
A Transcriptional Link between HER2, JAM-A and FOXA1 in Breast Cancer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Gene Silencing
2.3. RNA Extraction, Reverse Transcription and Quantitative Real-Time Polymerase Chain Reaction
2.4. In Silico Transcription Factor Binding Determination
2.5. Experimental Protein–DNA Binding Assays
2.6. Protein Extraction and Western Immunoblotting
2.7. Statistical Analysis
3. Results
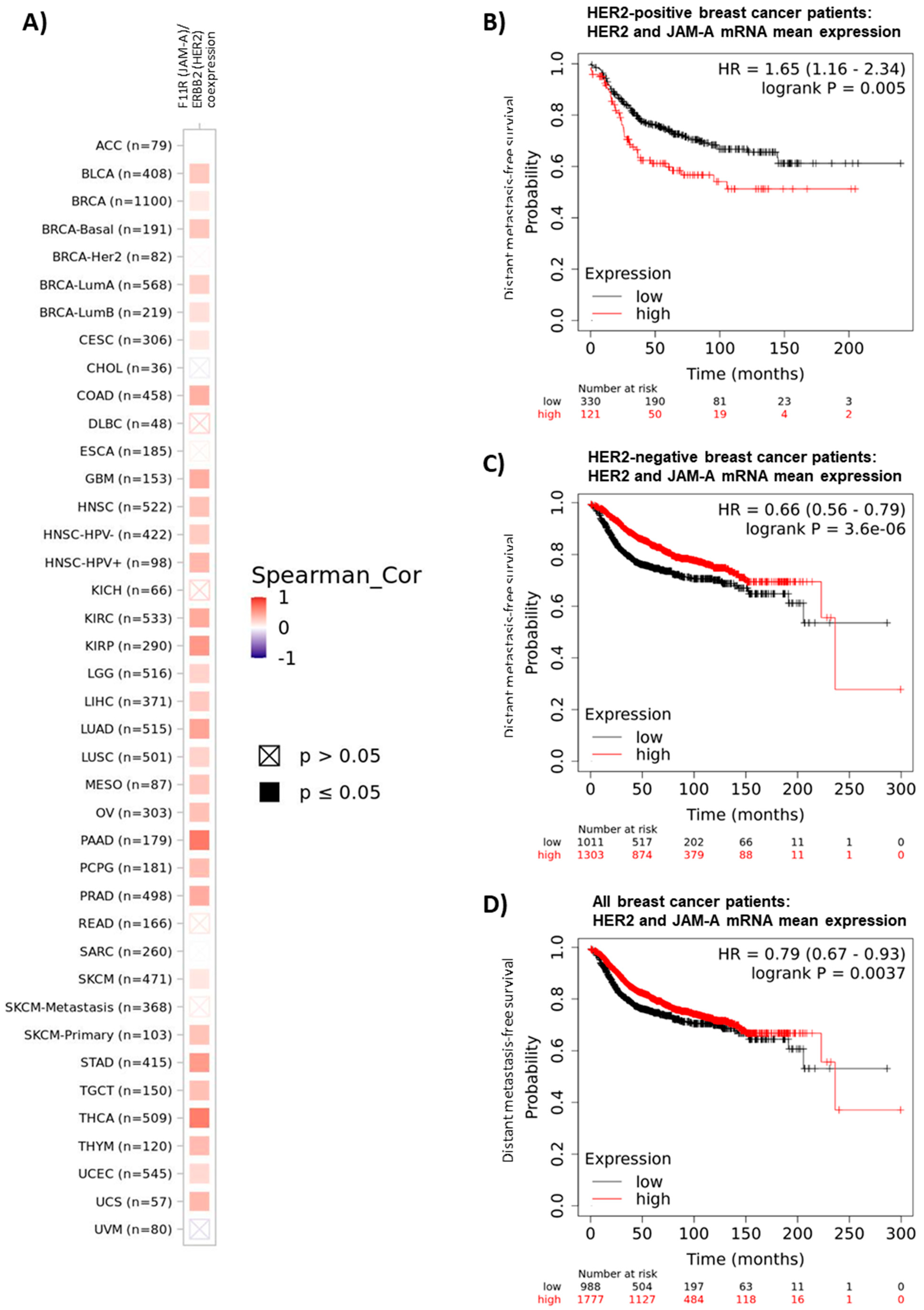
3.1. High Gene Expression of JAM-A and HER2 Correlate in Many Cancer Types
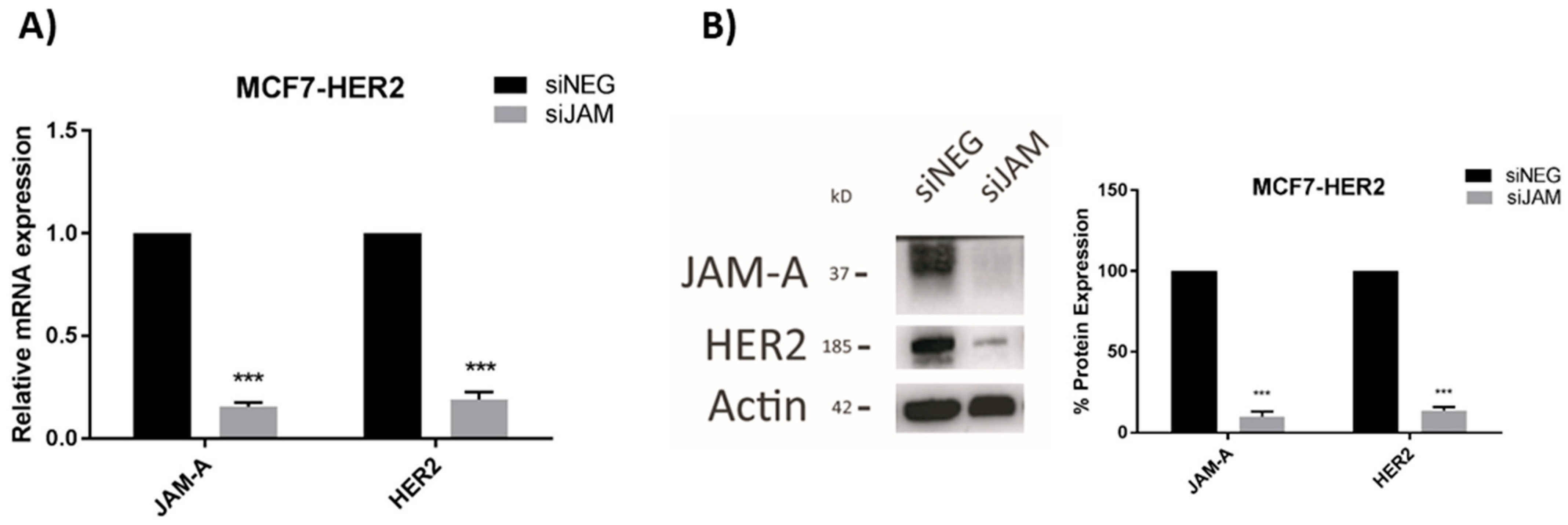
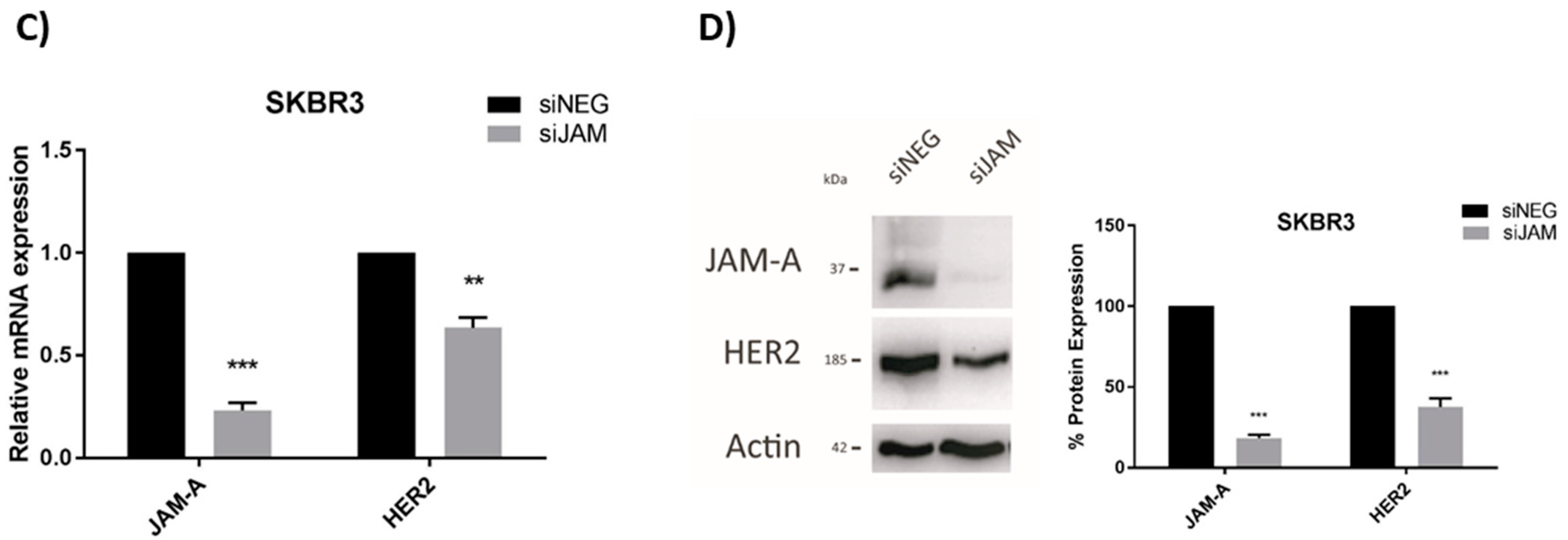
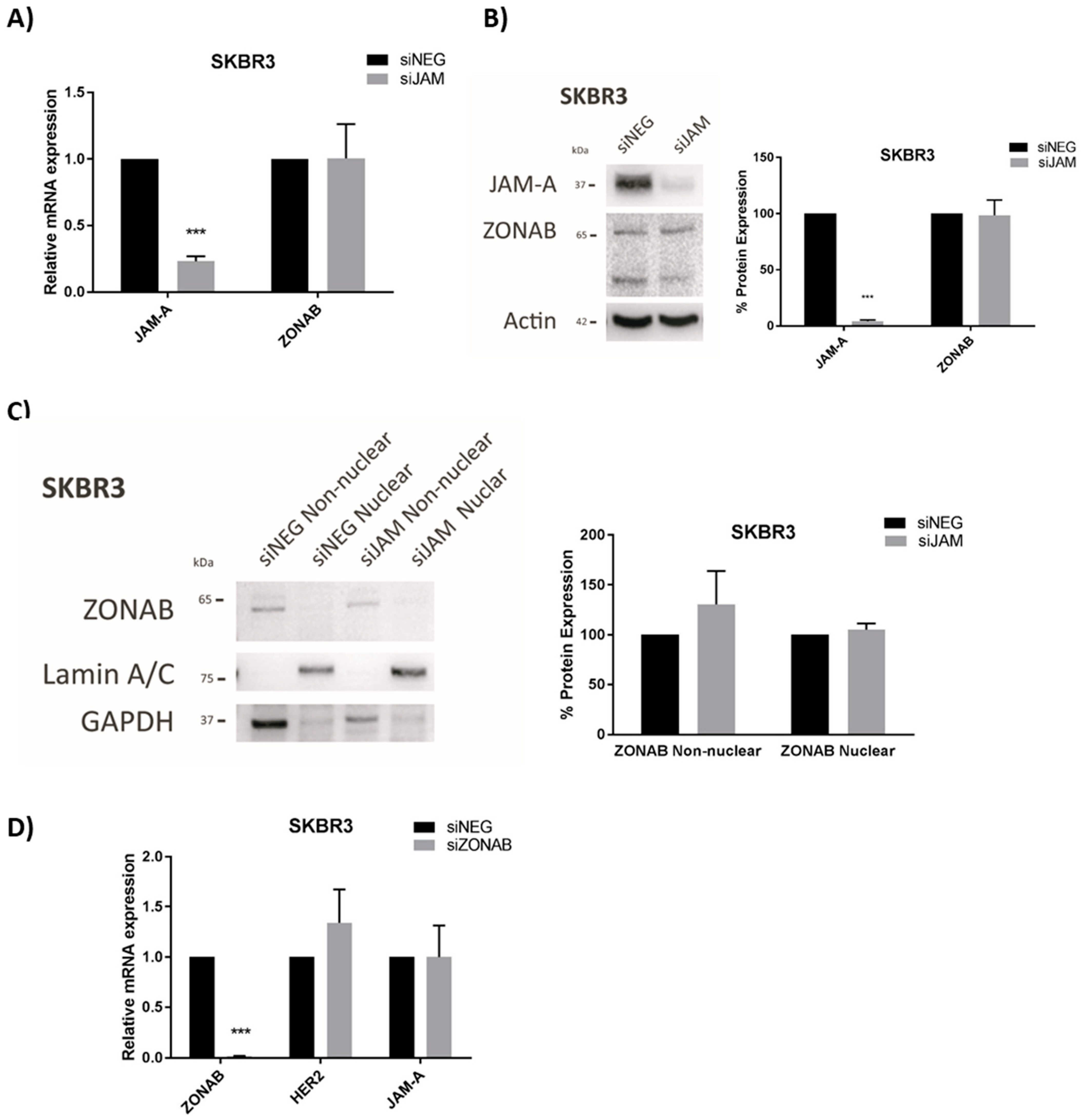
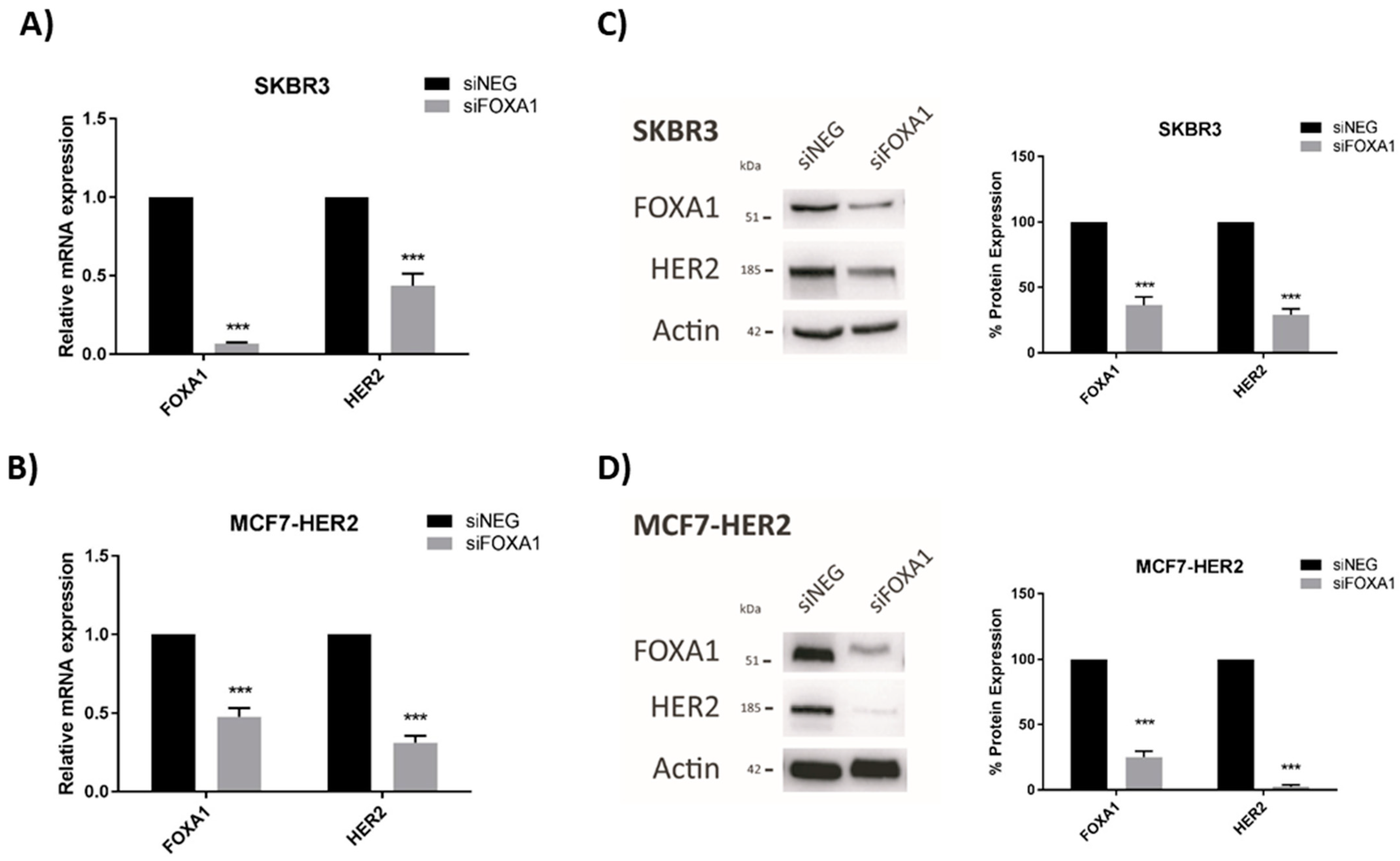
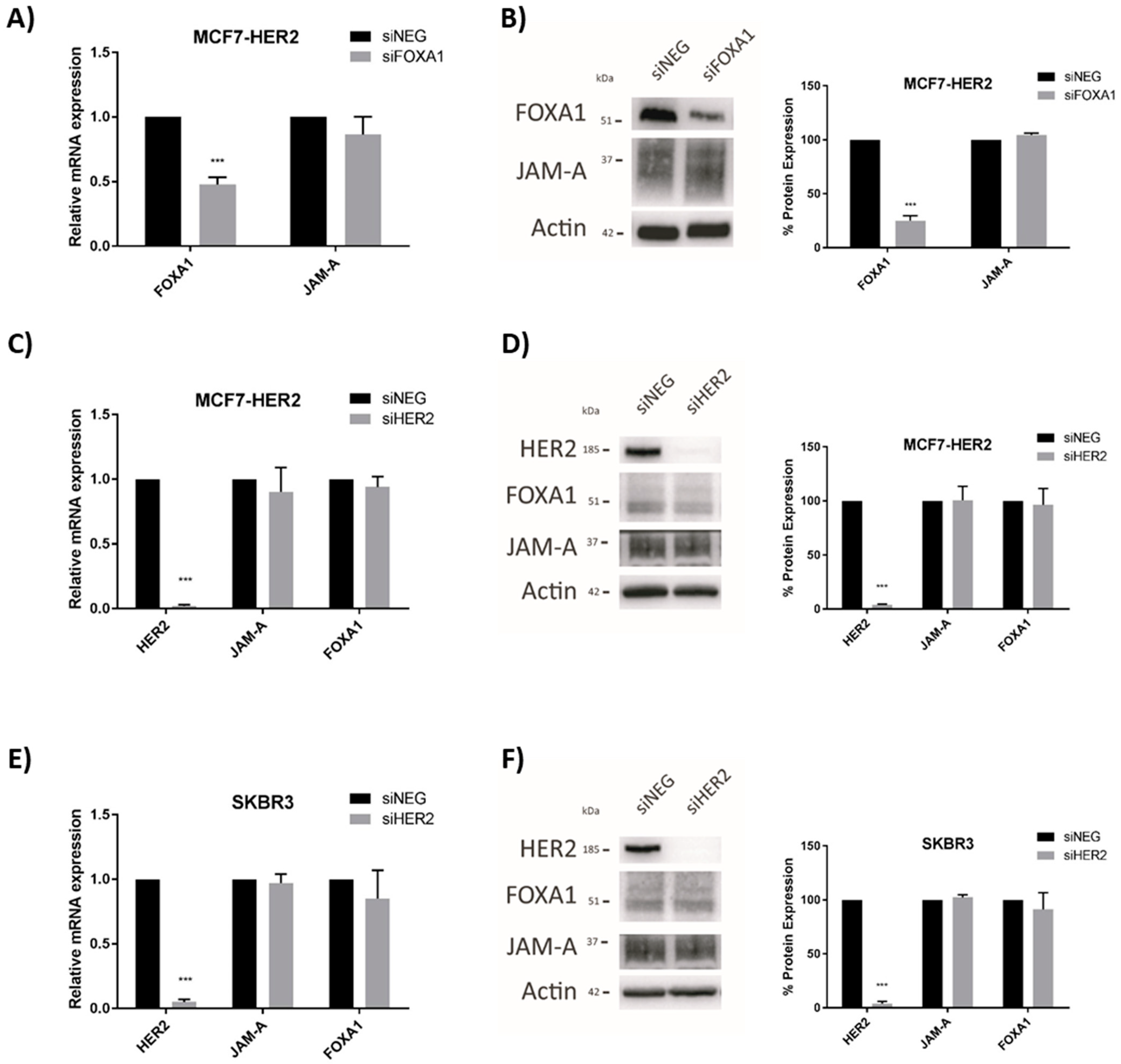
3.2. HER2 Gene and Protein Expression Levels Are Sensitive to Alterations in JAM-A or FOXA1 in Breast Cancer Cells
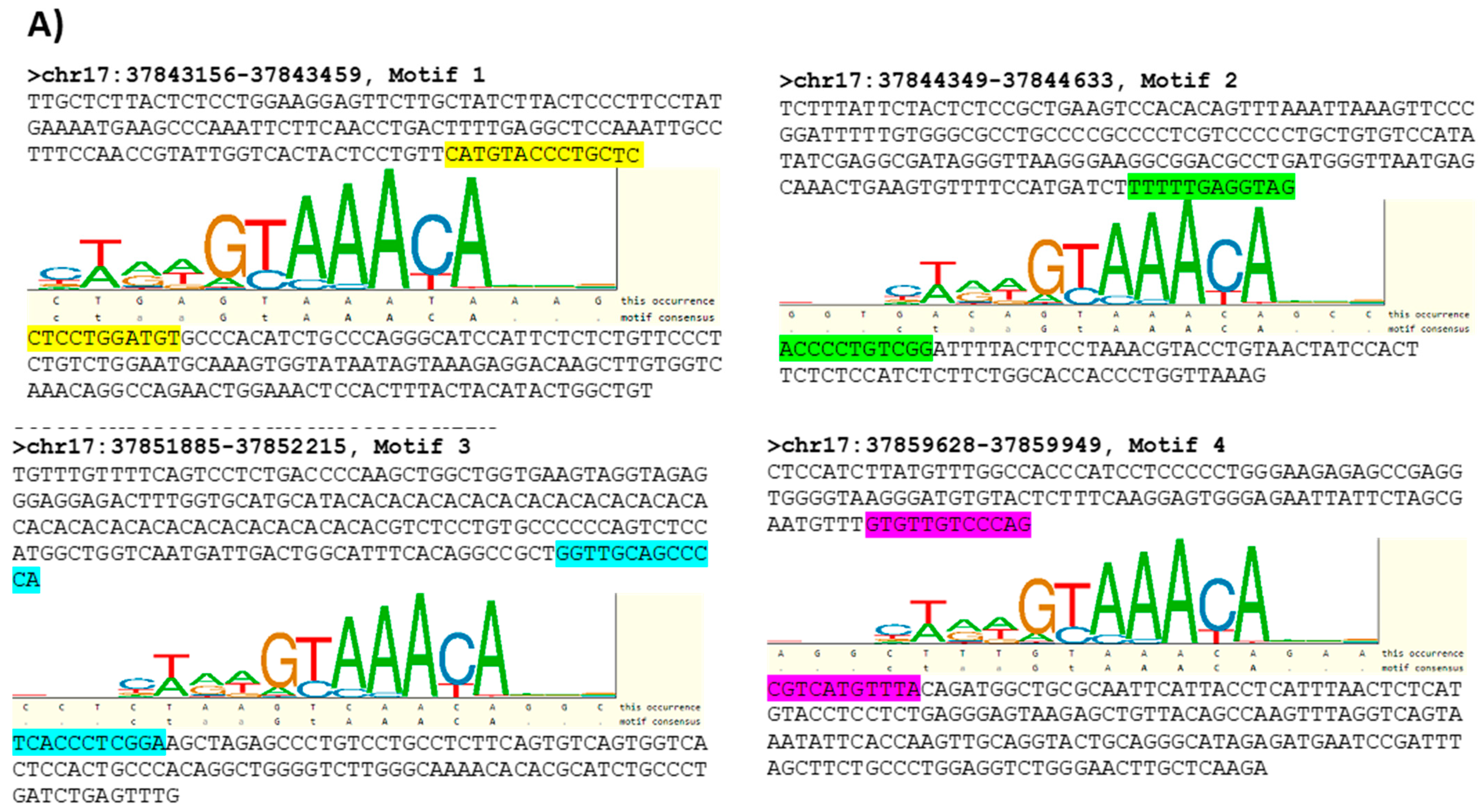
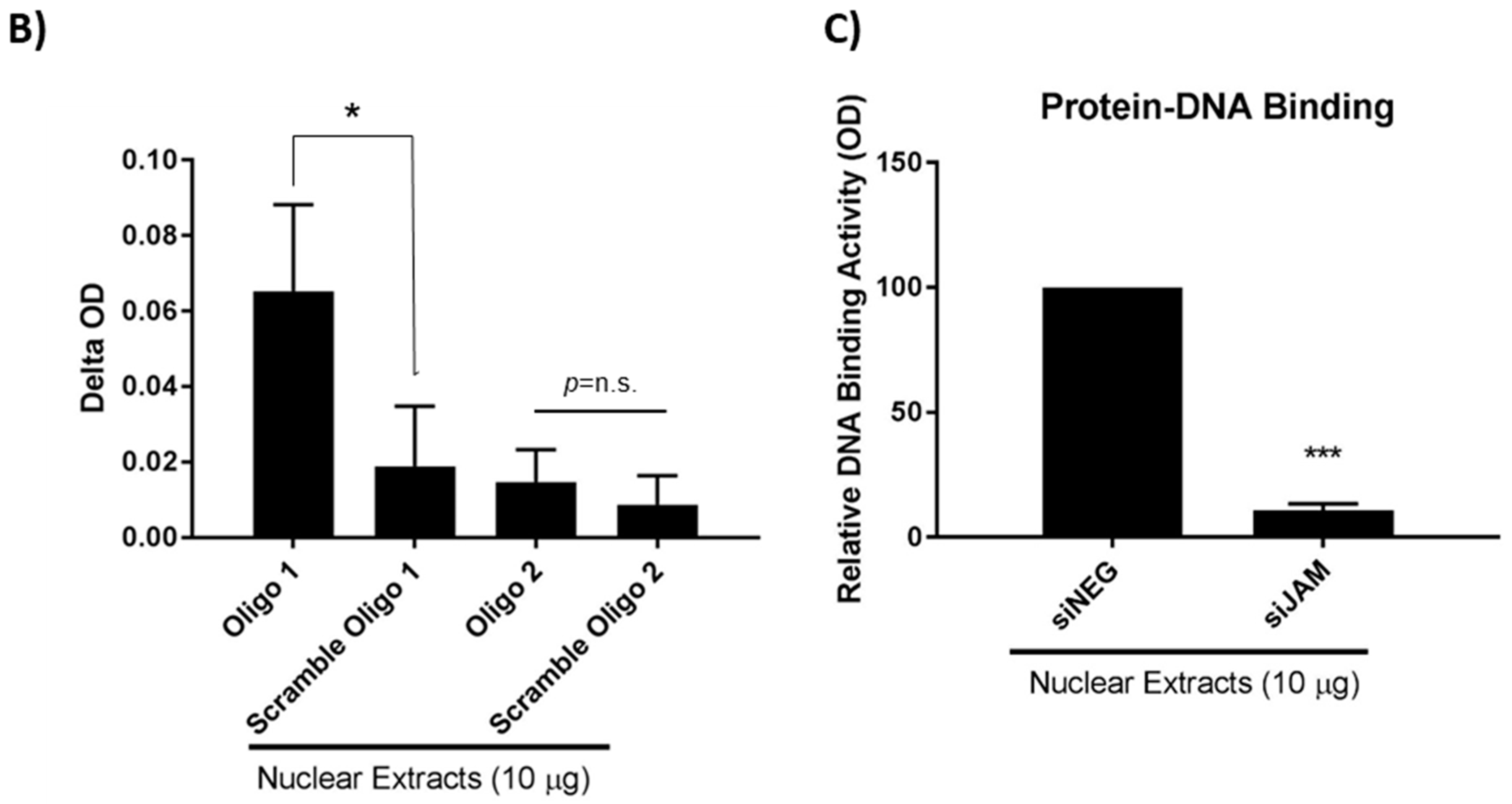
3.3. Characterizing FOXA1 Binding Sites in the HER2 Gene Proximal Promoter
3.4. Coincident High Gene Expression of JAM-A, HER2 and FOXA1 Is Associated with Poor Survival in HER2-Positive Breast and Gastric Cancer Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harries, M.; Smith, I. The development and clinical use of trastuzumab (Herceptin). Endocr. Relat. Cancer 2002, 9, 75–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slamon, D.; Eiermann, W.; Robert, N.; Pienkowski, T.; Martin, M.; Press, M.; Mackey, J.; Glaspy, J.; Chan, A.; Pawlicki, M.; et al. Adjuvant trastuzumab in HER2-positive breast cancer. N. Engl. J. Med. 2011, 365, 1273–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, K.; McSherry, E.A.; Hudson, L.; Kay, E.W.; Hill, A.D.; Young, L.S.; Hopkins, A.M. Junctional adhesion molecule-A is co-expressed with HER2 in breast tumors and acts as a novel regulator of HER2 protein degradation and signaling. Oncogene 2013, 32, 2799–2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prota, A.E.; Campbell, J.A.; Schelling, P.; Forrest, J.C.; Watson, M.J.; Peters, T.R.; Aurrand-Lions, M.; Imhof, B.A.; Dermody, T.S.; Stehle, T. Crystal structure of human junctional adhesion molecule 1: Implications for reovirus binding. Proc. Natl. Acad. Sci. USA 2003, 100, 5366–5371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Nusrat, A.; Schnell, F.J.; Reaves, T.A.; Walsh, S.; Pochet, M.; Parkos, C.A. Human junction adhesion molecule regulates tight junction resealing in epithelia. J. Cell Sci. 2000, 113, 2363–2374. [Google Scholar] [CrossRef]
- Garrido-Urbani, S.; Bradfield, P.F.; Imhof, B.A. Tight junction dynamics: The role of junctional adhesion molecules (JAMs). Cell Tissue Res. 2014, 355, 701–715. [Google Scholar] [CrossRef] [Green Version]
- McSherry, E.A.; McGee, S.F.; Jirstrom, K.; Doyle, E.M.; Brennan, D.J.; Landberg, G.; Dervan, P.A.; Hopkins, A.M.; Gallagher, W.M. JAM-A expression positively correlates with poor prognosis in breast cancer patients. Int. J. Cancer 2009, 125, 1343–1351. [Google Scholar] [CrossRef]
- Murakami, M.; Giampietro, C.; Giannotta, M.; Corada, M.; Torselli, I.; Orsenigo, F.; Cocito, A.; d’Ario, G.; Mazzarol, G.; Confalonieri, S.; et al. Abrogation of junctional adhesion molecule-A expression induces cell apoptosis and reduces breast cancer progression. PLoS ONE 2011, 6, e21242. [Google Scholar] [CrossRef] [Green Version]
- Rosager, A.M.; Sørensen, M.D.; Dahlrot, R.H.; Boldt, H.B.; Hansen, S.; Lathia, J.D.; Kristensen, B.W. Expression and prognostic value of JAM-A in gliomas. J. Neuro-Oncol. 2017, 135, 107–117. [Google Scholar] [CrossRef]
- Tian, Y.; Tian, Y.; Zhang, W.; Wei, F.; Yang, J.; Luo, X.; Zhou, T.; Hou, B.; Qian, S.; Deng, X.; et al. Junctional adhesion molecule-A, an epithelial-mesenchymal transition inducer, correlates with metastasis and poor prognosis in human nasopharyngeal cancer. Carcinogenesis 2015, 36, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Ikeo, K.; Oshima, T.; Shan, J.; Matsui, H.; Tomita, T.; Fukui, H.; Watari, J.; Miwa, H. Junctional adhesion molecule-A promotes proliferation and inhibits apoptosis of gastric cancer. Hepatogastroenterology 2015, 62, 540–545. [Google Scholar] [PubMed]
- Zhang, M.; Luo, W.; Huang, B.; Liu, Z.; Sun, L.; Zhang, Q.; Qiu, X.; Xu, K.; Wang, E. Overexpression of JAM-A in Non-Small Cell Lung Cancer Correlates with Tumor Progression. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leech, A.O.; Vellanki, S.H.; Rutherford, E.J.; Keogh, A.; Jahns, H.; Hudson, L.; O’Donovan, N.; Sabri, S.; Abdulkarim, B.; Sheehan, K.M.; et al. Cleavage of the extracellular domain of junctional adhesion molecule-A is associated with resistance to anti-HER2 therapies in breast cancer settings. Breast Cancer Res. 2018, 20, 140. [Google Scholar] [CrossRef] [PubMed]
- Marx, C.; Held, J.M.; Gibson, B.W.; Benz, C.C. ErbB2 trafficking and degradation associated with K48 and K63 polyubiquitination. Cancer Res. 2010, 70, 3709–3717. [Google Scholar] [CrossRef] [Green Version]
- Vellanki, S.H.; Cruz, R.G.B.; Jahns, H.; Hudson, L.; Sette, G.; Eramo, A.; Hopkins, A.M. Natural compound Tetrocarcin-A downregulates Junctional Adhesion Molecule-A in conjunction with HER2 and inhibitor of apoptosis proteins and inhibits tumor cell growth. Cancer Lett. 2019, 440–441, 23–34. [Google Scholar] [CrossRef]
- Scott, G.K.; Goga, A.; Bhaumik, D.; Berger, C.E.; Sullivan, C.S.; Benz, C.C. Coordinate suppression of ERBB2 and ERBB3 by enforced expression of micro-RNA miR-125a or miR-125b. J. Biol. Chem. 2007, 282, 1479–1486. [Google Scholar] [CrossRef] [Green Version]
- Leivonen, S.K.; Sahlberg, K.K.; Makela, R.; Due, E.U.; Kallioniemi, O.; Borresen-Dale, A.L.; Perala, M. High-throughput screens identify microRNAs essential for HER2 positive breast cancer cell growth. Mol. Oncol. 2014, 8, 93–104. [Google Scholar] [CrossRef]
- Cruz, R.G.B.; Madden, S.F.; Richards, C.E.; Vellanki, S.H.; Jahns, H.; Hudson, L.; Fay, J.; O’Farrell, N.; Sheehan, K.; Jirstrom, K.; et al. Human Epidermal Growth Factor Receptor-3 Expression Is Regulated at Transcriptional Level in Breast Cancer Settings by Junctional Adhesion Molecule-A via a Pathway Involving Beta-Catenin and FOXA1. Cancers 2021, 13, 871. [Google Scholar] [CrossRef]
- Ni, M.; Chen, Y.; Lim, E.; Wimberly, H.; Bailey, S.T.; Imai, Y.; Rimm, D.L.; Liu, X.S.; Brown, M. Targeting androgen receptor in estrogen receptor-negative breast cancer. Cancer Cell 2011, 20, 119–131. [Google Scholar] [CrossRef] [Green Version]
- Bernardo, G.M.; Bebek, G.; Ginther, C.L.; Sizemore, S.T.; Lozada, K.L.; Miedler, J.D.; Anderson, L.A.; Godwin, A.K.; Abdul-Karim, F.W.; Slamon, D.J.; et al. FOXA1 represses the molecular phenotype of basal breast cancer cells. Oncogene 2013, 32, 554–563. [Google Scholar] [CrossRef] [Green Version]
- Naderi, A.; Meyer, M.; Dowhan, D.H. Cross-regulation between FOXA1 and ErbB2 signaling in estrogen receptor-negative breast cancer. Neoplasia 2012, 14, 283–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browne, B.C.; Crown, J.; Venkatesan, N.; Duffy, M.J.; Clynes, M.; Slamon, D.; O’Donovan, N. Inhibition of IGF1R activity enhances response to trastuzumab in HER-2-positive breast cancer cells. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2011, 22, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Severson, E.A.; Jiang, L.; Ivanov, A.I.; Mandell, K.J.; Nusrat, A.; Parkos, C.A. Cis-dimerization Mediates Function of Junctional Adhesion Molecule A. Mol. Biol. Cell 2008, 19, 1862–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chazin, V.R.; Kaleko, M.; Miller, A.D.; Slamon, D.J. Transformation mediated by the human HER-2 gene independent of the epidermal growth factor receptor. Oncogene 1992, 7, 1859–1866. [Google Scholar]
- Pegram, M.D.; Finn, R.S.; Arzoo, K.; Beryt, M.; Pietras, R.J.; Slamon, D.J. The effect of HER-2/neu overexpression on chemotherapeutic drug sensitivity in human breast and ovarian cancer cells. Oncogene 1997, 15, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Wingender, E.; Dietze, P.; Karas, H.; Knüppel, R. TRANSFAC: A database on transcription factors and their DNA binding sites. Nucleic Acids Res. 1996, 24, 238–241. [Google Scholar] [CrossRef] [Green Version]
- Elnitski, L.; Riemer, C.; Petrykowska, H.; Florea, L.; Schwartz, S.; Miller, W.; Hardison, R. PipTools: A computational toolkit to annotate and analyze pairwise comparisons of genomic sequences. Genomics 2002, 80, 681–690. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Li, B.; Severson, E.; Pignon, J.C.; Zhao, H.; Li, T.; Novak, J.; Jiang, P.; Shen, H.; Aster, J.C.; Rodig, S.; et al. Comprehensive analyses of tumor immunity: Implications for cancer immunotherapy. Genome Biol. 2016, 17, 174. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Gyorffy, B. Survival analysis across the entire transcriptome identifies biomarkers with the highest prognostic power in breast cancer. Comput. Struct. Biotechnol. J. 2021, 19, 4101–4109. [Google Scholar] [CrossRef] [PubMed]
- Balda, M.S.; Matter, K. The tight junction protein ZO-1 and an interacting transcription factor regulate ErbB-2 expression. Embo J. 2000, 19, 2024–2033. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, G.; Martinez-Estrada, O.M.; Orsenigo, F.; Cordenonsi, M.; Citi, S.; Dejana, E. Interaction of junctional adhesion molecule with the tight junction components ZO-1, cingulin, and occludin. J. Biol. Chem. 2000, 275, 20520–20526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandell, K.J.; Parkos, C.A. The JAM family of proteins. Adv. Drug. Deliv. Rev. 2005, 57, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Czubak-Prowizor, K.; Babinska, A.; Swiatkowska, M. The F11 Receptor (F11R)/Junctional Adhesion Molecule-A (JAM-A) (F11R/JAM-A) in cancer progression. Mol. Cell Biochem. 2021, 477, 79–98. [Google Scholar] [CrossRef] [PubMed]
- Naik, M.U.; Naik, T.U.; Suckow, A.T.; Duncan, M.K.; Naik, U.P. Attenuation of junctional adhesion molecule-A is a contributing factor for breast cancer cell invasion. Cancer Res. 2008, 68, 2194–2203. [Google Scholar] [CrossRef] [Green Version]
- Leech, A.O.; Cruz, R.G.; Hill, A.D.; Hopkins, A.M. Paradigms lost-an emerging role for over-expression of tight junction adhesion proteins in cancer pathogenesis. Ann. Transl. Med. 2015, 3, 184. [Google Scholar] [CrossRef]
- Lanczky, A.; Nagy, A.; Bottai, G.; Munkacsy, G.; Szabo, A.; Santarpia, L.; Gyorffy, B. miRpower: A web-tool to validate survival-associated miRNAs utilizing expression data from 2178 breast cancer patients. Breast Cancer Res. Treat. 2016, 160, 439–446. [Google Scholar] [CrossRef]
- Engstrom, M.D.; Pfleger, B.F. Transcription control engineering and applications in synthetic biology. Synth. Syst. Biotechnol. 2017, 2, 176–191. [Google Scholar] [CrossRef]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef] [Green Version]
- Balda, M.S.; Garrett, M.D.; Matter, K. The ZO-1-associated Y-box factor ZONAB regulates epithelial cell proliferation and cell density. J. Cell Biol. 2003, 160, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, D.; Sircoulomb, F.; Imbert, J. A reason why the ERBB2 gene is amplified and not mutated in breast cancer. Cancer Cell Int. 2009, 9, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, Y.C.; Wang, Y.; Da Silva, N.; Kim, B.; Diao, R.Y.; Hill, E.; Brown, D.; Chan, H.C.; Breton, S. CFTR interacts with ZO-1 to regulate tight junction assembly and epithelial differentiation through the ZONAB pathway. J. Cell Sci. 2014, 127, 4396–4408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannenhalli, S.; Kaestner, K.H. The evolution of Fox genes and their role in development and disease. Nat. Rev. Genet. 2009, 10, 233–240. [Google Scholar] [CrossRef] [Green Version]
- Costa, R.H.; Grayson, D.R.; Darnell, J.E., Jr. Multiple hepatocyte-enriched nuclear factors function in the regulation of transthyretin and alpha 1-antitrypsin genes. Mol. Cell Biol. 1989, 9, 1415–1425. [Google Scholar]
- Kaestner, K.H. The FoxA factors in organogenesis and differentiation. Curr. Opin. Genet. Dev. 2010, 20, 527–532. [Google Scholar] [CrossRef] [Green Version]
- Bang, Y.J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Doane, A.S.; Danso, M.; Lal, P.; Donaton, M.; Zhang, L.; Hudis, C.; Gerald, W.L. An estrogen receptor-negative breast cancer subset characterized by a hormonally regulated transcriptional program and response to androgen. Oncogene 2006, 25, 3994–4008. [Google Scholar] [CrossRef] [Green Version]
- Farmer, P.; Bonnefoi, H.; Becette, V.; Tubiana-Hulin, M.; Fumoleau, P.; Larsimont, D.; Macgrogan, G.; Bergh, J.; Cameron, D.; Goldstein, D.; et al. Identification of molecular apocrine breast tumours by microarray analysis. Oncogene 2005, 24, 4660–4671. [Google Scholar] [CrossRef] [Green Version]
- Overdier, D.G.; Porcella, A.; Costa, R.H. The DNA-binding specificity of the hepatocyte nuclear factor 3/forkhead domain is influenced by amino-acid residues adjacent to the recognition helix. Mol. Cell Biol. 1994, 14, 2755–2766. [Google Scholar] [CrossRef]
- Cirillo, L.A.; Lin, F.R.; Cuesta, I.; Friedman, D.; Jarnik, M.; Zaret, K.S. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol. Cell 2002, 9, 279–289. [Google Scholar] [CrossRef]
- Bernardo, G.M.; Keri, R.A. FOXA1: A transcription factor with parallel functions in development and cancer. Biosci. Rep. 2012, 32, 113–130. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.B.; Sharma, A.; Smith, J.J.; Krishnan, M.; Chen, X.; Eschrich, S.; Washington, M.K.; Yeatman, T.J.; Beauchamp, R.D.; Dhawan, P. Claudin-1 up-regulates the repressor ZEB-1 to inhibit E-cadherin expression in colon cancer cells. Gastroenterology 2011, 141, 2140–2153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetsch, L.; Haeuw, J.F.; Beau-Larvor, C.; Gonzalez, A.; Zanna, L.; Malissard, M.; Lepecquet, A.M.; Robert, A.; Bailly, C.; Broussas, M.; et al. A novel role for junctional adhesion molecule-A in tumor proliferation: Modulation by an anti-JAM-A monoclonal antibody. Int. J. Cancer 2013, 132, 1463–1474. [Google Scholar] [CrossRef]
- Bednarek, R.; Selmi, A.; Wojkowska, D.; Karolczak, K.; Popielarski, M.; Stasiak, M.; Salifu, M.O.; Babinska, A.; Swiatkowska, M. Functional inhibition of F11 receptor (F11R/junctional adhesion molecule-A/JAM-A) activity by a F11R-derived peptide in breast cancer and its microenvironment. Breast Cancer Res. Treat. 2020, 179, 325–335. [Google Scholar] [CrossRef] [Green Version]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cruz, R.G.B.; Madden, S.F.; Brennan, K.; Hopkins, A.M. A Transcriptional Link between HER2, JAM-A and FOXA1 in Breast Cancer. Cells 2022, 11, 735. https://doi.org/10.3390/cells11040735
Cruz RGB, Madden SF, Brennan K, Hopkins AM. A Transcriptional Link between HER2, JAM-A and FOXA1 in Breast Cancer. Cells. 2022; 11(4):735. https://doi.org/10.3390/cells11040735
Chicago/Turabian StyleCruz, Rodrigo G. B., Stephen F. Madden, Kieran Brennan, and Ann M. Hopkins. 2022. "A Transcriptional Link between HER2, JAM-A and FOXA1 in Breast Cancer" Cells 11, no. 4: 735. https://doi.org/10.3390/cells11040735
APA StyleCruz, R. G. B., Madden, S. F., Brennan, K., & Hopkins, A. M. (2022). A Transcriptional Link between HER2, JAM-A and FOXA1 in Breast Cancer. Cells, 11(4), 735. https://doi.org/10.3390/cells11040735