
Early Cellular, Molecular, Morphological and Behavioral Changes in the Humanized Amyloid-Beta-Knock-In Mouse Model of Late-Onset Alzheimer’s Disease

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Humanized A-Beta-Knock-in (hAb-KI) Mice
2.2. Rotarod Test
2.3. Open-Field Test
2.4. Y-Maze Test
2.5. Morris-Water-Maze Test
2.6. qRT-PCR
2.7. Western-Blotting Analysis
2.8. Immunofluorescence Analysis
2.9. Dendritic Spine Analysis Using Golgi–cox Staining
2.10. Transmission Electron Microscopy (TEM) of Brain Mitochondria
2.11. Measurement of Soluble Aβ Levels
2.12. Mitochondrial Functional Assays
3. Results
3.1. Phenotypic Behavior
3.1.1. Rotarod Test
3.1.2. Morris Water Maze Test
3.1.3. Y-Maze Test
3.2. Gene-Expression Differences between hAbKI Mice and WT Mice
3.2.1. Mitochondrial-Dynamic Genes
3.2.2. Synaptic Genes
3.2.3. Biogenesis Genes
3.2.4. Mitophagy Genes
3.3. Immunoblotting Analysis
3.3.1. Mitochondrial-Dynamic Proteins
3.3.2. Mitochondrial Biogenesis
3.3.3. Dendritic and Synaptic Proteins
3.3.4. Mitophagy Proteins
3.3.5. Autophagy Proteins
3.3.6. Inflammatory Response Proteins
3.4. Immunofluorescence Analysis
3.5. Dendritic Spine Density
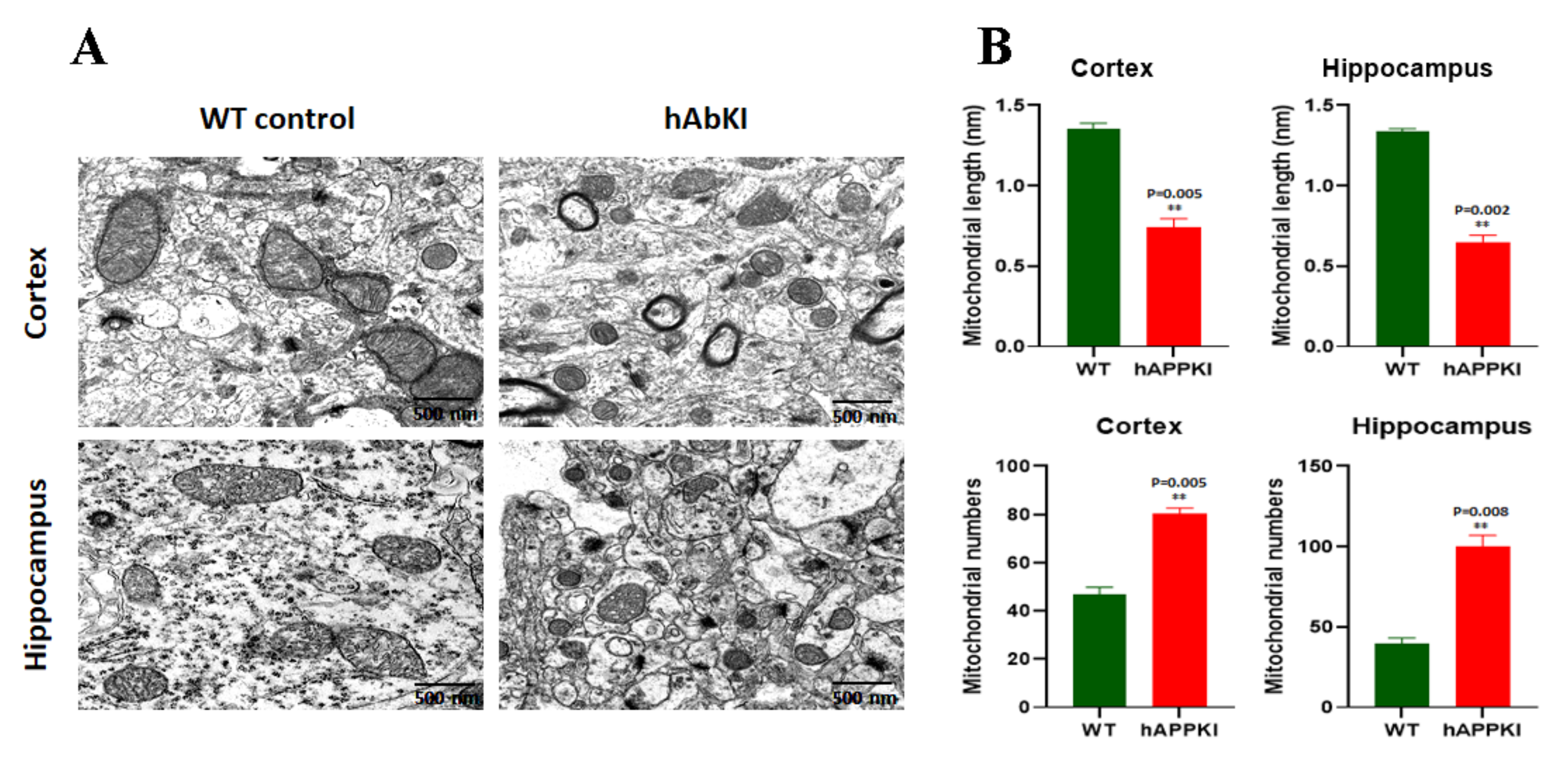
3.6. Transmission Electron Microscopy
3.7. Amyloid-Beta Levels
3.8. Mitochondrial Function
4. Discussion
4.1. mRNA Abundance and Protein Levels
4.2. Spine Density Changes and Synaptic Activity
4.3. Mitochondrial Morphology Changes
4.4. Human A-Beta Peptide Toxicity
4.5. Mitochondrial Function
4.6. Oxidative Stress
4.7. Inflammation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-beta in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 7, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 7, 789–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.H.; Beal, M.F. Amyloid-beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin; et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Morton, H.; Kshirsagar, S.; Orlov, E.; Bunquin, L.E.; Sawant, N.; Boleng, L.; George, M.; Basu, T.; Ramasubramanian, B.; Pradeepkiran, J.A.; et al. Defective mitophagy and synaptic degeneration in Alzheimer’s disease: Focus on aging, mitochondria and synapse. Free Radic. Biol. Med. 2021, 172, 652–667. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Hui, D.; Chalise, P.; Sharma, P.; Wang, X.; Andrews, S.J.; Pa, J.; Mahnken, J.D.; Morris, J.; Wilkins, H.M.; et al. Alzheimer’s Disease Neuroimaging Initiative (ADNI). Exploratory analysis of mtDNA haplogroups in two Alzheimer’s longitudinal cohorts. Alzheimers Dement. 2020, 16, 1164–1172. [Google Scholar] [CrossRef]
- Kumar, S.; Reddy, P.H. Are circulating microRNAs peripheral biomarkers for Alzheimer’s disease? Biochim. Biophys. Acta 2016, 1862, 1617–1627. [Google Scholar] [CrossRef]
- Gowda, P.; Reddy, P.H.; Kumar, S. Deregulated mitochondrial microRNAs in Alzheimer’s disease: Focus on synapse and mitochondria. Ageing Res. Rev. 2022, 73, 101529. [Google Scholar] [CrossRef]
- McGeer, P.L.; Rogers, J.; McGeer, E.G. Inflammation, Antiinflammatory Agents, and Alzheimer’s Disease: The Last 22 Years. J. Alzheimers Dis. 2016, 54, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 30, 12–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadigh-Eteghad, S.; Sabermarouf, B.; Majdi, A.; Talebi, M.; Farhoudi, M.; Mahmoudi, J. Amyloid-beta: A crucial factor in Alzheimer’s disease. Med. Princ. Pract. 2015, 24, 1–10. [Google Scholar] [CrossRef]
- Reddy, P.H. Abnormal tau, mitochondrial dysfunction, impaired axonal transport of mitochondria, and synaptic deprivation in Alzheimer’s disease. Brain Res. 2011, 1415, 136–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashrafian, H.; Zadeh, E.H.; Khan, R.H. Review on Alzheimer’s disease: Inhibition of amyloid-beta and tau tangle formation. Int. J. Biol. Macromol. 2021, 167, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Hondius, D.C.; Koopmans, F.; Leistner, C.; Pita-Illobre, D.; Peferoen-Baert, R.M.; Marbus, F.; Paliukhovich, I.; Li, K.W.; Rozemuller, A.J.M.; Hoozemans; et al. The proteome of granulovacuolar degeneration and neurofibrillary tangles in Alzheimer’s disease. Acta Neuropathol. 2021, 143, 341–358. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, A.; Reddy, P.H. Synaptic basis of Alzheimer’s disease: Focus on synaptic amyloid-beta, P-tau and mitochondria. Ageing Res. Rev. 2021, 65, 101208. [Google Scholar] [CrossRef]
- Sheladia, S.; Reddy, P.H. Age-Related Chronic Diseases and Alzheimer’s Disease in Texas: A Hispanic Focused Study. J. Alzheimers Dis. Rep. 2021, 5, 121–133. [Google Scholar] [CrossRef]
- Ayodele, T.; Rogaeva, E.; Kurup, J.T.; Beecham, G.; Reitz, C. Early-Onset Alzheimer’s Disease: What Is Missing in Research? Curr. Neurol. Neurosci. Rep. 2021, 21, 4. [Google Scholar] [CrossRef]
- Mao, P.; Reddy, P.H. Aging and amyloid-beta-induced oxidative DNA damage and mitochondrial dysfunction in Alzheimer’s disease: Implications for early intervention and therapeutics. Biochim. Biophys. Acta. 2011, 1812, 1359–1370. [Google Scholar] [CrossRef] [Green Version]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Mao, P.; Manczak, M.; Calkins, M.J.; Truong, Q.; Reddy, T.P.; Reddy, A.P.; Shirendeb, U.; Lo, H.H.; Rabinovitch, P.S.; Reddy, P.H. Mitochondria-targeted catalase reduces abnormal APP processing, amyloid β production and BACE1 in a mouse model of Alzheimer’s disease: Implications for neuroprotection and lifespan extension. Hum. Mol. Genet. 2012, 21, 2973–2990. [Google Scholar] [CrossRef]
- Safieh, M.; Korczyn, A.D.; Michaelson, D.M. ApoE4: An emerging therapeutic target for Alzheimer’s disease. BMC Med. 2019, 17, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, T.M.; Golde, T.E.; Lagier-Tourenne, C. Animal models of neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1370–1379. [Google Scholar] [CrossRef]
- Götz, J.; Bodea, L.G.; Goedert, M. Rodent models for Alzheimer disease. Nat. Rev. Neurosci. 2018, 19, 583–598. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Zheng, H. Practical considerations for choosing a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 89. [Google Scholar] [CrossRef] [PubMed]
- Baglietto-Vargas, D.; Forner, S.; Cai, L.; Martini, A.C.; Trujillo-Estrada, L.; Swarup, V.; Nguyen, M.M.T.; Do Huynh, K.; Javonillo, D.I.; Tran, K.M.; et al. Generation of a humanized Aβ expressing mouse demonstrating aspects of Alzheimer’s disease-like pathology. Nat. Commun. 2021, 12, 2421. [Google Scholar] [CrossRef]
- Kandimalla, R.; Manczak, M.; Fry, D.; Suneetha, Y.; Sesaki, H.; Reddy, P.H. Reduced dynamin-related protein 1 protects against phosphorylated Tau-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 4881–4897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijayan, M.; Bose, C.; Reddy, C.H. Protective effects of a small molecule inhibitor, DDQ against amyloid-beta in Alzheimer’s disease. Mitochondrion 2021, 59, 17–29. [Google Scholar] [CrossRef]
- Vijayan, M.; Bose, C.; Reddy, P.H. Anti-brain Aging Effects of Small Molecule Inhibitor DDQ. Mol. Neurobiol. 2021, 58, 3588–3600. [Google Scholar] [CrossRef]
- Kumar, S.; Morton, H.; Sawant, N.; Orlov, E.; Bunquin, L.E.; Pradeepkiran, J.A.; Alvir, R.; Reddy, P.H. MicroRNA-455-3p improves synaptic, cognitive functions and extends lifespan: Relevance to Alzheimer’s disease. Redox Biol. 2021, 48, 102182. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, M.; George, M.; Bunquin, L.E.; Bose, C.; Reddy, P.H. Protective effects of a small-molecule inhibitor DDQ against tau-induced toxicities in a transgenic tau mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2021, 24, ddab285. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Kandimalla, R.; Fry, D.; Sesaki, H.; Reddy, P.H. Protective effects of reduced dynamin-related protein 1 against amyloid-beta-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum. Mol. Genet. 2016, 23, 5148–5166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczak, M.; Kandimalla, R.; Yin, X.; Reddy, P.H. Hippocampal mutant APP and amyloid-beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 1332–1342. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H.; Yin, X.; Manczak, M.; Kumar, S.; Pradeepkiran, J.A.; Vijayan, M.; Reddy, A.P. Mutant APP and amyloid-beta-induced defective autophagy, mitophagy, mitochondrial structural and functional changes and synaptic damage in hippocampal neurons from Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 2502–2516. [Google Scholar] [CrossRef]
- Reddy, P.H. Mitochondrial medicine for aging and neurodegenerative diseases. Neuromol. Med. 2008, 10, 291–315. [Google Scholar] [CrossRef] [Green Version]
- Pradeepkiran, J.A.; Reddy, P.H. Defective mitophagy in Alzheimer’s disease. Ageing Res. Rev. 2020, 64, 101191. [Google Scholar] [CrossRef]
- Kandimalla, R.; Manczak, M.; Yin, X.; Wang, R.; Reddy, P.H. Hippocampal phosphorylated tau induced cognitive decline, dendritic spine loss and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2018, 27, 30–40. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H.; Mani, G.; Park, B.S.; Jacques, J.; Murdoch, G.; Whetsell, W., Jr.; Kaye, J.; Manczak, M. Differential loss of synaptic proteins in Alzheimer’s disease: Implications for synaptic dysfunction. J. Alzheimers Dis. 2005, 7, 103–117. [Google Scholar] [CrossRef]
- Kshirsagar, S.; Sawant, N.; Morton, H.; Reddy, A.P.; Reddy, P.H. Protective effects of mitophagy enhancers against amyloid-beta-induced mitochondrial and synaptic toxicities in Alzheimer disease. Hum. Mol. Genet. 2022, 31, 423–439. [Google Scholar] [CrossRef] [PubMed]
- Atwood, C.S.; Obrenovich, M.E.; Liu, T.; Chan, H.; Perry, G.; Smith, M.A.; Martins, R.N. Amyloid-beta: A chameleon walking in two worlds: A review of the trophic and toxic properties of amyloid-beta. Brain Res. Rev. 2003, 43, 1–16. [Google Scholar] [CrossRef]
- Atamna, H.; Frey, W.H., 2nd; Ko, N. Human and rodent amyloid-beta peptides differentially bind heme: Relevance to the human susceptibility to Alzheimer’s disease. Arch. Biochem. Biophys. 2009, 487, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | DNA Sequence (5′-3′) | PCR Product Size |
|---|---|---|
| Mitochondrial Structural Genes | ||
| Drp1 | Forward Primer ATGCCAGCAAGTCCACAGAA | 86 |
| Reverse Primer | ||
| Fis1 | Forward Primer CAAAGAGGAACAGCGGGACT | 95 |
| Reverse Primer ACAGCCCTCGCACATACTTT | ||
| Mfn1 | Forward Primer GCAGACAGCACATGGAGAGA | 83 |
| Reverse Primer GATCCGATTCCGAGCTTCCG | ||
| Mfn2 | Forward Primer TGCACCGCCATATAGAGGAAG | 78 |
| Reverse Primer TCTGCAGTGAACTGGCAATG | ||
| Opa1 | Forward Primer ACCTTGCCAGTTTAGCTCCC | 82 |
| Reverse Primer TTGGGACCTGCAGTGAAGAA | ||
| Mitochondrial Biogenesis Genes | ||
| PGC1α | Forward Primer GCAGTCGCAACATGCTCAAG | 83 |
| Reverse Primer GGGAACCCTTGGGGTCATTT | ||
| Nrf1 | Forward Primer AGAAACGGAAACGGCTCAT | 96 |
| Reverse Primer CATCCAACGTGGCTCTGAGT | ||
| Nrf2 | Forward Primer ATGGAGCAAGTTTGGCAGGA | 96 |
| Reverse Primer GCTGGGAACAGAGGTAGTAT | ||
| TFAM | Forward Primer TCCACAGAACAGCTACCCAA | 84 |
| Reverse Primer CCACAGGGCTGCAATTTTCC | ||
| Synaptic genes | ||
| Synaptophysin | Forward Primer CTGCGTTAAAGGGGGCACTA | 81 |
| Reverse Primer ACAGCCACGGTGACAAAGAA | ||
| PSD95 | Forward Primer CTTCATCCTTGCTGGGGGTC | 90 |
| Reverse Primer TTGCGGAGGTCAACACCATT | ||
| Synapsin 1 | Forward Primer TGAGGACATCAGTGTCGGGTAA | 64 |
| Reverse Primer GGCAATCTGCTCAAGCATAGC | ||
| Synapsin 2 | Forward Primer TCCCACTCATTGAGCAGACATACT | |
| Reverse Primer GGGAACGTAGGAAGCGTAAGC | ||
| Synaptobrevin 1 | Forward Primer TGCTGCCAAGCTAAAAAGGAA | 68 |
| Reverse Primer CAGATAGCTCCCAGCATGATCA | ||
| Synaptobrevin 2 | Forward Primer GGGACCAGAAGTTGTCGGAG | 89 |
| Reverse Primer CTTGAGCTTGGCTGCACTTG | ||
| Neurogranin | Forward Primer CTCCAAGCCAGACGACGATA | 83 |
| Reverse Primer AACTCGCCTGGATTTTGGCT | ||
| Mitophagy genes | ||
| PINK1 | Forward Primer CCATCGGGATCTCAAGTCCG | 70 |
| Reverse Primer GATCACTAGCCAGGGACAGC | ||
| Parkin | Forward Primer AGAGGTCCAGTTAAACCCACC | 90 |
| Reverse Primer GAGGGTTGCTTGTTTGCAGG | ||
| Autophagy genes | ||
| ATG5 | Forward Primer TCCATCCAAGGATGCGGTTG | 95 |
| Reverse Primer TCTGCATTTCGTTGATCACTTGAC | ||
| BCL2 | Forward Primer TCCTTCCAGCCTGAGAGCAA | 73 |
| Reverse Primer GCCTGAGAGGAGACGTCCTG | ||
| LC3B | Forward Primer TCCACTCCCATCTCCGAAGT | 94 |
| Reverse Primer TTGCTGTCCCGAATGTCTCC | ||
| Beta-actin | Forward Primer AGAAGCTGTGCTATGTTGCTCTA | 91 |
| Reverse Primer TCAGGCAGCTCATAGCTCTTC | ||
| Marker Primary Antibody—Species and Dilution | Purchased from Company, City and State | Secondary Antibody, Dilution | Purchased from Company, City and State |
|---|---|---|---|
| Drp1 Rabbit polyclonal 1:500 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
| Fis1 Rabbit polyclonal 1:500 | Protein Tech Group, Inc., Chicago, IL | Donkey anti-rabbit HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
| Mfn1 Rabbit polyclonal 1:400 | Abcam, Cambridge, MA | Donkey anti-rabbit HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
| Mfn2 Rabbit polyclonal 1:400 | Abcam, Cambridge, MA | Donkey anti-rabbit HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
| OPA1 Rabbit polyclonal 1:500 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
| SYN Rabbit monoclonal 1:400 | Abcam, Cambridge, MA | Donkey anti-rabbit HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
| PGC1a Rabbit polyclonal 1:500 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
| NRF1 Rabbit polyclonal 1:300 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
| NRF2 Rabbit polyclonal 1:300 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
| TFAM Rabbit polyclonal 1:300 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
| PINK1 Rabbit polyclonal 1:500 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
| Parkin Mouse polyclonal 1:500 | Novus Biological, Littleton, CO | Sheep anti-mouse HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
| ATG5 Rabbit Polyclonal 1: 1000 dilutions | Novus Biological, Littleton, CO | Donkey Anti-rabbit HRP 1:10,000 | GE Healthcare Amersham, Piscataway, NJ |
| LC3B Rabbit Polyclonal 1: 1000 dilutions | Novus Biological, Littleton, CO | Donkey Anti-rabbit HRP 1:10,000 | GE Healthcare Amersham, Piscataway, NJ |
| Beclin-1 Rabbit Polyclonal 1: 1000 dilutions | Novus Biological, Littleton, CO | Donkey Anti-rabbit HRP 1:10,000 | GE Healthcare Amersham, Piscataway, NJ |
| Bcl-2 Rabbit Polyclonal 1: 1000 dilutions | Novus Biological, Littleton, CO | Donkey Anti-rabbit HRP 1:10,000 | GE Healthcare Amersham, Piscataway, NJ |
| Iba1/AIF-1 2 Rabbit monoclonal 1: 1000 dilutions | Cell Signaling Technology, Inc., Danvers, MA | Donkey Anti-rabbit HRP 1:10,000 | GE Healthcare Amersham, Piscataway, NJ |
| Anti-NeuN Rabbit monoclonal 1: 1000 dilutions | Abcam, Cambridge, MA | Donkey Anti-rabbit HRP 1:10,000 | GE Healthcare Amersham, Piscataway, NJ |
| B-Actin Mouse Monoclonal 1:2000 | Millipore Sigma (Burlington, MA, USA) | Sheep anti-mouse HRP 1:10,000 | GE Healthcare Amersham, Piscataway, NJ |
| Marker Primary Antibody—Species and Dilution | Purchased from Company, City and State | Secondary Antibody, Dilution | Purchased from Company, City and State |
|---|---|---|---|
| Drp1 Rabbit polyclonal 1:100 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:200 | Invitrogen, Waltham, MA |
| Fis1 Rabbit polyclonal 1:100 | Protein Tech Group, Inc., Chicago, IL | Donkey anti-rabbit HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| Mfn1 Rabbit polyclonal 1:100 | Abcam, Cambridge, MA | Donkey anti-rabbit HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| Mfn2 Rabbit polyclonal 1:100 | Abcam, Cambridge, MA | Donkey anti-rabbit HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| OPA1 Rabbit polyclonal 1:100 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| SYN Rabbit monoclonal 1:400 | Abcam, Cambridge, MA | Donkey anti-rabbit HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| PGC1a Rabbit polyclonal 1:100 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| NRF1 Rabbit polyclonal 1:100 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| NRF2 Rabbit polyclonal 1:100 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| TFAM Rabbit polyclonal 1:100 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| PINK1 Rabbit polyclonal 1:100 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| Parkin Mouse polyclonal 1:100 | Novus Biological, Littleton, CO | Sheep anti-mouse HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| ATG5 Rabbit Polyclonal 1: 100 dilutions | Novus Biological, Littleton, CO | Donkey Anti-rabbit HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| LC3B Rabbit Polyclonal 1: 100 dilutions | Novus Biological, Littleton, CO | Donkey Anti-rabbit HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| Beclin-1 Rabbit Polyclonal 1: 100 dilutions | Novus Biological, Littleton, CO | Donkey Anti-rabbit HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| Bcl-2 Rabbit Polyclonal 1: 100 dilutions | Novus Biological, Littleton, CO | Donkey Anti-rabbit HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| Iba1/AIF-1 2 Rabbit monoclonal 1: 100 dilutions | Cell Signaling Technology, Inc., Danvers, MA | Donkey Anti-rabbit HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| Anti-NeuN Rabbit monoclonal 1: 100 dilutions | Abcam, Cambridge, MA | Donkey Anti-rabbit HRP 1:200 | GE Healthcare Amersham, Piscataway, NJ |
| Genes | mRNA Fold Change in AAPKI Mice | |
|---|---|---|
| Mitochondrial genes | Drp1 | 1.8 *** |
| Fis1 | 1.69 ** | |
| Mfn1 | −2 ** | |
| Mfn2 | −1.33 * | |
| OPA1 | −1.38 | |
| Biogenesis genes | PGC1 alpha | −1.7 * |
| Nrf1 | −1.4 | |
| Nrf2 | −1.36 | |
| TFAM | −2 * | |
| Synaptic genes | Synaptophysin | −2.04 ** |
| PSD95 | −1.1 * | |
| Snap25 | −2.27 ** | |
| Synapsin 1 | −1.4 * | |
| Synapsin 2 | −5 *** | |
| Synaptobrevin 1 | −1.26 | |
| Synaptobrevin 2 | −2.4 * | |
| Neurogranin | −1.54 * | |
| Mitophagy genes | PINK1 | −2 ** |
| Parkin | −1.16 | |
| Autophagy genes | ATG5 | −1.6 ** |
| BCL2 | −1.25 * | |
| LC3B | −1.33 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kshirsagar, S.; Alvir, R.V.; Hindle, A.; Kumar, S.; Vijayan, M.; Pradeepkiran, J.A.; Reddy, A.P.; Ramasubramanian, B.; Reddy, P.H. Early Cellular, Molecular, Morphological and Behavioral Changes in the Humanized Amyloid-Beta-Knock-In Mouse Model of Late-Onset Alzheimer’s Disease. Cells 2022, 11, 733. https://doi.org/10.3390/cells11040733
Kshirsagar S, Alvir RV, Hindle A, Kumar S, Vijayan M, Pradeepkiran JA, Reddy AP, Ramasubramanian B, Reddy PH. Early Cellular, Molecular, Morphological and Behavioral Changes in the Humanized Amyloid-Beta-Knock-In Mouse Model of Late-Onset Alzheimer’s Disease. Cells. 2022; 11(4):733. https://doi.org/10.3390/cells11040733
Chicago/Turabian StyleKshirsagar, Sudhir, Rainier Vladlen Alvir, Ashly Hindle, Subodh Kumar, Murali Vijayan, Jangampalli Adi Pradeepkiran, Arubala P. Reddy, Bhagavathi Ramasubramanian, and P. Hemachandra Reddy. 2022. "Early Cellular, Molecular, Morphological and Behavioral Changes in the Humanized Amyloid-Beta-Knock-In Mouse Model of Late-Onset Alzheimer’s Disease" Cells 11, no. 4: 733. https://doi.org/10.3390/cells11040733
APA StyleKshirsagar, S., Alvir, R. V., Hindle, A., Kumar, S., Vijayan, M., Pradeepkiran, J. A., Reddy, A. P., Ramasubramanian, B., & Reddy, P. H. (2022). Early Cellular, Molecular, Morphological and Behavioral Changes in the Humanized Amyloid-Beta-Knock-In Mouse Model of Late-Onset Alzheimer’s Disease. Cells, 11(4), 733. https://doi.org/10.3390/cells11040733