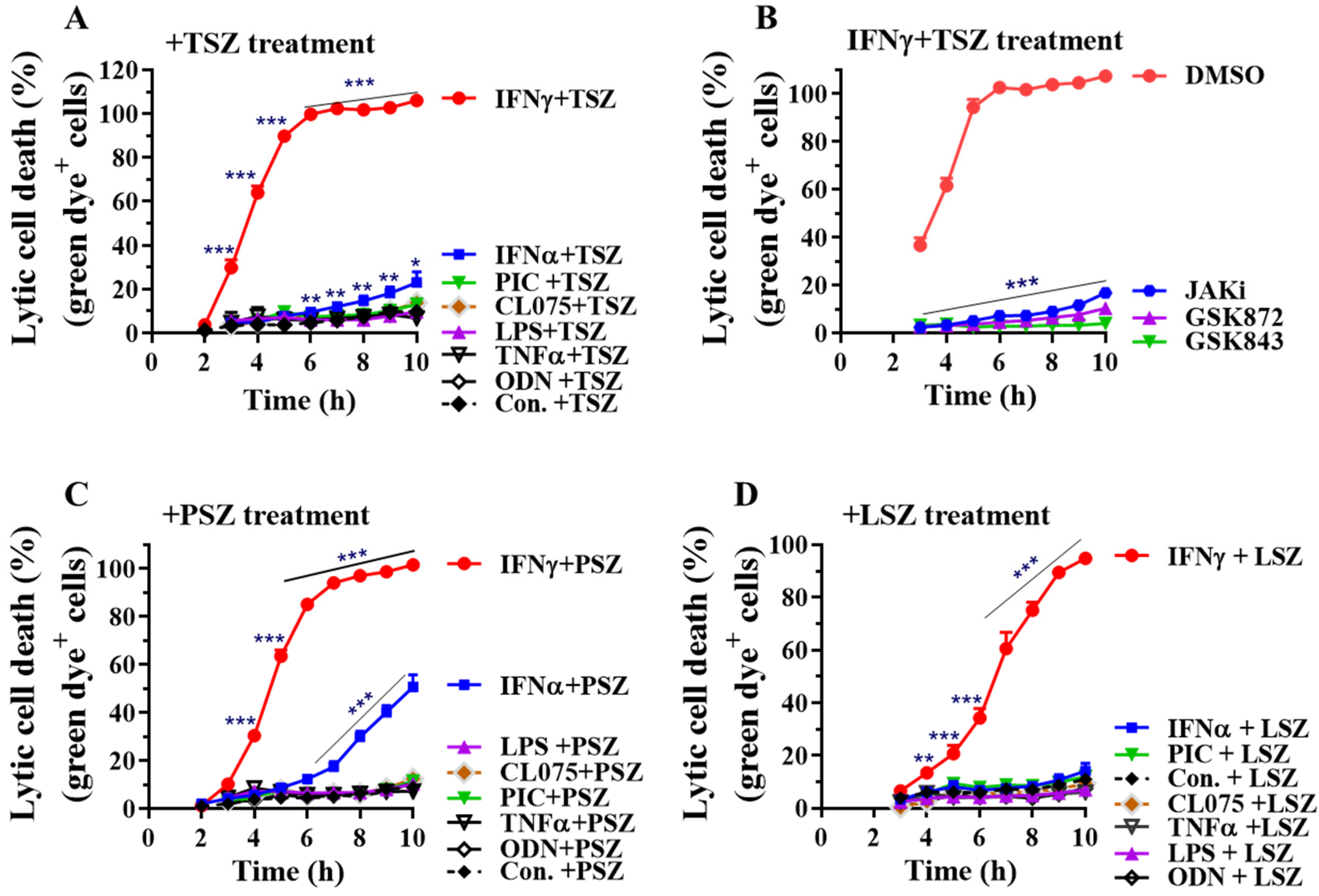
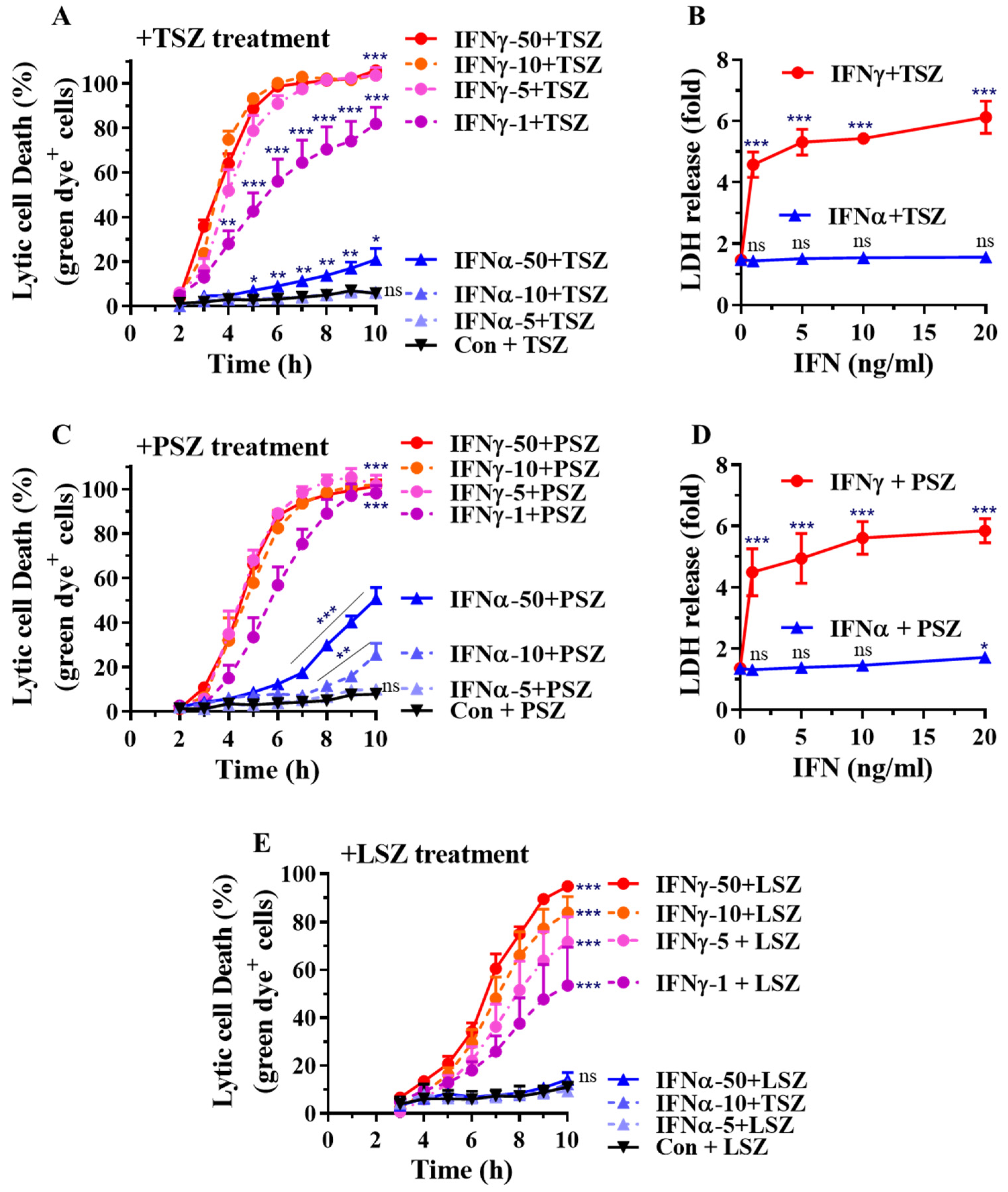
3.1. IFNγ among Inflammatory Mediators Preferentially Promotes Necroptosis in Primary Alveolar Epithelial Cells
3.2. IFNγ Preferentially Promotes Necroptosis in Primary Airway Epithelial Cells
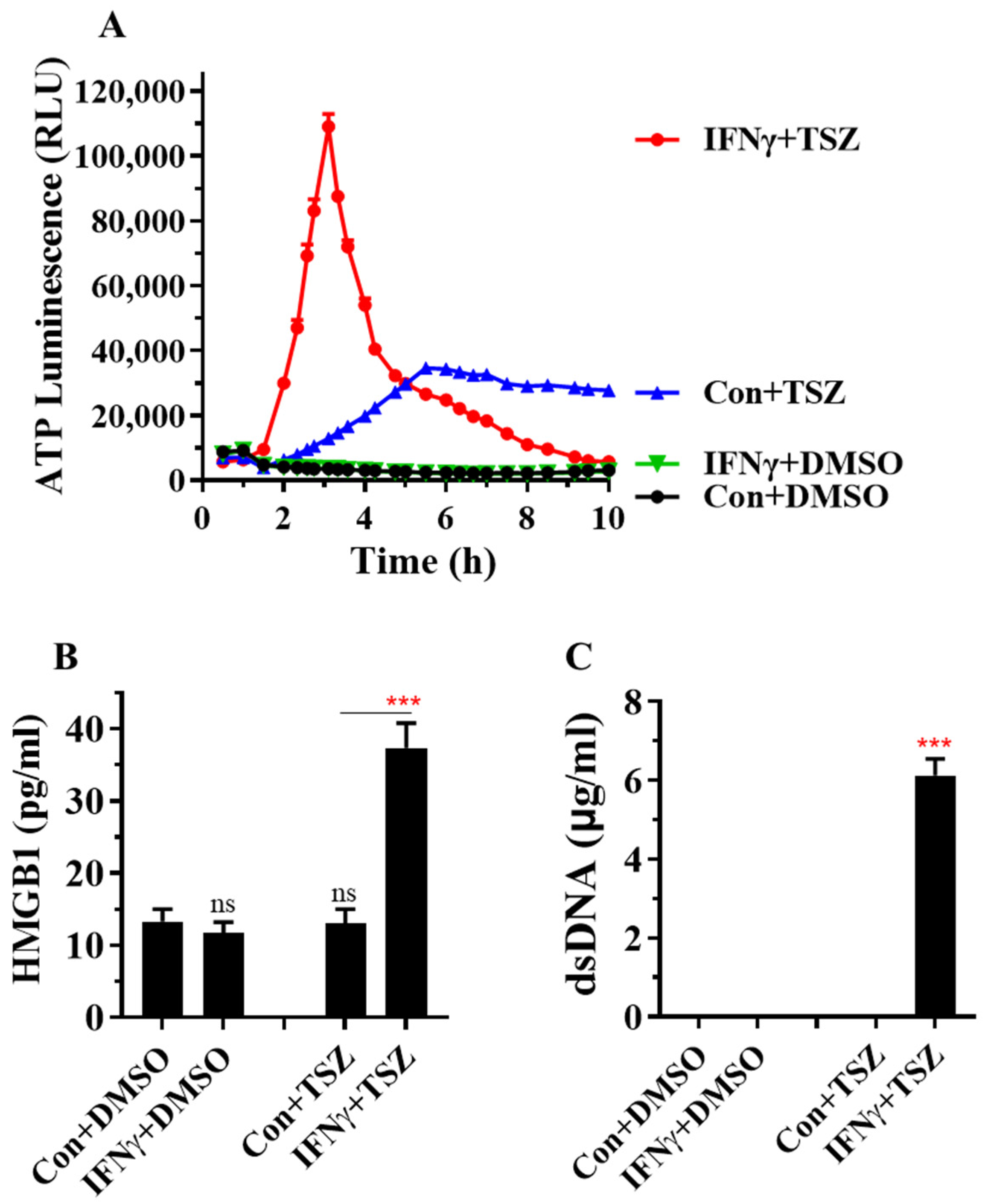
3.3. IFNγ Greatly Accelerates the Release of DAMPs from AECs
3.4. MLKL Is Predominantly Induced by IFNγ in Primary Lung Epithelial Cells and MLKL Silencing or Inhibition Largely Suppresses the IFNγ-Promoted Necroptosis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Manicone, A.M. Role of the pulmonary epithelium and inflammatory signals in acute lung injury. Expert Rev. Clin. Immunol. 2009, 5, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Fan, E.; Brodie, D.; Slutsky, A.S. Acute Respiratory Distress Syndrome: Advances in Diagnosis and Treatment. JAMA 2018, 319, 698–710. [Google Scholar] [CrossRef]
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology, Patterns of Care, and Mortality for Patients with Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 2016, 315, 788–800. [Google Scholar] [CrossRef]
- Li, L.; Huang, Q.; Wang, D.C.; Ingbar, D.H.; Wang, X. Acute lung injury in patients with COVID-19 infection. Clin. Transl. Med. 2020, 10, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Camporota, L.; Cronin, J.N.; Busana, M.; Gattinoni, L.; Formenti, F. Pathophysiology of coronavirus-19 disease acute lung injury. Curr. Opin. Crit. Care 2022, 28, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Peritore, A.F.; D’Amico, R.; Siracusa, R.; Cordaro, M.; Fusco, R.; Gugliandolo, E.; Genovese, T.; Crupi, R.; Di Paola, R.; Cuzzocrea, S.; et al. Management of Acute Lung Injury: Palmitoylethanolamide as a New Approach. Int. J. Mol. Sci. 2021, 22, 5533. [Google Scholar] [CrossRef]
- Faust, H.; Mangalmurti, N.S. Collateral damage: Necroptosis in the development of lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 318, L215–L225. [Google Scholar] [CrossRef] [PubMed]
- Mizumura, K.; Maruoka, S.; Gon, Y.; Choi, A.M.; Hashimoto, S. The role of necroptosis in pulmonary diseases. Respir. Investig. 2016, 54, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Fan, E.K.Y.; Fan, J. Regulation of alveolar macrophage death in acute lung inflammation. Respir. Res. 2018, 19, 50. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.E.; Price, D.R.; Ryter, S.W.; Choi, A.M.K. Necroptosis: A crucial pathogenic mediator of human disease. JCI Insight 2019, 4, e128834. [Google Scholar] [CrossRef]
- Kaczmarek, A.; Vandenabeele, P.; Krysko, D.V. Necroptosis: The release of damage-associated molecular patterns and its physiological relevance. Immunity 2013, 38, 209–223. [Google Scholar] [CrossRef] [Green Version]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Wallach, D.; Kang, T.B.; Dillon, C.P.; Green, D.R. Programmed necrosis in inflammation: Toward identification of the effector molecules. Science 2016, 352, aaf2154. [Google Scholar] [CrossRef]
- He, S.; Wang, X. RIP kinases as modulators of inflammation and immunity. Nat. Immunol. 2018, 19, 912–922. [Google Scholar] [CrossRef]
- Shashaty, M.G.S.; Reilly, J.P.; Faust, H.E.; Forker, C.M.; Ittner, C.A.G.; Zhang, P.X.; Hotz, M.J.; Fitzgerald, D.; Yang, W.; Anderson, B.J.; et al. Plasma receptor interacting protein kinase-3 levels are associated with acute respiratory distress syndrome in sepsis and trauma: A cohort study. Crit. Care 2019, 23, 235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, H.; Kinjo, T.; Arakaki, W.; Miyagi, K.; Tateyama, M.; Fujita, J. Serum levels of receptor-interacting protein kinase-3 in patients with COVID-19. Crit. Care 2020, 24, 484. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.C.; Schenck, E.J.; Siempos, I.I.; Cloonan, S.M.; Finkelsztein, E.J.; Pabon, M.A.; Oromendia, C.; Ballman, K.V.; Baron, R.M.; Fredenburgh, L.E.; et al. Circulating RIPK3 levels are associated with mortality and organ failure during critical illness. JCI Insight 2018, 3, e99692. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, T.; Li, H.; Liu, Q.; Zhang, Z.; Xie, W.; Feng, Y.; Socorburam, T.; Wu, G.; Xia, Z.; et al. Receptor Interacting Protein 3-Mediated Necroptosis Promotes Lipopolysaccharide-Induced Inflammation and Acute Respiratory Distress Syndrome in Mice. PLoS ONE 2016, 11, e0155723. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Wang, S.; Fu, R.; Zhou, M.; Zhang, T.; Pan, W.; Yang, N.; Huang, Y. RIP3 dependent NLRP3 inflammasome activation is implicated in acute lung injury in mice. J. Transl. Med. 2018, 16, 233. [Google Scholar] [CrossRef]
- Lin, B.; Jin, Z.; Chen, X.; Zhao, L.; Weng, C.; Chen, B.; Tang, Y.; Lin, L. Necrostatin1 protects mice from acute lung injury by suppressing necroptosis and reactive oxygen species. Mol. Med. Rep. 2020, 21, 2171–2181. [Google Scholar]
- Siempos, I.I.; Ma, K.C.; Imamura, M.; Baron, R.M.; Fredenburgh, L.E.; Huh, J.W.; Moon, J.S.; Finkelsztein, E.J.; Jones, D.S.; Lizardi, M.T.; et al. RIPK3 mediates pathogenesis of experimental ventilator-induced lung injury. JCI Insight 2018, 3, e97102. [Google Scholar] [CrossRef]
- Hansen, L.W.; Jacob, A.; Yang, W.L.; Bolognese, A.C.; Prince, J.; Nicastro, J.M.; Coppa, G.F.; Wang, P. Deficiency of receptor-interacting protein kinase 3 (RIPK3) attenuates inflammation and organ injury in neonatal sepsis. J. Pediatr. Surg. 2018, 53, 1699–1705. [Google Scholar] [CrossRef]
- Sharma, A.; Matsuo, S.; Yang, W.L.; Wang, Z.; Wang, P. Receptor-interacting protein kinase 3 deficiency inhibits immune cell infiltration and attenuates organ injury in sepsis. Crit. Care 2014, 18, R142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolognese, A.C.; Yang, W.L.; Hansen, L.W.; Denning, N.L.; Nicastro, J.M.; Coppa, G.F.; Wang, P. Inhibition of necroptosis attenuates lung injury and improves survival in neonatal sepsis. Surgery 2018, 164, 110–116. [Google Scholar] [CrossRef]
- Santos, L.D.; Antunes, K.H.; Muraro, S.P.; de Souza, G.F.; da Silva, A.G.; de Souza Felipe, J.; Zanetti, L.C.; Czepielewski, R.S.; Magnus, K.; Scotta, M.; et al. TNF-mediated alveolar macrophage necroptosis drives disease pathogenesis during Respiratory Syncytial Virus infection. Eur. Respir. J. 2020, 57, 2003764. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Juarbe, N.; Bradley, K.M.; Shenoy, A.T.; Gilley, R.P.; Reyes, L.F.; Hinojosa, C.A.; Restrepo, M.I.; Dube, P.H.; Bergman, M.A.; Orihuela, C.J. Pore-forming toxin-mediated ion dysregulation leads to death receptor-independent necroptosis of lung epithelial cells during bacterial pneumonia. Cell Death Differ. 2017, 24, 917–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Juarbe, N.; Gilley, R.P.; Hinojosa, C.A.; Bradley, K.M.; Kamei, A.; Gao, G.; Dube, P.H.; Bergman, M.A.; Orihuela, C.J. Pore-Forming Toxins Induce Macrophage Necroptosis during Acute Bacterial Pneumonia. PLoS Pathog. 2015, 11, e1005337. [Google Scholar] [CrossRef]
- Gonzalez-Juarbe, N.; Riegler, A.N.; Jureka, A.S.; Gilley, R.P.; Brand, J.D.; Trombley, J.E.; Scott, N.R.; Platt, M.P.; Dube, P.H.; Petit, C.M.; et al. Influenza-Induced Oxidative Stress Sensitizes Lung Cells to Bacterial-Toxin-Mediated Necroptosis. Cell Rep. 2020, 32, 108062. [Google Scholar] [CrossRef]
- Cui, Y.L.; Qiu, L.H.; Zhou, S.Y.; Li, L.F.; Qian, Z.Z.; Liu, X.M.; Zhang, H.L.; Ren, X.B.; Wang, Y.Q. Necroptosis as a potential therapeutic target in multiple organ dysfunction syndrome. Oncotarget 2017, 8, 56980–56990. [Google Scholar] [CrossRef]
- Qing, D.Y.; Conegliano, D.; Shashaty, M.G.; Seo, J.; Reilly, J.P.; Worthen, G.S.; Huh, D.; Meyer, N.J.; Mangalmurti, N.S. Red blood cells induce necroptosis of lung endothelial cells and increase susceptibility to lung inflammation. Am. J. Respir. Crit. Care Med. 2014, 190, 1243–1254. [Google Scholar] [CrossRef]
- Tang, H.; Zhao, Z.J.; Landon, E.J.; Inagami, T. Regulation of calcium-sensitive tyrosine kinase Pyk2 by angiotensin II in endothelial cells. Roles of Yes tyrosine kinase and tyrosine phosphatase SHP-2. J. Biol. Chem. 2000, 275, 8389–8396. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Jitkaew, S.; Cai, Z.; Choksi, S.; Li, Q.; Luo, J.; Liu, Z.G. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 5322–5327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; Wang, L.; Miao, L.; Wang, T.; Du, F.; Zhao, L.; Wang, X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009, 137, 1100–1111. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, W.J.; Sridharan, H.; Huang, C.; Mandal, P.; Upton, J.W.; Gough, P.J.; Sehon, C.A.; Marquis, R.W.; Bertin, J.; Mocarski, E.S. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 2013, 288, 31268–31279. [Google Scholar] [CrossRef] [Green Version]
- Pedranzini, L.; Dechow, T.; Berishaj, M.; Comenzo, R.; Zhou, P.; Azare, J.; Bornmann, W.; Bromberg, J. Pyridone 6, a pan-Janus-activated kinase inhibitor, induces growth inhibition of multiple myeloma cells. Cancer Res. 2006, 66, 9714–9721. [Google Scholar] [CrossRef] [Green Version]
- Mandal, P.; Berger, S.B.; Pillay, S.; Moriwaki, K.; Huang, C.; Guo, H.; Lich, J.D.; Finger, J.; Kasparcova, V.; Votta, B.; et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol. Cell 2014, 56, 481–495. [Google Scholar] [CrossRef] [Green Version]
- Hildebrand, J.M.; Tanzer, M.C.; Lucet, I.S.; Young, S.N.; Spall, S.K.; Sharma, P.; Pierotti, C.; Garnier, J.M.; Dobson, R.C.; Webb, A.I.; et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc. Natl. Acad. Sci. USA 2014, 111, 15072–15077. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Zhang, Y.; Cui, M.; Jin, L.; Wang, Y.; Lv, F.; Liu, Y.; Zheng, W.; Shang, H.; Zhang, J.; et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat. Med. 2016, 22, 175–182. [Google Scholar] [CrossRef]
- Zhou, T.; DeRoo, E.; Yang, H.; Stranz, A.; Wang, Q.; Ginnan, R.; Singer, H.A.; Liu, B. MLKL and CaMKII Are Involved in RIPK3-Mediated Smooth Muscle Cell Necroptosis. Cells 2021, 10, 2397. [Google Scholar] [CrossRef]
- Ishida, A.; Kameshita, I.; Okuno, S.; Kitani, T.; Fujisawa, H. A novel highly specific and potent inhibitor of calmodulin-dependent protein kinase II. Biochem. Biophys. Res. Commun. 1995, 212, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Bloom, O.; Zhang, M.; Vishnubhakat, J.M.; Ombrellino, M.; Che, J.; Frazier, A.; Yang, H.; Ivanova, S.; Borovikova, L.; et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science 1999, 285, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, H.; Czura, C.J.; Sama, A.E.; Tracey, K.J. HMGB1 as a late mediator of lethal systemic inflammation. Am. J. Respir. Crit. Care Med. 2001, 164, 1768–1773. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Czura, C.J.; Tracey, K.J. The cytokine activity of HMGB1. J. Leukoc. Biol. 2005, 78, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Klaver, D.; Thurnher, M. Control of Macrophage Inflammation by P2Y Purinergic Receptors. Cells 2021, 10, 1098. [Google Scholar] [CrossRef]
- Pelleg, A. Extracellular adenosine 5′-triphosphate in pulmonary disorders. Biochem. Pharmacol. 2021, 187, 114319. [Google Scholar] [CrossRef]
- Maelfait, J.; Liverpool, L.; Rehwinkel, J. Nucleic Acid Sensors and Programmed Cell Death. J. Mol. Biol. 2020, 432, 552–568. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef]
- Fenimore, J. Regulation of IFN-gamma Expression. Adv. Exp. Med. Biol. 2016, 941, 1–19. [Google Scholar] [PubMed]
- Gomez, J.C.; Yamada, M.; Martin, J.R.; Dang, H.; Brickey, W.J.; Bergmeier, W.; Dinauer, M.C.; Doerschuk, C.M. Mechanisms of interferon-gamma production by neutrophils and its function during Streptococcus pneumoniae pneumonia. Am. J. Respir. Cell Mol. Biol. 2015, 52, 349–364. [Google Scholar] [CrossRef] [Green Version]
- Schoenborn, J.R.; Wilson, C.B. Regulation of interferon-gamma during innate and adaptive immune responses. Adv. Immunol. 2007, 96, 41–101. [Google Scholar]
- Zheng, C.; Yin, S.; Yang, Y.; Yu, Y.; Xie, X. CD24 aggravates acute liver injury in autoimmune hepatitis by promoting IFN-gamma production by CD4(+) T cells. Cell. Mol. Immunol. 2018, 15, 260–271. [Google Scholar] [CrossRef]
- Gadotti, A.C.; de Castro Deus, M.; Telles, J.P.; Wind, R.; Goes, M.; Garcia Charello Ossoski, R.; de Padua, A.M.; de Noronha, L.; Moreno-Amaral, A.; Baena, C.P.; et al. IFN-gamma is an independent risk factor associated with mortality in patients with moderate and severe COVID-19 infection. Virus Res. 2020, 289, 198171. [Google Scholar] [CrossRef]
- Ghazavi, A.; Ganji, A.; Keshavarzian, N.; Rabiemajd, S.; Mosayebi, G. Cytokine profile and disease severity in patients with COVID-19. Cytokine 2021, 137, 155323. [Google Scholar] [CrossRef]
- Theron, M.; Huang, K.J.; Chen, Y.W.; Liu, C.C.; Lei, H.Y. A probable role for IFN-gamma in the development of a lung immunopathology in SARS. Cytokine 2005, 32, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Montalvo Villalba, M.C.; Valdes Ramirez, O.; Mune Jimenez, M.; Arencibia Garcia, A.; Martinez Alfonso, J.; Gonzalez Baez, G.; Roque Arrieta, R.; Rosell Simon, D.; Alvarez Gainza, D.; Sierra Vazquez, B.; et al. Interferon gamma, TGF-beta1 and RANTES expression in upper airway samples from SARS-CoV-2 infected patients. Clin. Immunol. 2020, 220, 108576. [Google Scholar] [CrossRef] [PubMed]
- Akbari, H.; Tabrizi, R.; Lankarani, K.B.; Aria, H.; Vakili, S.; Asadian, F.; Noroozi, S.; Keshavarz, P.; Faramarz, S. The role of cytokine profile and lymphocyte subsets in the severity of coronavirus disease 2019 (COVID-19): A systematic review and meta-analysis. Life Sci. 2020, 258, 118167. [Google Scholar] [CrossRef] [PubMed]
- Bos, L.D.; Schouten, L.R.; van Vught, L.A.; Wiewel, M.A.; Ong, D.S.Y.; Cremer, O.; Artigas, A.; Martin-Loeches, I.; Hoogendijk, A.J.; van der Poll, T.; et al. Identification and validation of distinct biological phenotypes in patients with acute respiratory distress syndrome by cluster analysis. Thorax 2017, 72, 876–883. [Google Scholar] [CrossRef]
- Mock, J.R.; Tune, M.K.; Dial, C.F.; Torres-Castillo, J.; Hagan, R.S.; Doerschuk, C.M. Effects of IFN-gamma on immune cell kinetics during the resolution of acute lung injury. Physiol. Rep. 2020, 8, e14368. [Google Scholar] [CrossRef]
- Aoyagi, T.; Yamamoto, N.; Hatta, M.; Tanno, D.; Miyazato, A.; Ishii, K.; Suzuki, K.; Nakayama, T.; Taniguchi, M.; Kunishima, H.; et al. Activation of pulmonary invariant NKT cells leads to exacerbation of acute lung injury caused by LPS through local production of IFN-gamma and TNF-alpha by Gr-1+ monocytes. Int. Immunol. 2011, 23, 97–108. [Google Scholar] [CrossRef]
- Doherty, G.M.; Lange, J.R.; Langstein, H.N.; Alexander, H.R.; Buresh, C.M.; Norton, J.A. Evidence for IFN-gamma as a mediator of the lethality of endotoxin and tumor necrosis factor-alpha. J. Immunol. 1992, 149, 1666–1670. [Google Scholar] [PubMed]
- Heinzel, F.P. The role of IFN-gamma in the pathology of experimental endotoxemia. J. Immunol. 1990, 145, 2920–2924. [Google Scholar] [PubMed]
- Car, B.D.; Eng, V.M.; Schnyder, B.; Ozmen, L.; Huang, S.; Gallay, P.; Heumann, D.; Aguet, M.; Ryffel, B. Interferon gamma receptor deficient mice are resistant to endotoxic shock. J. Exp. Med. 1994, 179, 1437–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohler, J.; Heumann, D.; Garotta, G.; LeRoy, D.; Bailat, S.; Barras, C.; Baumgartner, J.D.; Glauser, M.P. IFN-gamma involvement in the severity of gram-negative infections in mice. J. Immunol. 1993, 151, 916–921. [Google Scholar] [PubMed]
- Liu, Q.; Xie, W.; Wang, Y.; Chen, S.; Han, J.; Wang, L.; Gui, P.; Wu, Q. JAK2/STAT1-mediated HMGB1 translocation increases inflammation and cell death in a ventilator-induced lung injury model. Lab. Investig. 2019, 99, 1810–1821. [Google Scholar] [CrossRef]
- Yamada, M.; Kubo, H.; Kobayashi, S.; Ishizawa, K.; Sasaki, H. Interferon-gamma: A key contributor to hyperoxia-induced lung injury in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 287, L1042–L1047. [Google Scholar] [CrossRef]
- Moskophidis, D.; Kioussis, D. Contribution of virus-specific CD8+ cytotoxic T cells to virus clearance or pathologic manifestations of influenza virus infection in a T cell receptor transgenic mouse model. J. Exp. Med. 1998, 188, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Ostler, T.; Davidson, W.; Ehl, S. Virus clearance and immunopathology by CD8(+) T cells during infection with respiratory syncytial virus are mediated by IFN-gamma. Eur. J. Immunol. 2002, 32, 2117–2123. [Google Scholar] [CrossRef]
- Ramana, C.V.; DeBerge, M.P.; Kumar, A.; Alia, C.S.; Durbin, J.E.; Enelow, R.I. Inflammatory impact of IFN-gamma in CD8+ T cell-mediated lung injury is mediated by both Stat1-dependent and -independent pathways. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L650–L657. [Google Scholar] [CrossRef] [Green Version]
- Wlodarczyk, M.F.; Kraft, A.R.; Chen, H.D.; Kenney, L.L.; Selin, L.K. Anti-IFN-gamma and peptide-tolerization therapies inhibit acute lung injury induced by cross-reactive influenza A-specific memory T cells. J. Immunol. 2013, 190, 2736–2746. [Google Scholar] [CrossRef]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-alpha and IFN-gamma Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149–168. [Google Scholar] [CrossRef] [PubMed]
- Tamada, N.; Tojo, K.; Yazawa, T.; Goto, T. Necrosis Rather Than Apoptosis is the Dominant form of Alveolar Epithelial Cell Death in Lipopolysaccharide-Induced Experimental Acute Respiratory Distress Syndrome Model. Shock 2020, 54, 128–139. [Google Scholar] [CrossRef]
- Aeffner, F.; Bolon, B.; Davis, I.C. Mouse Models of Acute Respiratory Distress Syndrome: A Review of Analytical Approaches, Pathologic Features, and Common Measurements. Toxicol. Pathol. 2015, 43, 1074–1092. [Google Scholar] [CrossRef] [PubMed]
- Thapa, R.J.; Basagoudanavar, S.H.; Nogusa, S.; Irrinki, K.; Mallilankaraman, K.; Slifker, M.J.; Beg, A.A.; Madesh, M.; Balachandran, S. NF-kappaB protects cells from gamma interferon-induced RIP1-dependent necroptosis. Mol. Cell Biol. 2011, 31, 2934–2946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thapa, R.J.; Nogusa, S.; Chen, P.; Maki, J.L.; Lerro, A.; Andrake, M.; Rall, G.F.; Degterev, A.; Balachandran, S. Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc. Natl. Acad. Sci. USA 2013, 110, E3109–E3118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; Kwon, J.Y.; Kim, S.Y.; Jung, K.; Cho, M.L. Interferon-gamma regulates inflammatory cell death by targeting necroptosis in experimental autoimmune arthritis. Sci. Rep. 2017, 7, 10133. [Google Scholar] [CrossRef] [PubMed]
- Hao, Q.; Kundu, S.; Kleam, J.; Zhao, Z.J.; Idell, S.; Tang, H. Enhanced RIPK3 kinase activity-dependent lytic cell death in M1 but not M2 macrophages. Mol. Immunol. 2021, 129, 86–93. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, Q.; Shetty, S.; Tucker, T.A.; Idell, S.; Tang, H. Interferon-γ Preferentially Promotes Necroptosis of Lung Epithelial Cells by Upregulating MLKL. Cells 2022, 11, 563. https://doi.org/10.3390/cells11030563
Hao Q, Shetty S, Tucker TA, Idell S, Tang H. Interferon-γ Preferentially Promotes Necroptosis of Lung Epithelial Cells by Upregulating MLKL. Cells. 2022; 11(3):563. https://doi.org/10.3390/cells11030563
Chicago/Turabian StyleHao, Qin, Sreerama Shetty, Torry A. Tucker, Steven Idell, and Hua Tang. 2022. "Interferon-γ Preferentially Promotes Necroptosis of Lung Epithelial Cells by Upregulating MLKL" Cells 11, no. 3: 563. https://doi.org/10.3390/cells11030563
APA StyleHao, Q., Shetty, S., Tucker, T. A., Idell, S., & Tang, H. (2022). Interferon-γ Preferentially Promotes Necroptosis of Lung Epithelial Cells by Upregulating MLKL. Cells, 11(3), 563. https://doi.org/10.3390/cells11030563