
Discordant Genome Assemblies Drastically Alter the Interpretation of Single-Cell RNA Sequencing Data Which Can Be Mitigated by a Novel Integration Method

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Husbandry
2.2. Astyanax Mexicanus Heart Dissection and Digestion
2.3. 3′ UTR Extension
- Identify transcripts without 3′ UTR annotation in the Ensembl GTF file
- Compare fragment coverage over 100 bp flanking the terminal exon
- If median 3′ coverage > median 5′ coverage, extend last exon 100 bp in the 3′ direction and repeat steps 2 & 3 until no further extension occurs.
2.4. 10x Single-Cell RNA-Sequencing and Analysis
3. Results
3.1. Datasets Generated from the Same Sample Change in Their Fundamental Structure Depending on Genome Assembly and Annotation
3.2. Dimensional Reduction and Data Visualization Is Robust to Differences in Genome Assembly
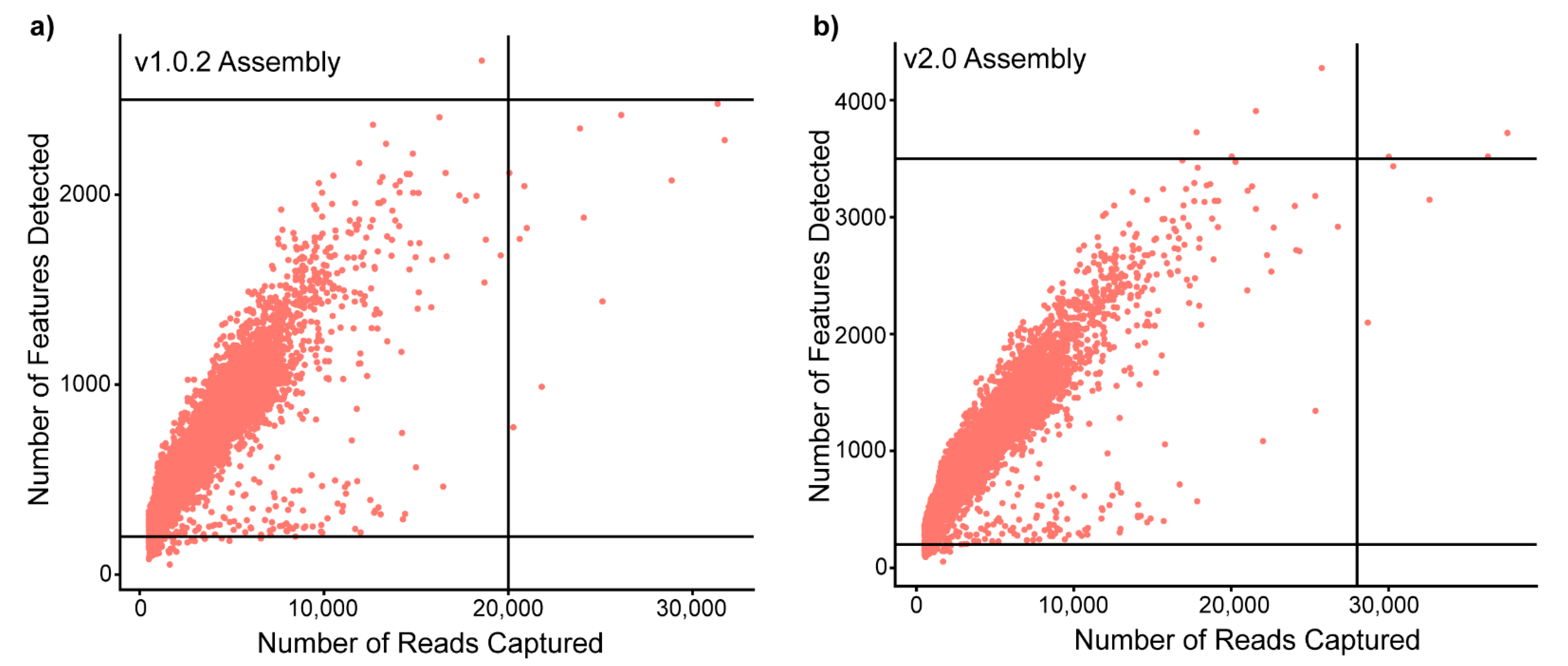
3.3. Incomplete Reference Genomes, When Used in Isolation, Create Problems in Cell-Type Identification and Differential Gene Expression Analysis and Have the Potential to Miss Data
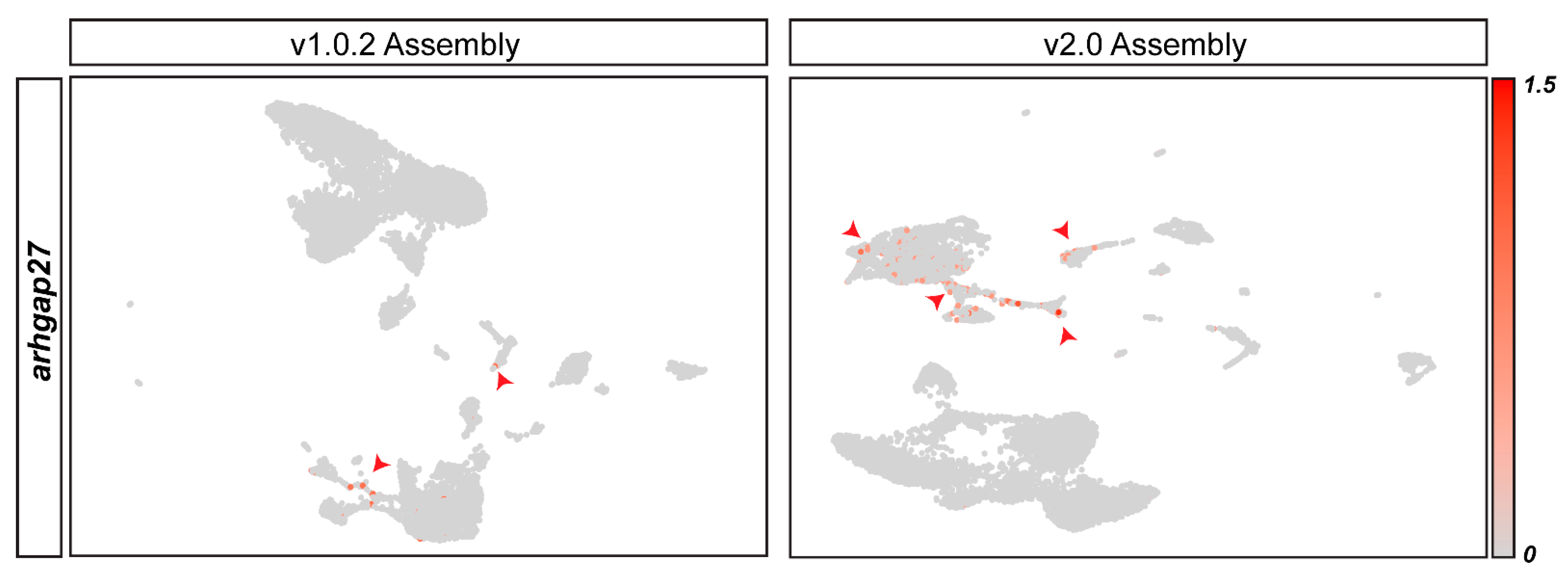
3.4. Genome Assembly Alignment Can Distort Data Interpretation from Specific Cell Types Due to the Problems Created by Underlying Differences in Genome Assemblies
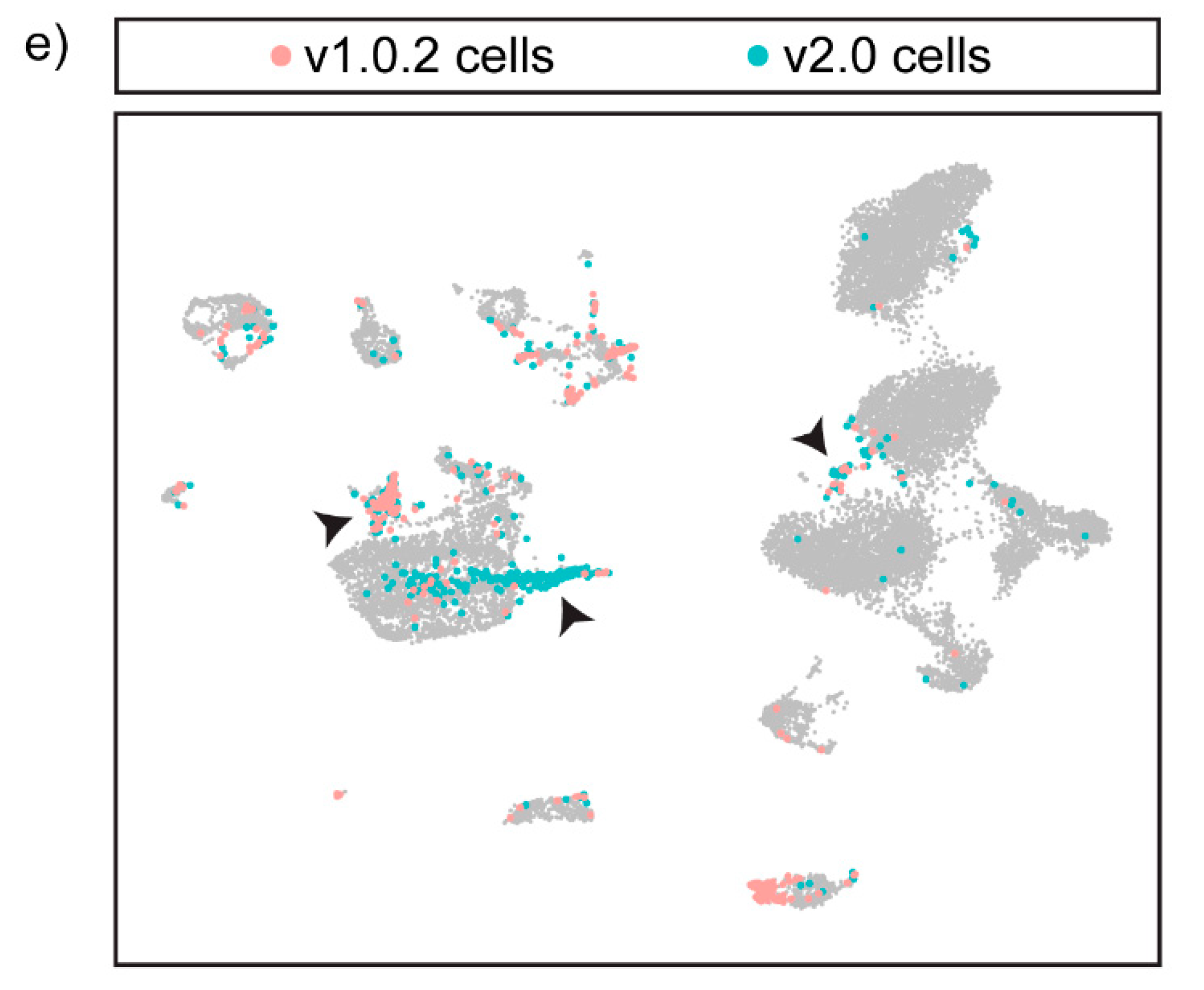
3.5. Integration of the Two Datasets Improves Cell-Type Annotation and Maximises the Information That Can Be Obtained from a Single-Cell Dataset
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Haque, A.; Engel, J.; Teichmann, S.A.; Lönnberg, T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 2017, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Schaum, N.; Karkanias, J.; Neff, N.F.; May, A.P.; Quake, S.R.; Wyss-Coray, T.; Darmanis, S.; Batson, J.; Botvinnik, O.; Chen, M.B.; et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018, 562, 367–372. [Google Scholar]
- Shao, Y.; Wang, X.-B.; Zhang, J.-J.; Li, M.-L.; Wu, S.-S.; Ma, X.-Y.; Wang, X.; Zhao, H.-F.; Li, Y.; Zhu, H.H.; et al. Genome and single-cell RNA-sequencing of the earthworm Eisenia andrei identifies cellular mechanisms underlying regeneration. Nat. Commun. 2020, 11, 2656. [Google Scholar] [CrossRef] [PubMed]
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 96. [Google Scholar] [CrossRef] [Green Version]
- Waterston, R.H.; Pachter, L. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562. [Google Scholar]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar]
- Sulston, J.; Du, Z.; Thomas, K.; Wilson, R.; Hillier, L.; Staden, R.; Halloran, N.; Green, P.; Thierry-Mieg, J.; Qiu, L.; et al. The C. elegans genome sequencing project: A beginning. Nature 1992, 356, 37–41. [Google Scholar] [CrossRef]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Rice, E.S.; Green, R. New Approaches for Genome Assembly and Scaffolding. Annu. Rev. Anim. Biosci. 2019, 7, 17–40. [Google Scholar] [CrossRef]
- Armstrong, J.; Fiddes, I.T.; Diekhans, M.; Paten, B. Whole-Genome Alignment and Comparative Annotation. Annu. Rev. Anim. Biosci. 2019, 7, 41–64. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, W.R. Astyanax surface and cave fish morphs. Evodevo 2020, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Gross, J.B. The complex origin of Astyanax cavefish. BMC Evol. Biol. 2012, 12, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, J.B.; Meyer, B.; Perkins, M. The rise of Astyanax cavefish. Dev. Dyn. 2015, 244, 1031–1038. [Google Scholar] [CrossRef]
- Herman, A.; Brandvain, Y.; Weagley, J.; Jeffery, W.R.; Keene, A.C.; Kono, T.J.Y.; Bilandžija, H.; Borowsky, R.; Espinasa, L.; O’Quin, K.; et al. The role of gene flow in rapid and repeated evolution of cave-related traits in Mexican tetra, Astyanax mexicanus. Mol. Ecol. 2018, 27, 4397–4416. [Google Scholar] [CrossRef]
- Fumey, J.; Hinaux, H.; Noirot, C.; Thermes, C.; Rétaux, S.; Casane, D. Evidence for late Pleistocene origin of Astyanax mexicanus cavefish. BMC Evol. Biol. 2018, 18, 43. [Google Scholar] [CrossRef]
- O’Quin, K.E.; Yoshizawa, M.; Doshi, P.; Jeffery, W.R. Quantitative genetic analysis of retinal degeneration in the blind cavefish. PLoS ONE 2013, 8, e57281. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, Y.; Jeffery, W.R. Central role for the lens in cave fish eye degeneration. Science 2000, 289, 631–633. [Google Scholar] [CrossRef]
- Ma, L.; Ng, M.; van der Weele, C.M.; Yoshizawa, M.; Jeffery, W.R. Dual roles of the retinal pigment epithelium and lens in cavefish eye degeneration. J. Exp. Zool. Part B Mol. Dev. Evol. 2020, 334, 438–449. [Google Scholar] [CrossRef]
- Ma, L.; Gore, A.V.; Castranova, D.; Shi, J.; Ng, M.; Tomins, K.A.; van der Weele, C.M.; Weinstein, B.M.; Jeffery, W.R. A hypomorphic cystathionine β-synthetase gene contributes to cavefish eye loss by disrupting optic vasculature. Nat. Commun. 2020, 11, 2772. [Google Scholar] [CrossRef]
- Riddle, M.R.; Aspiras, A.C.; Gaudenz, K.; Peuß, R.; Sung, J.Y.; Martineau, B.; Peavey, M.; Box, A.C.; Tabin, J.A.; McGaugh, S.; et al. Insulin resistance in cavefish as an adaptation to a nutrient-limited environment. Nature 2018, 555, 647–651. [Google Scholar] [CrossRef] [PubMed]
- McGaugh, S.E.; Gross, J.B.; Aken, B.; Blin, M.; Borowsky, R.; Chalopin, D.; Hinaux, H.; Jeffery, W.R.; Keene, A.C.; Ma, L.; et al. The cavefish genome reveals candidate genes for eye loss. Nat. Commun. 2014, 5, 5307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warren, W.C.; Boggs, T.E.; Borowsky, R.; Carlson, B.M.; Ferrufino, E.; Gross, J.B.; Hillier, L.; Hu, Z.; Keene, A.C.; Kenzior, A.; et al. A chromosome-level genome of Astyanax mexicanus surface fish for comparing population-specific genetic differences contributing to trait evolution. Nat. Commun. 2021, 12, 1447. [Google Scholar] [CrossRef]
- Sander, V.; Su, G.; Jopling, C.; Morera, C.; Belmonte, J.C.I. Isolation and in vitro culture of primary cardiomyocytes from adult zebrafish hearts. Nat. Protoc. 2013, 8, 800–809. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Willaim, M.M., III; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data Resource Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef] [PubMed]
- Hafemeister, C.; Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019, 20, 296. [Google Scholar] [CrossRef] [Green Version]
- Zappia, L.; Oshlack, A. Clustering trees: A visualization for evaluating clusterings at multiple resolutions. GigaScience 2018, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Grinnell, F. Fibroblasts, myofibroblasts, and wound contraction. J. Cell Biol. 1994, 124, 401–404. [Google Scholar] [CrossRef]
- Schürch, W.; Seemayer, T.A.; Gabbiani, G. The myofibroblast: A quarter century after its discovery. Am. J. Surg. Pathol. 1998, 22, 141–147. [Google Scholar] [CrossRef]
- Gabbiani, G.; Ryan, G.B.; Majno, G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia 1971, 27, 549–550. [Google Scholar] [CrossRef]
- Stockdale, W.; Lemieux, M.E.; Killen, A.C.; Zhao, J.; Hu, Z.; Riepsaame, J.; Hamilton, N.; Kudoh, T.; Riley, P.R.; van Aerle, R.; et al. Heart Regeneration in the Mexican Cavefish. Cell Rep. 2018, 25, 1997–2007.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafer, M.E.R.; Sawh, A.N.; Schier, A.F. Gene family evolution underlies cell-type diversification in the hypothalamus of teleosts. Nat. Ecol. Evol. 2021, 6, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, J.; Persons, J.L.; Peuß, R.; Hassan, H.; Kenzior, A.; Xiong, S.; Olsen, L.; Maldonado, E.; Kowalko, J.E.; Rohner, N. Comparative transcriptome analysis of wild and lab populations of Astyanax mexicanus uncovers differential effects of environment and morphotype on gene expression. J. Exp. Zool. Part B Mol. Dev. Evol. 2020, 334, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Peuß, R.; Box, A.C.; Chen, S.; Wang, Y.; Tsuchiya, D.; Persons, J.L.; Kenzior, A.; Maldonado, E.; Krishnan, J.; Scharsack, J.P.; et al. Adaptation to low parasite abundance affects immune investment and immunopathological responses of cavefish. Nat. Ecol. Evol. 2020, 4, 1416–1430. [Google Scholar] [CrossRef] [PubMed]
- Boudjema, I.; Kang, D.; Séverine, B.; Elodie, J.; Romain, F.; Qiaowei, P.; Jorge, T.; Céline, L.; Adrien, C.; Lisa, G.; et al. A supernumerary ‘B-sex’ chromosome drives male sex determination in the Pachón cavefish, Astyanax mexicanus. Curr. Biol. 2021, 31, 4800–4809.e9. [Google Scholar]
- Butler, J.; MacCallum, I.; Kleber, M.; Shlyakhter, I.A.; Belmonte, M.K.; Lander, E.S.; Nusbaum, C.; Jaffe, D.B. ALLPATHS: De novo assembly of whole-genome shotgun microreads. Genome Res. 2008, 18, 810–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assembly Readouts | v1.0.2 Assembly | v2.0 Assembly |
|---|---|---|
| Matrix Dimensions | 25,489 genes, 8870 cells | 27,420 genes, 9018 cells |
| Reads Mapped to Genome | 72.6% | 74.1% |
| Reads Mapped to Transcriptome | 41.7% | 52.4% |
| Median Reads/Cell | 30,901 | 30,394 |
| Median Genes/Cell | 794 | 1201 |
| Assembly-specific cells | 16 | 225 |
| Assembly-specific genes | 4311 | 5638 |
| nCount vs. nFeature Correlation | 0.84 | 0.88 |
| Quality Control Thresholds | nFeatures: 200–2500 | nFeatures: 200–3500 |
| nCounts: <20,000 | nCounts <28,000 | |
| % Cells Passed Filtering | 98.2% | 98.9% |
| Cell Numbers Post-Filtering | 8717 cells | 8926 cells |
| % Genes Passed Filtering | 64.3% | 63.9% |
| Gene Numbers Post-Filtering | 16,408 genes | 17,528 genes |
| PCA Dimensions | 30 | 30 |
| Clustering Resolution | 1.25 | 1.25 |
| Number of Clusters Found | 26 | 27 |
| Cell Type | v1.0.2 Assembly | v2.0 Assembly |
|---|---|---|
| Cardiomyocytes | ENSAMXG00005008576 * | myh7l * |
| actc1a | ENSAMXG00000004797 * | |
| tnnc1a | nme2b.1 * | |
| cox6a2 * | TNNC1 | |
| aldoaa | aldoab | |
| zgc:193541 | idh2 | |
| ENSAMXG00005013223 * | cox7b | |
| IDH2 | slc25a5 | |
| atp5mc3a | cox7c | |
| atp5if1a * | tnnt2b | |
| Endothelium | ENSAMXG00005007750 * | lyve1a |
| ENSAMXG00005016906 * | ENSAMXG00000041928 * | |
| ENSAMXG00005003412 * | krt18a.1 | |
| ENSAMXG00005021204 * | rgs5b | |
| plpp2a | ENSAMXG00000036379 * | |
| ENSAMXG00005022026 * | il13ra2 | |
| krt5 | sat1a.2 | |
| Ucmaa * | serpinh1b | |
| ENSAMXG00005012084 * | her6 | |
| ENSAMXG00005004741 * | ENSAMXG00000035697 * | |
| Fibroblasts | thbs1a | ccl25b |
| tcf21 | rbp4 | |
| lxn | apoeb | |
| kcne4 | pmp22a | |
| mustn1a | tagln | |
| ENSAMXG00005006660 * | TCF21 | |
| pltp | dcn | |
| clec19a | col1a2 | |
| BAMBI * | anxa1a (ENSAMXG00000035597) | |
| hmx4 | sostdc1a | |
| Neutrophils | ENSAMXG00005022612 * | ENSAMXG00000006427 * |
| c6ast1 | mmp9(ENSAMXG00000007722) | |
| ENSAMXG00005012967 * | ENSAMXG00000035474 * | |
| ENSAMXG00005007030 * | LECT2 | |
| ENSAMXG00005022013 * | c6ast1 | |
| adam8a | ENSAMXG00000037167 * | |
| ltb4r | cebp1 | |
| ENSAMXG00005015365 * | ENSAMXG00000001798 * | |
| mmp9 | Scinlb | |
| ENSAMXG00005024801 * | ENSAMXG00000034260 * | |
| T cells | pfn1 | ENSAMXG00000036068 * |
| laptm5 | pfn1 | |
| cxcr4b | laptm5 | |
| ENSAMXG00005014236 * | ctsl.1 (ENSAMXG00000029871) | |
| coro1a | cxcr4b | |
| ENSAMXG00005012967 * | rac2 * | |
| rgs13 | dusp2 | |
| PTPRC | rgs13 | |
| runx3 | cotl1 | |
| ENSAMXG00005022013 * | ENSAMXG00000001798 * | |
| B cells | ENSAMXG00005001652 * | ENSAMXG00000033936 * |
| ENSAMXG00005007434 * | zgc:194275 | |
| cd37 | ENSAMXG00000029163 * | |
| ENSAMXG00005014280 * | ENSAMXG00000038512 * | |
| si:dkey-24p1.1 | ENSAMXG00000006777 * | |
| ENSAMXG00005006484 * | cd37 | |
| ENSAMXG00005014291 * | ENSAMXG00000036191 * | |
| ENSAMXG00005012813 * | ENSAMXG00000034153 * | |
| ENSAMXG00005000610 * | ENSAMXG00000043949 * | |
| ENSAMXG00005002435 * | ENSAMXG00000043088 * | |
| Erythrocytes | hbaa2 | ENSAMXG00000029151 * |
| ENSAMXG00005017042 * | hbaa2 (ENSAMXG00000029181) | |
| wu:fj16a03 | ENSAMXG00000037273 * | |
| cahz | hbba2 | |
| nt5c2l1 | HBE1 (ENSAMXG00000037475) * | |
| ENSAMXG00005020328 * | si:ch211-250g4.3 * | |
| ENSAMXG00005017060 * | si:ch211-103n10.5 | |
| mt2.2 | Cahz | |
| zgc:163057 * | slc4a1a | |
| ENSAMXG00005017061 * | wu:fj16a03 | |
| Epicardium | ENSAMXG00005022849 * | ENSAMXG00000036050 * |
| tcf21 | TCF21 | |
| ENSAMXG00005007716 * | Cfd | |
| ENSAMXG00005012482 * | c3a.1 * | |
| zgc:158846 | ENSAMXG00000036137 * | |
| ENSAMXG00005022791 * | igfbp5a | |
| ENSAMXG00005009039 * | stmn1a | |
| ENSAMXG00005008245 * | wt1b | |
| ENSAMXG00005022313 * | glis2a | |
| wt1b | slc29a1a | |
| Smooth Muscle | si:dkey-57k2.6 | CASP6 |
| ENSAMXG00005003735 * | Tagln | |
| thbs1b | angptl7 | |
| ENSAMXG00005011018 * | ENSAMXG00000031755 * | |
| TPM1 | acta2 * | |
| anxa1a | si:dkey-57k2.6 | |
| ITIH3 | rbp4 | |
| ENSAMXG00005005681 * | sox9b | |
| ENSAMXG00005018089 * | anxa1a (ENSAMXG00000035597) | |
| thbs4a | thbs1b | |
| Dendritic Cells/Macrophages | ENSAMXG00005011614 * | ENSAMXG00000036068 * |
| ENSAMXG00005002001 * | ccl35.1 * | |
| ENSAMXG00005021693 * | ENSAMXG00000037572 * | |
| ccl34a.3 * | cd74a | |
| cxcr4b | ENSAMXG00000004394 * | |
| ENSAMXG00005014236 * | cxcl8a | |
| cd74a | cxcr4b | |
| ENSAMXG00005001734 * | ENSAMXG00000042210 * | |
| ENSAMXG00005009773 * | si:dkey-5n18.1 | |
| c1qb | il1b (ENSAMXG00000035729) * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Potts, H.G.; Lemieux, M.E.; Rice, E.S.; Warren, W.; Choudhury, R.P.; Mommersteeg, M.T.M. Discordant Genome Assemblies Drastically Alter the Interpretation of Single-Cell RNA Sequencing Data Which Can Be Mitigated by a Novel Integration Method. Cells 2022, 11, 608. https://doi.org/10.3390/cells11040608
Potts HG, Lemieux ME, Rice ES, Warren W, Choudhury RP, Mommersteeg MTM. Discordant Genome Assemblies Drastically Alter the Interpretation of Single-Cell RNA Sequencing Data Which Can Be Mitigated by a Novel Integration Method. Cells. 2022; 11(4):608. https://doi.org/10.3390/cells11040608
Chicago/Turabian StylePotts, Helen G., Madeleine E. Lemieux, Edward S. Rice, Wesley Warren, Robin P. Choudhury, and Mathilda T. M. Mommersteeg. 2022. "Discordant Genome Assemblies Drastically Alter the Interpretation of Single-Cell RNA Sequencing Data Which Can Be Mitigated by a Novel Integration Method" Cells 11, no. 4: 608. https://doi.org/10.3390/cells11040608
APA StylePotts, H. G., Lemieux, M. E., Rice, E. S., Warren, W., Choudhury, R. P., & Mommersteeg, M. T. M. (2022). Discordant Genome Assemblies Drastically Alter the Interpretation of Single-Cell RNA Sequencing Data Which Can Be Mitigated by a Novel Integration Method. Cells, 11(4), 608. https://doi.org/10.3390/cells11040608