
p53 in Proximal Tubules Mediates Chronic Kidney Problems after Cisplatin Treatment


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Treatment
2.2. Animals and Treatment
2.3. Reagents and Antibodies
2.4. Immunofluorescence
2.5. Immunoblot Analysis
2.6. Morphological Examination of Apoptosis
2.7. RNA Isolation and Quantitative Real-Time PCR
2.8. Renal Function and Transcutaneous Measurement of GFR
2.9. Histological Staining
2.10. Image Quantification
2.11. Statistics Analysis
3. Results
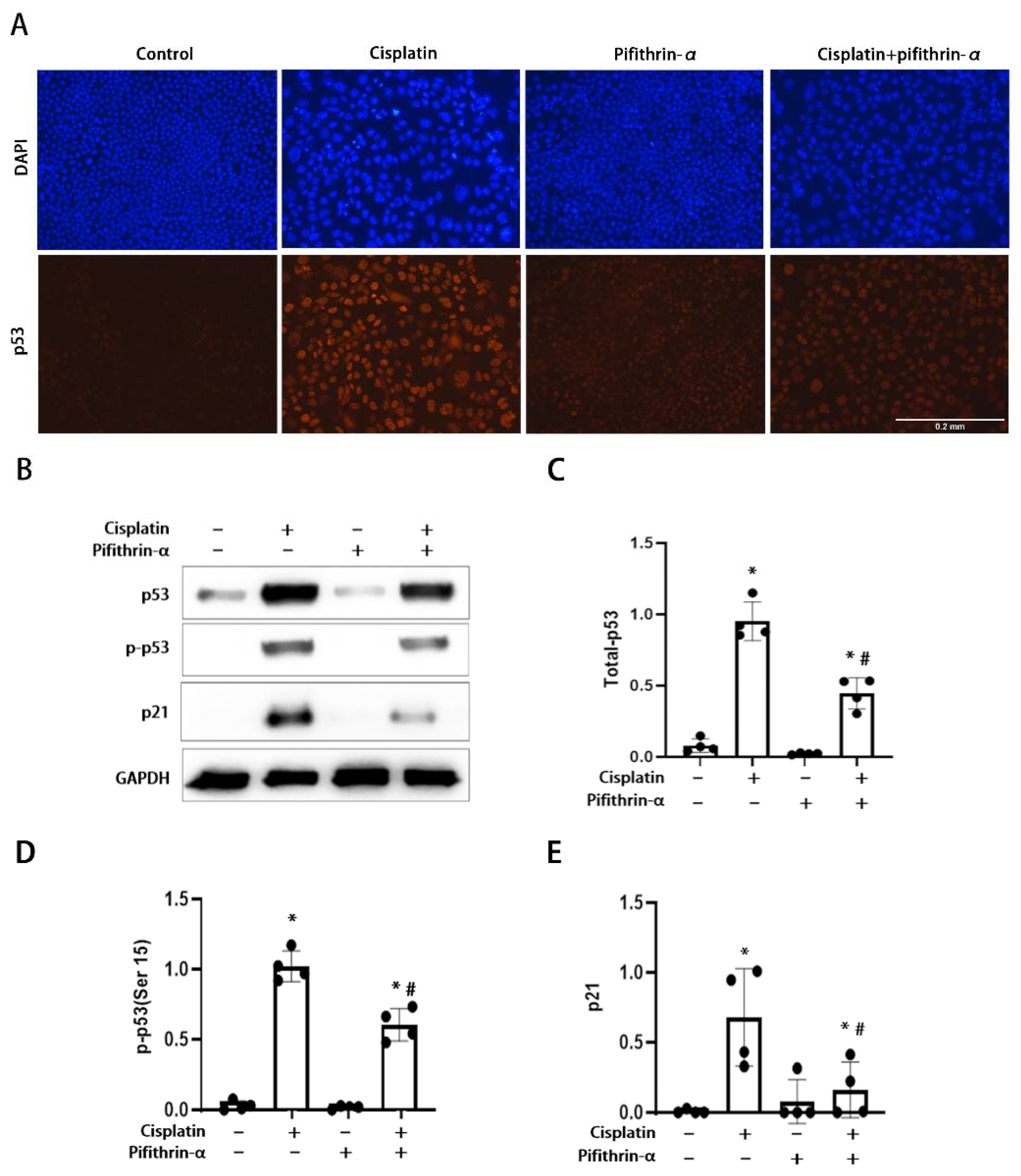
3.1. Activation of p53 in RLDC-Treated BUMPT Cells and Its Inhibition by Pifithrin-α
3.2. Pifithrin-α Attenuates Apoptosis in RLDC-Treated BUMPT Cells
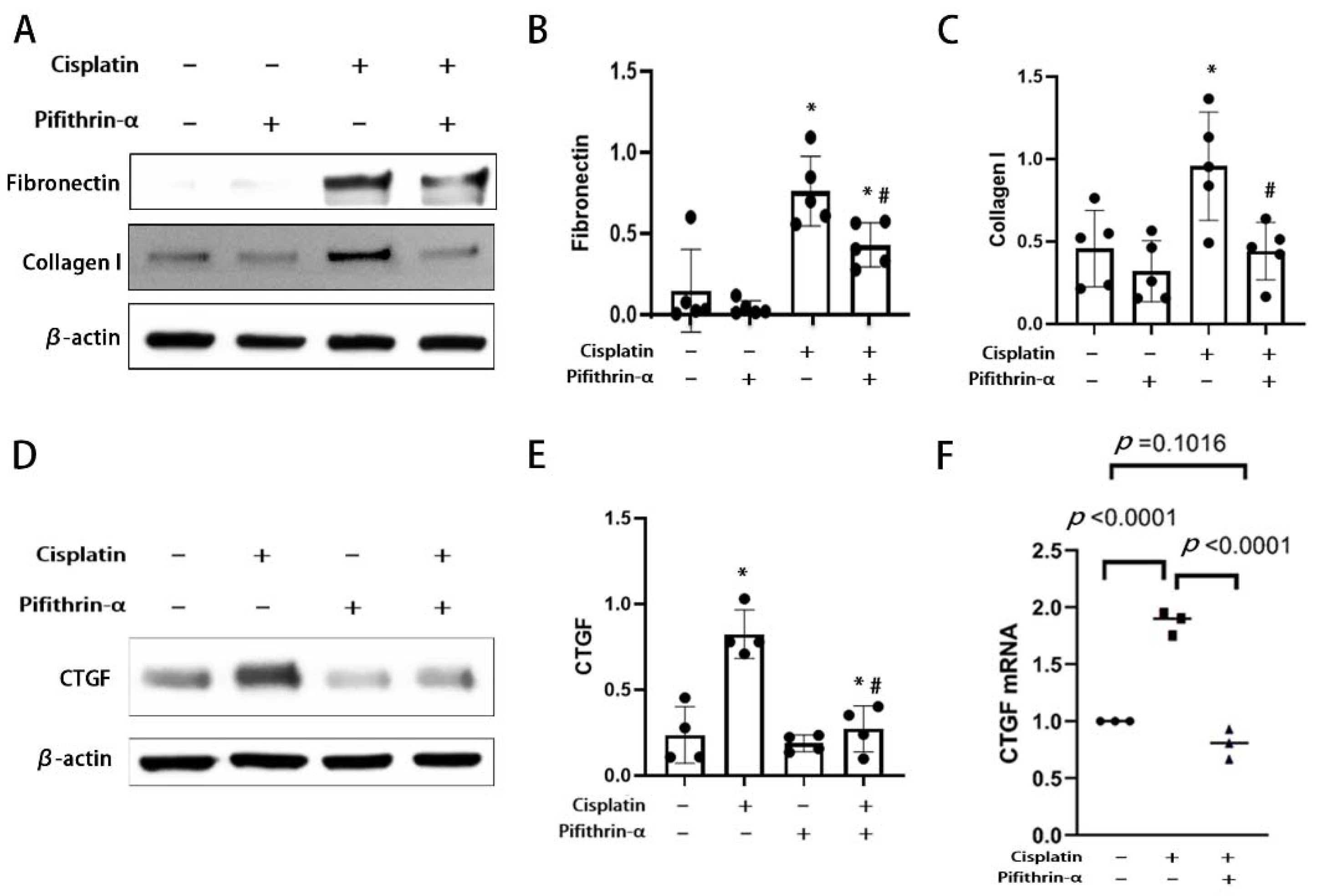
3.3. Pifithrin-α Reduces RLDC-Induced Fibrotic Changes in BUMPT Cells
3.4. RLDC-Induced Renal Fibrosis Is Attenuated in PT-p53-KO Mice
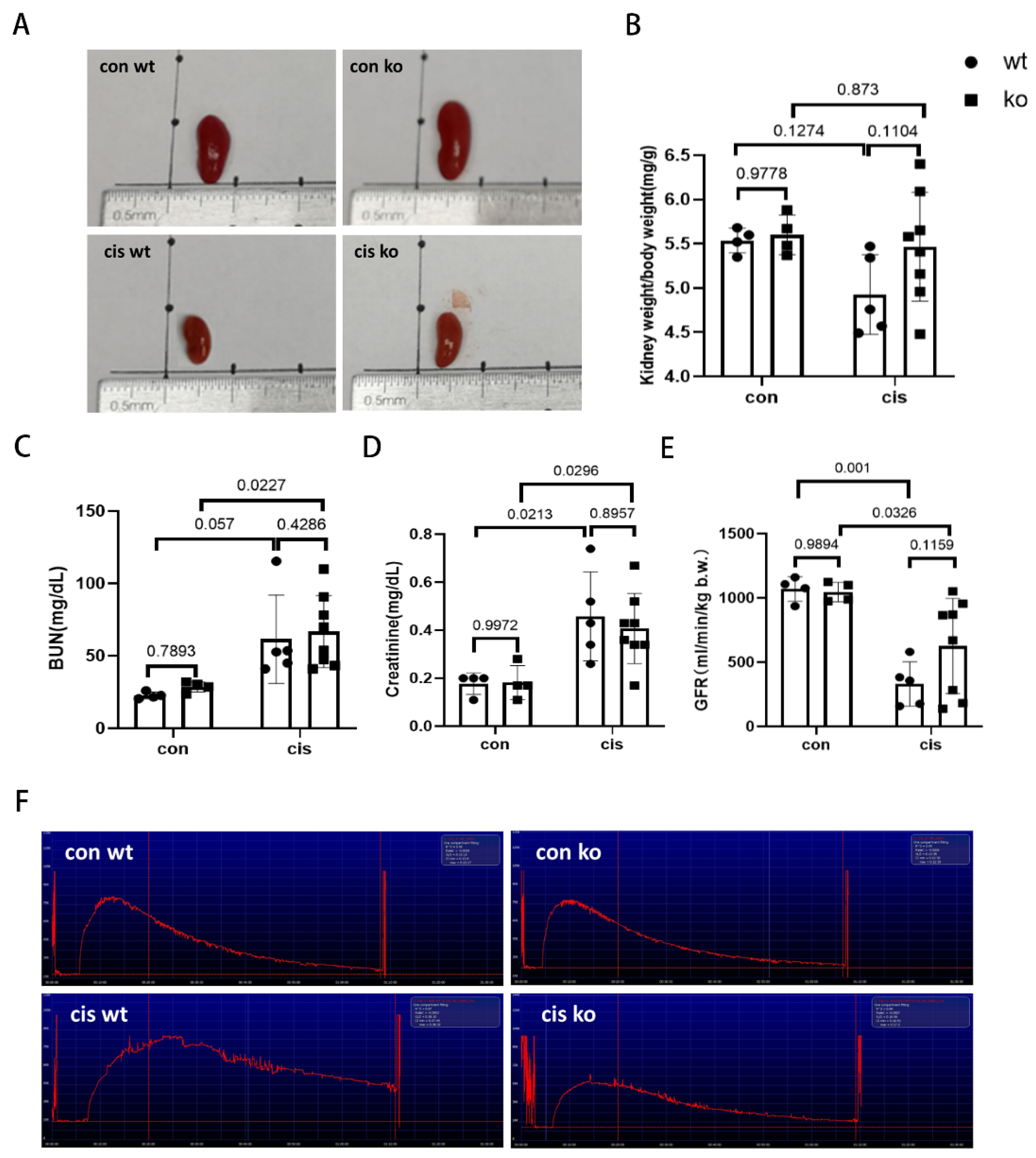
3.5. RLDC-Induced Kidney Atrophy and Renal Function Decline Are Moderately Suppressed in PT-p53-KO Mice
3.6. Proximal Tubular p53 Deficiency Suppresses Tubular Degeneration and Induces Renal Cell Proliferation in RLDC-Treated Mice
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug. Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, T.; Nazneen, A.; Abid, M.R.; Razzaque, M.S. Cisplatin-associated nephrotoxicity and pathological events. Contrib. Nephrol. 2005, 148, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Cepeda, V.; Fuertes, M.A.; Castilla, J.; Alonso, C.; Quevedo, C.; Perez, J.M. Biochemical mechanisms of cisplatin cytotoxicity. Anticancer Agents Med. Chem. 2007, 7, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Atherton, S.S. Tumor necrosis factor-alpha in cisplatin nephrotoxicity: A homebred foe? Kidney Int. 2007, 72, 5–7. [Google Scholar] [CrossRef] [Green Version]
- Burns, C.V.; Edwin, S.B.; Szpunar, S.; Forman, J. Cisplatin-induced nephrotoxicity in an outpatient setting. Pharmacotherapy 2021, 41, 184–190. [Google Scholar] [CrossRef]
- Zhang, D.; Liu, Y.; Wei, Q.; Huo, Y.; Liu, K.; Liu, F.; Dong, Z. Tubular p53 regulates multiple genes to mediate AKI. J. Am. Soc. Nephrol. 2014, 25, 2278–2289. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, K.; Zhou, L.; Mi, Q.S.; Huang, S.; She, J.X.; Dong, Z. MicroRNA-34a is induced via p53 during cisplatin nephrotoxicity and contributes to cell survival. Mol. Med. 2010, 16, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.; Liu, F.; Guan, B.; Luo, Z.; Lin, J.; Fang, W.; Liu, L.; Zuo, W. p53 induces miR-199a-3p to suppress mechanistic target of rapamycin activation in cisplatin-induced acute kidney injury. J. Cell Biochem. 2019, 120, 17625–17634. [Google Scholar] [CrossRef]
- Lu, Q.B.; Du, Q.; Wang, H.P.; Tang, Z.H.; Wang, Y.B.; Sun, H.J. Salusin-beta mediates tubular cell apoptosis in acute kidney injury: Involvement of the PKC/ROS signaling pathway. Redox Biol. 2020, 30, 101411. [Google Scholar] [CrossRef]
- Morigi, M.; Perico, L.; Rota, C.; Longaretti, L.; Conti, S.; Rottoli, D.; Novelli, R.; Remuzzi, G.; Benigni, A. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Investig. 2015, 125, 715–726. [Google Scholar] [CrossRef] [Green Version]
- Torres, R.; Velazquez, H.; Chang, J.J.; Levene, M.J.; Moeckel, G.; Desir, G.V.; Safirstein, R. Three-Dimensional Morphology by Multiphoton Microscopy with Clearing in a Model of Cisplatin-Induced CKD. J. Am. Soc. Nephrol. 2016, 27, 1102–1112. [Google Scholar] [CrossRef]
- Sharp, C.N.; Doll, M.A.; Dupre, T.V.; Shah, P.P.; Subathra, M.; Siow, D.; Arteel, G.E.; Megyesi, J.; Beverly, L.J.; Siskind, L.J. Repeated administration of low-dose cisplatin in mice induces fibrosis. Am. J. Physiol. Renal. Physiol. 2016, 310, F560–F568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katagiri, D.; Hamasaki, Y.; Doi, K.; Negishi, K.; Sugaya, T.; Nangaku, M.; Noiri, E. Interstitial renal fibrosis due to multiple cisplatin treatments is ameliorated by semicarbazide-sensitive amine oxidase inhibition. Kidney Int. 2016, 89, 374–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, L.M.; Lever, J.M.; Traylor, A.M.; Chen, B.; Yang, Z.; Esman, S.K.; Jiang, Y.; Cutter, G.R.; Boddu, R.; George, J.F.; et al. Divergent effects of AKI to CKD models on inflammation and fibrosis. Am. J. Physiol. Renal. Physiol. 2018, 315, F1107–F1118. [Google Scholar] [CrossRef]
- Su, H.; Ye, C.; Lei, C.T.; Tang, H.; Zeng, J.Y.; Yi, F.; Zhang, C. Subcellular trafficking of tubular MDM2 implicates in acute kidney injury to chronic kidney disease transition during multiple low-dose cisplatin exposure. FASEB J. 2020, 34, 1620–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Cai, J.; Li, F.; Liu, Z.; Shu, S.; Wang, Y.; Liu, Y.; Tang, C.; Dong, Z. Chronic effects of repeated low-dose cisplatin treatment in mouse kidneys and renal tubular cells. Am. J. Physiol. Renal. Physiol. 2019, 317, F1582–F1592. [Google Scholar] [CrossRef]
- Pabla, N.; Dong, G.; Jiang, M.; Huang, S.; Kumar, M.V.; Messing, R.O.; Dong, Z. Inhibition of PKCdelta reduces cisplatin-induced nephrotoxicity without blocking chemotherapeutic efficacy in mouse models of cancer. J. Clin. Investig. 2011, 121, 2709–2722. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Wei, Q.; Wang, J.; Du, Q.; Yu, J.; Zhang, L.; Dong, Z. Regulation of PUMA-alpha by p53 in cisplatin-induced renal cell apoptosis. Oncogene 2006, 25, 4056–4066. [Google Scholar] [CrossRef] [Green Version]
- Megyesi, J.; Safirstein, R.L.; Price, P.M. Induction of p21WAF1/CIP1/SDI1 in kidney tubule cells affects the course of cisplatin-induced acute renal failure. J. Clin. Investig. 1998, 101, 777–782. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Dong, Z. Regulation and pathological role of p53 in cisplatin nephrotoxicity. J. Pharmacol. Exp. Ther. 2008, 327, 300–307. [Google Scholar] [CrossRef]
- Jiang, M.; Wei, Q.; Pabla, N.; Dong, G.; Wang, C.Y.; Yang, T.; Smith, S.B.; Dong, Z. Effects of hydroxyl radical scavenging on cisplatin-induced p53 activation, tubular cell apoptosis and nephrotoxicity. Biochem. Pharmacol. 2007, 73, 1499–1510. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.; Ma, Z.; Zhu, J.; Liu, Z.; Liu, Y.; Liu, Y.; Cai, J.; Dong, Z. P53 in kidney injury and repair: Mechanism and therapeutic potentials. Pharmacol. Ther. 2019, 195, 5–12. [Google Scholar] [CrossRef]
- Jiang, M.; Yi, X.; Hsu, S.; Wang, C.Y.; Dong, Z. Role of p53 in cisplatin-induced tubular cell apoptosis: Dependence on p53 transcriptional activity. Am. J. Physiol. Renal. Physiol. 2004, 287, F1140–F1147. [Google Scholar] [CrossRef]
- Wei, Q.; Dong, G.; Yang, T.; Megyesi, J.; Price, P.M.; Dong, Z. Activation and involvement of p53 in cisplatin-induced nephrotoxicity. Am. J. Physiol. Renal. Physiol. 2007, 293, F1282–F1291. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010, 16, 535–543. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Xu, X.; Li, H.; Chen, J.; Xiang, X.; Dong, Z.; Zhang, D. p53 induces miR199a-3p to suppress SOCS7 for STAT3 activation and renal fibrosis in UUO. Sci. Rep. 2017, 7, 43409. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Pan, J.; Li, H.; Long, J.; Fang, F.; Chen, J.; Zhu, X.; Xiang, X.; Zhang, D. lncRNA ZEB1-AS1 Was Suppressed by p53 for Renal Fibrosis in Diabetic Nephropathy. Mol. Ther. Nucleic Acids 2018, 12, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Ying, Y.; Kim, J.; Westphal, S.N.; Long, K.E.; Padanilam, B.J. Targeted deletion of p53 in the proximal tubule prevents ischemic renal injury. J. Am. Soc. Nephrol. 2014, 25, 2707–2716. [Google Scholar] [CrossRef] [Green Version]
- Dagher, P.C.; Mai, E.M.; Hato, T.; Lee, S.Y.; Anderson, M.D.; Karozos, S.C.; Mang, H.E.; Knipe, N.L.; Plotkin, Z.; Sutton, T.A. The p53 inhibitor pifithrin-alpha can stimulate fibrosis in a rat model of ischemic acute kidney injury. Am. J. Physiol. Renal. Physiol. 2012, 302, F284–F291. [Google Scholar] [CrossRef] [Green Version]
- Sinha, D.; Wang, Z.; Price, V.R.; Schwartz, J.H.; Lieberthal, W. Chemical anoxia of tubular cells induces activation of c-Src and its translocation to the zonula adherens. Am. J. Physiol. Renal. Physiol. 2003, 284, F488–F497. [Google Scholar] [CrossRef]
- Wei, Q.; Dong, G.; Franklin, J.; Dong, Z. The pathological role of Bax in cisplatin nephrotoxicity. Kidney Int. 2007, 72, 53–62. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Li, L.; Livingston, M.J.; Zhang, D.; Mi, Q.; Zhang, M.; Ding, H.F.; Huo, Y.; Mei, C.; Dong, Z. p53/microRNA-214/ULK1 axis impairs renal tubular autophagy in diabetic kidney disease. J. Clin. Investig. 2020, 130, 5011–5026. [Google Scholar] [CrossRef]
- Dong, Z.; Wang, J. Hypoxia selection of death-resistant cells. A role for Bcl-X(L). J. Biol. Chem. 2004, 279, 9215–9221. [Google Scholar] [CrossRef] [Green Version]
- Wei, Q.; Sun, H.; Song, S.; Liu, Y.; Liu, P.; Livingston, M.J.; Wang, J.; Liang, M.; Mi, Q.S.; Huo, Y.; et al. MicroRNA-668 represses MTP18 to preserve mitochondrial dynamics in ischemic acute kidney injury. J. Clin. Investig. 2018, 128, 5448–5464. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, K.; Wei, Q.; Pabla, N.; Dong, G.; Mi, Q.S.; Liang, M.; Mei, C.; Dong, Z. MicroRNA-687 Induced by Hypoxia-Inducible Factor-1 Targets Phosphatase and Tensin Homolog in Renal Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. 2015, 26, 1588–1596. [Google Scholar] [CrossRef]
- Jiang, M.; Wei, Q.; Dong, G.; Komatsu, M.; Su, Y.; Dong, Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012, 82, 1271–1283. [Google Scholar] [CrossRef] [Green Version]
- Sohn, D.; Graupner, V.; Neise, D.; Essmann, F.; Schulze-Osthoff, K.; Janicke, R.U. Pifithrin-alpha protects against DNA damage-induced apoptosis downstream of mitochondria independent of p53. Cell Death Differ. 2009, 16, 869–878. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Zhang, L. The transcriptional targets of p53 in apoptosis control. Biochem. Biophys. Res. Commun. 2005, 331, 851–858. [Google Scholar] [CrossRef]
- Megyesi, J.; Udvarhelyi, N.; Safirstein, R.L.; Price, P.M. The p53-independent activation of transcription of p21 WAF1/CIP1/SDI1 after acute renal failure. Am. J. Physiol. 1996, 271, F1211–F1216. [Google Scholar] [CrossRef]
- Kodama, T.; Takehara, T.; Hikita, H.; Shimizu, S.; Shigekawa, M.; Tsunematsu, H.; Li, W.; Miyagi, T.; Hosui, A.; Tatsumi, T.; et al. Increases in p53 expression induce CTGF synthesis by mouse and human hepatocytes and result in liver fibrosis in mice. J. Clin. Investig. 2011, 121, 3343–3356. [Google Scholar] [CrossRef] [Green Version]
- Kinashi, H.; Falke, L.L.; Nguyen, T.Q.; Bovenschen, N.; Aten, J.; Leask, A.; Ito, Y.; Goldschmeding, R. Connective tissue growth factor regulates fibrosis-associated renal lymphangiogenesis. Kidney Int. 2017, 92, 850–863. [Google Scholar] [CrossRef] [Green Version]
- Venkatachalam, M.A.; Weinberg, J.M.; Kriz, W.; Bidani, A.K. Failed Tubule Recovery, AKI-CKD Transition, and Kidney Disease Progression. J. Am. Soc. Nephrol. 2015, 26, 1765–1776. [Google Scholar] [CrossRef] [Green Version]
- Samarakoon, R.; Dobberfuhl, A.D.; Cooley, C.; Overstreet, J.M.; Patel, S.; Goldschmeding, R.; Meldrum, K.K.; Higgins, P.J. Induction of renal fibrotic genes by TGF-beta1 requires EGFR activation, p53 and reactive oxygen species. Cell Signal. 2013, 25, 2198–2209. [Google Scholar] [CrossRef]
- Mao, L.; Liu, L.; Zhang, T.; Wu, X.; Zhang, T.; Xu, Y. MKL1 mediates TGF-beta-induced CTGF transcription to promote renal fibrosis. J. Cell Physiol. 2020, 235, 4790–4803. [Google Scholar] [CrossRef]
- Ferenbach, D.A.; Bonventre, J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef]
- Wang, X.; Simpson, E.R.; Brown, K.A. p53: Protection against Tumor Growth beyond Effects on Cell Cycle and Apoptosis. Cancer Res. 2015, 75, 5001–5007. [Google Scholar] [CrossRef] [Green Version]
- Engeland, K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018, 25, 114–132. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.J. p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 2020, 20, 471–480. [Google Scholar] [CrossRef]
- Olivier, M.; Eeles, R.; Hollstein, M.; Khan, M.A.; Harris, C.C.; Hainaut, P. The IARC TP53 database: New online mutation analysis and recommendations to users. Hum. Mutat. 2002, 19, 607–614. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, S.; Hu, X.; Ma, Z.; Wei, Q.; Xiang, X.; Li, S.; Wen, L.; Liang, Y.; Dong, Z. p53 in Proximal Tubules Mediates Chronic Kidney Problems after Cisplatin Treatment. Cells 2022, 11, 712. https://doi.org/10.3390/cells11040712
Fu S, Hu X, Ma Z, Wei Q, Xiang X, Li S, Wen L, Liang Y, Dong Z. p53 in Proximal Tubules Mediates Chronic Kidney Problems after Cisplatin Treatment. Cells. 2022; 11(4):712. https://doi.org/10.3390/cells11040712
Chicago/Turabian StyleFu, Shuangshuang, Xiaoru Hu, Zhengwei Ma, Qingqing Wei, Xiaohong Xiang, Siyao Li, Lu Wen, Yumei Liang, and Zheng Dong. 2022. "p53 in Proximal Tubules Mediates Chronic Kidney Problems after Cisplatin Treatment" Cells 11, no. 4: 712. https://doi.org/10.3390/cells11040712
APA StyleFu, S., Hu, X., Ma, Z., Wei, Q., Xiang, X., Li, S., Wen, L., Liang, Y., & Dong, Z. (2022). p53 in Proximal Tubules Mediates Chronic Kidney Problems after Cisplatin Treatment. Cells, 11(4), 712. https://doi.org/10.3390/cells11040712