
Metabolic Contribution and Cerebral Blood Flow Regulation by Astrocytes in the Neurovascular Unit


{kind=link}
{kind=link}
Abstract
:1. Introduction
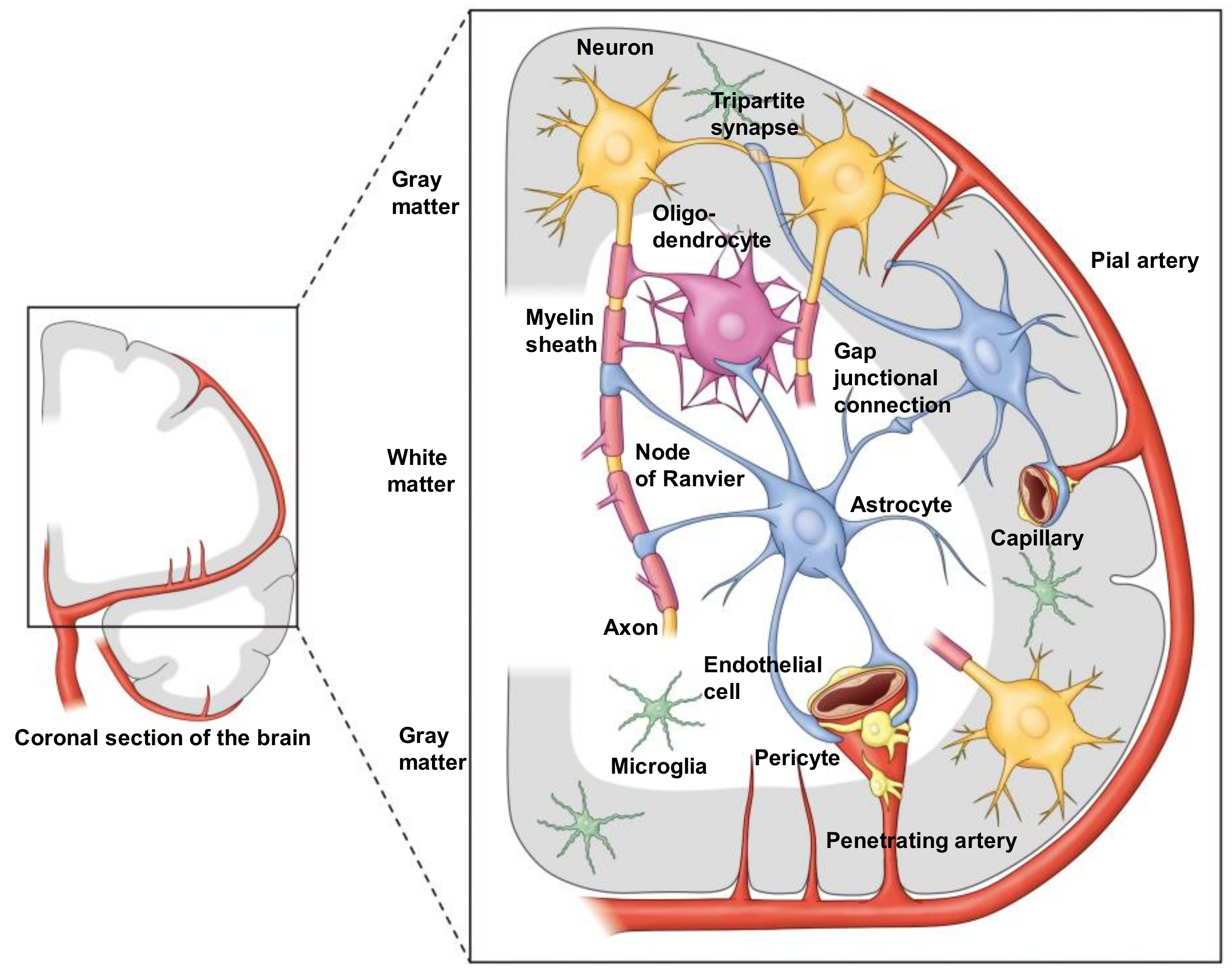
2. Construction of the Virtual Concept of an NVU
3. Current Status of Research on the NVU and the Positioning of Astrocytes
4. Anatomical Features of Astrocytes in the NVU
5. Regulation of Cerebral Blood Flow by Astrocytes in the NVU
6. Group I Metabolic Glutamate Receptors in the Astrocytes
7. Epoxyeicosatrienoic Acids
8. Constriction of the Cerebral Microvessels by Astrocytes
9. Functional Hyperemia at the Capillary Level
10. Glymphatic System and Cerebral Circulation and Metabolism
11. Glucose Metabolic Compartmentalization Formed by Cell Types in the NVU
12. Astrocyte–Neuron Lactate Shuttle Model
13. Link between Glucose Metabolism, Lipid Metabolism and Ketone Bodies
14. D-Serine and L-Serine
15. Mitochondrial Transfer of Astrocytes and Neurons
16. Conclusions and Issues That Remain to Be Resolved
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 3PGDH | 3-phosphoglyceratedehydrogenase |
| 20-HETE | 20-hydroxyeicosatetraenoic acid |
| AA | arachidonic acid |
| AMP | adenosine monophosphate |
| AMPK | AMP-activated protein kinase |
| ANLS | astrocyte–neuron lactate shuttle |
| ATP | adenosine triphosphate |
| AQP4 | aquaporin 4 |
| CBF | cerebral blood flow |
| CMRglc | cerebral metabolic rate of glucose |
| CMRO2 | cerebral metabolic rate of oxygen |
| COX | cyclooxygenase |
| CYP | cytochrome P450 |
| DAG | diacylglycerol |
| DS | D-serine |
| ECM | extracellular matrix |
| EDHFs | endothelium-derived hyperpolarizing factors |
| EETs | epoxyeicosatrienoic acids |
| ER | endoplasmic reticulum |
| GFAP | glial fibrillary acidic protein |
| GS | glymphatic system |
| IP3 | inositol-1,4,5-triphosphate |
| iPS | induced pluripotent stem cells |
| LS | L-serine |
| MCA | middle cerebral artery |
| MCTs | monocarboxylate transporters |
| mGluRs | metabotropic glutamate receptors |
| MRS | magnetic resonance spectroscopy |
| NINDS | National Institute of Neurological Disorders and Stroke |
| NMDA | N-methyl-D-aspartate |
| nNOS | neuronal nitric oxide synthase |
| NVU | neurovascular unit |
| OPCs | oligodendrocyte precursor cells |
| PDHC | pyruvate dehydrogenase complex |
| PLA2 | phospholipase A2 |
| PLC | phospholipase C |
| PPP | pentose-phosphate pathway |
| PSD-95 | postsynaptic density protein 95 |
| rt-PA | recombinant tissue plasminogen activator |
| SRR | serine racemase |
| TCA | tricarboxylic acid |
| WT | wild-type |
References
- Bernier, L.-P.; Brunner, C.; Cottarelli, A.; Balbi, M. Location Matters: Navigating Regional Heterogeneity of the Neurovascular Unit. Front. Cell. Neurosci. 2021, 15, 696540. [Google Scholar] [CrossRef] [PubMed]
- Naranjo, O.; Osborne, O.; Torices, S.; Toborek, M. In Vivo Targeting of the Neurovascular Unit: Challenges and Advancements. Cell. Mol. Neurobiol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, C.; Donato, L.; Alibrandi, S.; Scimone, C.; D’Angelo, R.; Sidoti, A. Oxidative Stress and the Neurovascular Unit. Life 2021, 11, 767. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, S.; Iadecola, C. Revisiting the neurovascular unit. Nat. Neurosci. 2021, 24, 1198–1209. [Google Scholar] [CrossRef]
- Kugler, E.C.; Greenwood, J.; MacDonald, R.B. The “Neuro-Glial-Vascular” Unit: The Role of Glia in Neurovascular Unit Formation and Dysfunction. Front. Cell Dev. Biol. 2021, 9, 732820. [Google Scholar] [CrossRef]
- National Institute of Neurological Disorders and Stroke. Report of the Stroke Progress Review Group. 2002; pp. 1–116. Available online: https://www.stroke.nih.gov/documents/SPRG_report_042002_508C.pdf (accessed on 18 February 2022).
- Del Zoppo, G.J. Toward the neurovascular unit. A journey in clinical translation: 2012 Thomas Willis Lecture. Stroke 2013, 44, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Ji, C.; Shao, A. Neurovascular Unit Dysfunction and Neurodegenerative Disorders. Front. Neurosci. 2020, 14, 334. [Google Scholar] [CrossRef]
- Ivanidze, J.; Skafida, M.; Pandya, S.; Patel, D.; Osborne, J.R.; Raj, A.; Gupta, A.; Henchcliffe, C.; Dyke, J.P. Molecular Imaging of Striatal Dopaminergic Neuronal Loss and the Neurovascular Unit in Parkinson Disease. Front. Neurosci. 2020, 14, 528809. [Google Scholar] [CrossRef]
- Soto-Rojas, L.O.; Pacheco-Herrero, M.; Martínez-Gómez, P.A.; Campa-Córdoba, B.B.; Apátiga-Pérez, R.; Villegas-Rojas, M.M.; Harrington, C.R.; de la Cruz, F.; Garcés-Ramírez, L.; Luna-Muñoz, J. The Neurovascular Unit Dysfunction in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 2022. [Google Scholar] [CrossRef]
- Apátiga-Pérez, R.; Soto-Rojas, L.O.; Campa-Córdoba, B.B.; Luna-Viramontes, N.I.; Cuevas, E.; Villanueva-Fierro, I.; Ontiveros-Torres, M.A.; Bravo-Muñoz, M.; Flores-Rodríguez, P.; Garcés-Ramirez, L.; et al. Neurovascular dysfunction and vascular amyloid accumulation as early events in Alzheimer’s disease. Metab. Brain Dis. 2021, 37, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Balasa, R.; Barcutean, L.; Mosora, O.; Manu, D. Reviewing the Significance of Blood–Brain Barrier Disruption in Multiple Sclerosis Pathology and Treatment. Int. J. Mol. Sci. 2021, 22, 8370. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Mok, H.; Yeo, W.-S.; Ahn, J.-H.; Choi, Y.K. Role of ginseng in the neurovascular unit of neuroinflammatory diseases focused on the blood-brain barrier. J. Ginseng Res. 2021, 45, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Lo, E.H.; Dalkara, T.; Moskowitz, M.A. Mechanisms, challenges and opportunities in stroke. Nat. Rev. Neurosci. 2003, 4, 399–414. [Google Scholar] [CrossRef] [PubMed]
- Del Zoppo, G.J. Inflammation and the neurovascular unit in the setting of focal cerebral ischemia. Neuroscience 2009, 158, 972–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Zoppo, G.J. Relationship of neurovascular elements to neuron injury during ischemia. Cerebrovasc. Dis. 2009, 27, 65–76. [Google Scholar] [CrossRef] [Green Version]
- Moskowitz, M.A.; Lo, E.H.; Iadecola, C. The Science of Stroke: Mechanisms in Search of Treatments. Neuron 2010, 67, 181–198. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S. Neuroprotective Function of High Glycolytic Activity in Astrocytes: Common Roles in Stroke and Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 6568. [Google Scholar] [CrossRef]
- Takahashi, S. Metabolic compartmentalization between astroglia and neurons in physiological and pathophysiological conditions of the neurovascular unit. Neuropathology 2020, 40, 121–137. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Izawa, Y.; Suzuki, N. Astrogliopathy as a loss of astroglial protective function against glycoxidative stress under hyperglycemia. Rinsho Shinkeigaku 2012, 52, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Clarke, D.D.; Sokoloff, L. Circulation and energy metabolism of the brain. In Basic Neurochemistry: Molecular, Cellular, and Medical Aspects, 6th ed.; Siegel, G., Agranoff, B., Albers, R.W., Fisher, S., Eds.; Lippincott-Raven: Philadelphia, PA, USA, 1999; pp. 637–669. [Google Scholar]
- Dienel, G.A. Energy metabolism in the brain. In From Molecules to Networks: An Introduction to Cellular and Molecular Neuroscience, 2nd ed.; Byrne, J.H., Roberts, J.L., Eds.; Academic Press: London, UK, 2009; pp. 49–110. [Google Scholar]
- Hartmann, D.A.; Berthiaume, A.-A.; Grant, R.I.; Harrill, S.A.; Koski, T.; Tieu, T.; McDowell, K.P.; Faino, A.V.; Kelly, A.L.; Shih, A.Y. Brain capillary pericytes exert a substantial but slow influence on blood flow. Nat. Neurosci. 2021, 24, 633–645. [Google Scholar] [CrossRef]
- Hartmann, D.A.; Coelho-Santos, V.; Shih, A.Y. Pericyte Control of Blood Flow Across Microvascular Zones in the Central Nervous System. Annu. Rev. Physiol. 2021, 84. [Google Scholar] [CrossRef] [PubMed]
- Eltanahy, A.M.; Koluib, Y.A.; Gonzales, A. Pericytes: Intrinsic Transportation Engineers of the CNS Microcirculation. Front. Physiol. 2021, 12, 719701. [Google Scholar] [CrossRef] [PubMed]
- Zambach, S.A.; Cai, C.; Helms, H.C.C.; Hald, B.O.; Dong, Y.; Fordsmann, J.C.; Nielsen, R.M.; Hu, J.; Lønstrup, M.; Brodin, B.; et al. Precapillary sphincters and pericytes at first-order capillaries as key regulators for brain capillary perfusion. Proc. Natl. Acad. Sci. USA 2021, 118, e2023749118. [Google Scholar] [CrossRef]
- Grubb, S.; Lauritzen, M.; Aalkjær, C. Brain capillary pericytes and neurovascular coupling. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2021, 254, 110893. [Google Scholar] [CrossRef]
- Nakada, T.; Kwee, I.L. Fluid Dynamics Inside the Brain Barrier: Current Concept of Interstitial Flow, Glymphatic Flow, and Cerebrospinal Fluid Circulation in the Brain. Neuroscience 2019, 25, 155–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakada, T.; Kwee, I.L.; Igarashi, H.; Suzuki, Y. Aquaporin-4 Functionality and Virchow-Robin Space Water Dynamics: Physiological Model for Neurovascular Coupling and Glymphatic Flow. Int. J. Mol. Sci. 2017, 18, 1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakada, T. Virchow-Robin space and aquaporin-4: New insights on an old friend. Croat. Med. J. 2014, 55, 328–336. [Google Scholar] [CrossRef]
- Ding, Z.; Guo, S.; Luo, L.; Zheng, Y.; Gan, S.; Kang, X.; Wu, X.; Zhu, S. Emerging Roles of Microglia in Neuro-vascular Unit: Implications of Microglia-Neurons Interactions. Front. Cell Neurosci. 2021, 15, 706025. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Marsh, S.E.; Stevens, B. Microglia and Astrocytes in Disease: Dynamic Duo or Partners in Crime? Trends Immunol. 2020, 41, 820–835. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Iizumi, T.; Takahashi, S.; Mashima, K.; Minami, K.; Izawa, Y.; Abe, T.; Hishiki, T.; Suematsu, M.; Kajimura, M.; Suzuki, N. A possible role of microglia-derived nitric oxide by lipopolysaccharide in activation of astroglial pentose-phosphate pathway via the Keap1/Nrf2 system. J. Neuroinflamm. 2016, 13, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charvériat, M.; Mouthon, F.; Rein, W.; Verkhratsky, A. Connexins as therapeutic targets in neurological and neuropsychiatric disorders. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166098. [Google Scholar] [CrossRef] [PubMed]
- Stephan, J.; Eitelmann, S.; Zhou, M. Approaches to Study Gap Junctional Coupling. Front. Cell. Neurosci. 2021, 15, 640406. [Google Scholar] [CrossRef]
- Lorenzini, L.; Fernandez, M.; Baldassarro, V.A.; Bighinati, A.; Giuliani, A.; Calzà, L.; Giardino, L. White Matter and Neuroprotection in Alzheimer’s Dementia. Molecules 2020, 25, 503. [Google Scholar] [CrossRef] [Green Version]
- Psychogios, K.; Tsivgoulis, G. Intravenous thrombolysis for acute ischemic stroke: Why not? Curr. Opin. Neurol. 2021, 35, 10–17. [Google Scholar] [CrossRef]
- Yang, S.H.; Liu, R. Four Decades of Ischemic Penumbra and Its Implication for Ischemic Stroke. Transl. Stroke Res. 2021, 12, 937–945. [Google Scholar] [CrossRef]
- Lyden, P.D. Cerebroprotection for Acute Ischemic Stroke: Looking Ahead. Stroke 2021, 52, 3033–3044. [Google Scholar] [CrossRef]
- Lyden, P.; Buchan, A.; Boltze, J.; Fisher, M. Top Priorities for Cerebroprotective Studies-A Paradigm Shift: Report From STAIR XI. Stroke 2021, 52, 3063–3071. [Google Scholar] [CrossRef]
- Ganesh, A.; Ospel, J.M.; Menon, B.K.; Demchuk, A.M.; McTaggart, R.A.; Nogueira, R.G.; Poppe, A.Y.; Almekhlafi, M.A.; Hanel, R.A.; Thomalla, G.; et al. Assessment of Discrepancies Between Follow-up Infarct Volume and 90-Day Outcomes Among Patients With Ischemic Stroke Who Received Endovascular Therapy. JAMA Netw. Open 2021, 4, e2132376. [Google Scholar] [CrossRef]
- Zhou, X.-F. ESCAPE-NA1 Trial Brings Hope of Neuroprotective Drugs for Acute Ischemic Stroke: Highlights of the Phase 3 Clinical Trial on Nerinetide. Neurosci. Bull. 2021, 37, 579–581. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.D.; Goyal, M.; Menon, B.K.; Nogueira, R.G.; McTaggart, R.A.; Demchuk, A.M.; Poppe, A.Y.; Buck, B.H.; Field, T.S.; Dowlatshahi, D.; et al. Efficacy and safety of nerinetide for the treatment of acute ischaemic stroke (ESCAPE-NA1): A multicentre, double-blind, randomised controlled trial. Lancet 2020, 395, 878–887. [Google Scholar] [CrossRef]
- Ballarin, B.; Tymianski, M. Discovery and development of NA-1 for the treatment of acute ischemic stroke. Acta Pharmacol. Sin. 2018, 39, 661–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.J.; Tymianski, M. Targeting NMDA receptors in stroke: New hope in neuroprotection. Mol. Brain 2018, 11, 15. [Google Scholar] [CrossRef]
- Del Zoppo, G.J.; Mabuchi, T. Cerebral microvessel responses to focal ischemia. J. Cereb. Blood Flow Metab. 2003, 23, 879–894. [Google Scholar] [CrossRef]
- Mabuchi, T.; Lucero, J.; Feng, A.; Koziol, J.A.; Del Zoppo, G.J. Focal Cerebral Ischemia Preferentially Affects Neurons Distant from Their Neighboring Microvessels. J. Cereb. Blood Flow Metab. 2005, 25, 257–266. [Google Scholar] [CrossRef]
- Panickar, K.S.; Norenberg, M.D. Astrocytes in cerebral ischemic injury: Morphological and general considerations. Glia 2005, 50, 287–298. [Google Scholar] [CrossRef]
- Seifert, G.; Schilling, K.; Steinhäuser, C. Astrocyte dysfunction in neurological disorders: A molecular perspective. Nat. Rev. Neurosci. 2006, 7, 194–206. [Google Scholar] [CrossRef]
- Frantseva, M.V.; Kokarovtseva, L.; Velazquez, J.L.P. Ischemia-Induced Brain Damage Depends on Specific Gap-Junctional Coupling. J. Cereb. Blood Flow Metab. 2002, 22, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Paulson, O.B.; Newman, E.A. Does the Release of Potassium from Astrocyte Endfeet Regulate Cerebral Blood Flow? Science 1987, 237, 896–898. [Google Scholar] [CrossRef] [Green Version]
- Mosso, A. Sulla circolazione del sangue nel cervello dell’uomo [On the circulation of blood in the human brain]. R. Accad. Lincei. 1880, 5, 237–358. [Google Scholar]
- Roy, C.S.; Sherrington, C.S. On the Regulation of the Blood-supply of the Brain. J. Physiol. 1890, 11, 85–158. [Google Scholar] [CrossRef] [PubMed]
- Sokoloff, L.; Kety, S.S. Regulation of cerebral circulation. Physiol. Rev. Suppl. 1960, 4, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Zonta, M.; Angulo, M.C.; Gobbo, S.; Rosengarten, B.; Hossmann, K.A.; Pozzan, T.; Carmignoto, G. Neuron-toastrocyte signaling is central to the dynamic control of brain microcirculation. Nat. Neurosci. 2003, 6, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Zonta, M.; Sebelin, A.; Gobbo, S.; Fellin, T.; Pozzan, T.; Carmignoto, G. Glutamate-mediated cytosolic calcium oscillations regulate a pulsatile prostaglandin release from cultured rat astrocytes. J. Physiol. 2003, 553, 407–414. [Google Scholar] [CrossRef]
- Harder, D.R.; Alkayed, N.J.; Lange, A.R.; Gebremedhin, D.; Roman, R.J. Functional hyperemia in the brain: Hypothesis for astrocyte-derived vasodilator metabolites. Stroke 1998, 29, 229–234. [Google Scholar] [CrossRef]
- Mulligan, S.J.; MacVicar, B. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature 2004, 431, 195–199. [Google Scholar] [CrossRef]
- MacVicar, B.; Newman, E.A. Astrocyte Regulation of Blood Flow in the Brain. Cold Spring Harb. Perspect. Biol. 2015, 7, a020388. [Google Scholar] [CrossRef]
- Gordon, G.R.J.; Howarth, C.; MacVicar, B.A.; Roach, R.C.; Hackett, P.H.; Wagner, P.D. Bidirectional Control of Blood Flow by Astrocytes: A Role for Tissue Oxygen and Other Metabolic Factors. Adv. Exp. Med. Biol. 2016, 903, 209–219. [Google Scholar]
- Gordon, G.R.J.; Howarth, C.; MacVicar, B.A. Bidirectional control of arteriole diameter by astrocytes. Exp. Physiol. 2011, 96, 393–399. [Google Scholar] [CrossRef]
- Salman, M.M.; Kitchen, P.; Iliff, J.J.; Bill, R.M. Aquaporin 4 and glymphatic flow have central roles in brain fluid homeostasis. Nat. Rev. Neurosci. 2021, 22, 650–651. [Google Scholar] [CrossRef]
- Igarashi, H.; Suzuki, Y.; Kwee, I.L.; Nakada, T. Water influx into cerebrospinal fluid is significantly reduced in senile plaque bearing transgenic mice, supporting beta-amyloid clearance hypothesis of Alzheimer’s disease. Neurol Res. 2014, 36, 1094–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igarashi, H.; Tsujita, M.; Kwee, I.L.; Nakada, T. Water influx into cerebrospinal fluid is primarily controlled by aquaporin-4, not by aquaporin-1: 17O JJVCPE MRI study in knockout mice. Neuroreport 2014, 25, 39–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yankova, G.; Bogomyakova, O.; Tulupov, A. The glymphatic system and meningeal lymphatics of the brain: New understanding of brain clearance. Rev. Neurosci. 2021, 32, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Hablitz, L.M.; Nedergaard, M. The glymphatic system. Curr. Biol. 2021, 31, R1371–R1375. [Google Scholar] [CrossRef]
- Bèchet NB, Shanbhag NC, Lundgaard I: Glymphatic pathways in the gyrencephalic brain. J. Cereb. Blood Flow Metab. 2021, 41, 2264–2279. [CrossRef]
- Klostranec, J.M.; Vucevic, D.; Bhatia, K.D.; Kortman, H.G.J.; Krings, T.; Murphy, K.P.; Terbrugge, K.G.; Mikulis, D.J. Current Concepts in Intracranial Interstitial Fluid Transport and the Glymphatic System: Part I—Anatomy and Physiology. Radiology 2021, 301, 502–514. [Google Scholar] [CrossRef]
- Klostranec, J.M.; Vucevic, D.; Bhatia, K.D.; Kortman, H.G.J.; Krings, T.; Murphy, K.P.; Terbrugge, K.G.; Mikulis, D.J. Current Concepts in Intracranial Interstitial Fluid Transport and the Glymphatic System: Part II—Imaging Techniques and Clinical Applications. Radiology 2021, 301, 516–532. [Google Scholar] [CrossRef]
- Fox, P.; Raichle, M.E. Focal physiological uncoupling of cerebral blood flow and oxidative metabolism during somatosensory stimulation in human subjects. Proc. Natl. Acad. Sci. USA 1986, 83, 1140–1144. [Google Scholar] [CrossRef] [Green Version]
- Fox, P.T.; Raichle, M.E.; Mintun, M.A.; Dence, C. Nonoxidative Glucose Consumption During Focal Physiologic Neural Activity. Science 1988, 241, 462–464. [Google Scholar] [CrossRef]
- Prichard, J.; Rothman, D.; Novotny, E.; Petroff, O.; Kuwabara, T.; Avison, M.; Howseman, A.; Hanstock, C.; Shulman, R. Lactate rise detected by 1H NMR in human visual cortex during physiologic stimulation. Proc. Natl. Acad. Sci. USA 1991, 88, 5829–5831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, S.; Driscoll, B.F.; Law, M.J.; Sokoloff, L. Role of sodium and potassium ions in regulation of glucose metabolism in cultured astroglia. Proc. Natl. Acad. Sci. USA 1995, 92, 4616–4620. [Google Scholar] [CrossRef] [Green Version]
- Sokoloff, L.; Takahashi, S.; Gotoh, J.; Driscoll, B.F.; Law, M.J. Contribution of astroglia to functionally activated energy metabolism. Dev. Neurosci. 1996, 18, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Souza, D.G.; Almeida, R.F.; Souza, D.O.; Zimmer, E.R. The astrocyte biochemistry. Semin. Cell Dev. Biol. 2019, 95, 142–150. [Google Scholar] [CrossRef]
- Bordone, M.; Salman, M.M.; Titus, H.E.; Amini, E.; Andersen, J.V.; Chakraborti, B.; Diuba, A.V.; Dubouskaya, T.G.; Ehrke, E.; De Freitas, A.E.; et al. The energetic brain—A review from students to students. J. Neurochem. 2019, 151, 139–165. [Google Scholar] [CrossRef]
- Aubert, A.; Costalat, R.; Magistretti, P.J.; Pellerin, L. Brain lactate kinetics: Modeling evidence for neuronal lactate uptake upon activation. Proc. Natl. Acad. Sci. USA 2005, 102, 16448–16453. [Google Scholar] [CrossRef] [Green Version]
- Laughton, J.D.; Bittar, P.; Charnay, Y.; Pellerin, L.; Kovari, E.; Magistretti, P.J.; Bouras, C. Metabolic compartmentalization in the human cortex and hippocampus: Evidence for a cell- and region-specific localization of lactate dehydrogenase 5 and pyruvate dehydrogenase. BMC Neurosci. 2007, 8, 35. [Google Scholar] [CrossRef] [Green Version]
- Pellerin, L.; Bouzier-Sore, A.-K.; Aubert, A.; Serres, S.; Merle, M.; Costalat, R.; Magistretti, P.J. Activity-dependent regulation of energy metabolism by astrocytes: An update. Glia 2007, 55, 1251–1262. [Google Scholar] [CrossRef]
- Jolivet, R.; Allaman, I.; Pellerin, L.; Magistretti, P.J.; Weber, B. Comment on Recent Modeling Studies of Astrocyte–Neuron Metabolic Interactions. J. Cereb. Blood Flow Metab. 2010, 30, 1982–1986. [Google Scholar] [CrossRef] [Green Version]
- Pellerin, L.; Magistretti, P.J. Sweet sixteen for ANLS. J. Cereb. Blood Flow Metab. 2012, 32, 1152–1166. [Google Scholar] [CrossRef]
- Mazuel, L.; Blanc, J.; Repond, C.; Bouchaud, V.; Raffard, G.; Deglon, N.; Bonvento, G.; Pellerin, L.; Bouzier-Sore, A.-K. A neuronal MCT2 knockdown in the rat somatosensory cortex reduces both the NMR lactate signal and the BOLD response during whisker stimulation. PLoS ONE 2017, 12, e0174990. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L. Neuroenergetics: Astrocytes Have a Sweet Spot for Glucose. Curr. Biol. 2018, 28, R1258–R1260. [Google Scholar] [CrossRef] [Green Version]
- Dienel, G.A.; Hertz, L. Glucose and lactate metabolism during brain activation. J. Neurosci. Res. 2001, 66, 824–838. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L.; Dienel, G.A. Lactate transport and transporters: General principles and functional roles in brain cells. J. Neurosci. Res. 2005, 79, 11–18. [Google Scholar] [CrossRef]
- Hertz, L.; Peng, L.; Dienel, G.A. Energy metabolism in astrocytes: High rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J. Cereb. Blood Flow Metab. 2007, 27, 219–249. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A.; Ball, K.K.; Cruz, N.F. A glycogen phosphorylase inhibitor selectively enhances local rates of glucose utilization in brain during sensory stimulation of conscious rats: Implications for glycogen turnover. J. Neurochem. 2007, 102, 466–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dienel, G.A. Astrocytes are ’good scouts’: Being prepared also helps neighboring neurons. J. Cereb. Blood Flow Metab. 2010, 30, 1893–1894. [Google Scholar] [CrossRef] [Green Version]
- Dienel, G.A. Brain lactate metabolism: The discoveries and the controversies. J. Cereb. Blood Flow Metab. 2012, 32, 1107–1138. [Google Scholar] [CrossRef] [Green Version]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: The role of glucose in physiologicaland pathological brain function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Dienel, G.A.; Cruz, N.F. Contributions of glycogen to astrocytic energetics during brain activation. Metab. Brain Dis. 2015, 30, 281–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dienel, G.A. The metabolic trinity, glucose–glycogen–lactate, links astrocytes and neurons in brain energetics, signaling, memory, and gene expression. Neurosci. Lett. 2017, 637, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A. Lack of appropriate stoichiometry: Strong evidence against an energetically important astrocyte-neuron lactate shuttle in brain. J. Neurosci. Res. 2017, 95, 2103–2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dienel, G.A. Brain glucose metabolism: Integration ofenergetics with function. Physiol. Rev. 2019, 99, 949–1045. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S. Lactate and ketone bodies act as energy substrates as well as signal molecules in the brain. In Psychology and Pathophysiological Outcomes of Eating; IntechOpen: London, UK, 2021; pp. 1–20. [Google Scholar]
- Jensen, N.J.; Wodschow, H.Z.; Nilsson, M.; Rungby, J. Effects of Ketone Bodies on Brain Metabolism and Function in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8767. [Google Scholar] [CrossRef]
- Takahashi, S.; Iizumi, T.; Mashima, K.; Abe, T.; Suzuki, N. Roles and Regulation of Ketogenesis in Cultured Astroglia and Neurons Under Hypoxia and Hypoglycemia. ASN Neuro 2014, 6, 1759091414550997. [Google Scholar] [CrossRef]
- Ploux, E.; Freret, T.; Billard, J.M. D-serine in physiological and pathological brain aging. Biochim. Biophys. Acta Proteins Proteom. 2021, 1869, 140542. [Google Scholar] [CrossRef]
- Abe, T.; Suzuki, M.; Sasabe, J.; Takahashi, S.; Unekawa, M.; Mashima, K.; Iizumi, T.; Hamase, K.; Konno, R.; Aiso, S.; et al. Cellular Origin and Regulation of D-and L-Serine in in Vitro and in Vivo Models of Cerebral Ischemia. J. Cereb. Blood Flow Metab. 2014, 34, 1928–1935. [Google Scholar] [CrossRef] [Green Version]
- Maugard, M.; Vigneron, P.A.; Bolaños, J.P.; Bonvento, G. l-Serine links metabolism with neurotransmission. Prog Neurobiol. 2021, 197, 101896. [Google Scholar] [CrossRef]
- Ye, L.; Sun, Y.; Jiang, Z.; Wang, G. L-Serine, an Endogenous Amino Acid, Is a Potential Neuroprotective Agent for Neurological Disease and Injury. Front. Mol. Neurosci. 2021, 14, 726665. [Google Scholar] [CrossRef]
- Le Douce, J.; Maugard, M.; Veran, J.; Matos, M.; Jégo, P.; Vigneron, P.A.; Faivre, E.; Toussay, X.; Vandenberghe, M.; Balbastre, Y.; et al. Impairment of Glycolysis-Derived l-Serine Production in Astrocytes Contributes to Cognitive Deficits in Alzheimer’s Disease. Cell Metab. 2020, 31, 503–517.e8. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Nakamura, Y.; Li, W.; Hamanaka, G.; Arai, K.; Lo, E.H.; Hayakawa, K. Effects of O-GlcNAcylation on functional mitochondrial transfer from astrocytes. J. Cereb. Blood Flow Metab. 2021, 41, 1523–1535. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Lo, E.H.; Hayakawa, K. Placental Mitochondria Therapy for Cerebral Ischemia-Reperfusion Injury in Mice. Stroke 2020, 51, 3142–3146. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Chan, S.J.; Mandeville, E.T.; Park, J.H.; Bruzzese, M.; Montaner, J.; Arai, K.; Rosell, A.; Lo, E.H. Protective Effects of Endothelial Progenitor Cell-Derived Extracellular Mitochondria in Brain Endothelium. Stem Cells 2018, 36, 1404–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayakawa, K.; Bruzzese, M.; Chou, S.H.-Y.; Ning, M.; Ji, X.; Lo, E.H. Extracellular Mitochondria for Therapy and Diagnosis in Acute Central Nervous System Injury. JAMA Neurol. 2018, 75, 119–122. [Google Scholar] [CrossRef]
- Chou, S.H.-Y.; Lan, J.; Esposito, E.; Ning, M.; Balaj, L.; Ji, X.; Lo, E.H.; Hayakawa, K. Extracellular Mitochondria in Cerebrospinal Fluid and Neurological Recovery After Subarachnoid Hemorrhage. Stroke 2017, 48, 2231–2237. [Google Scholar] [CrossRef]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Corrigendum: Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 539, 123. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-H.; Lo, E.H.; Hayakawa, K. Endoplasmic Reticulum Interaction Supports Energy Production and Redox Homeostasis in Mitochondria Released from Astrocytes. Transl. Stroke Res. 2021, 12, 1045–1054. [Google Scholar] [CrossRef]
- Arai, K.; Bonsack, B.; Borlongan, M.; Lo, E.H. Brief overview: Protective roles of astrocyte-derived pentraxin-3 in blood-brain barrier integrity. Brain Circ. 2019, 5, 145–149. [Google Scholar] [CrossRef]
- Cheng, X.Y.; Biswas, S.; Li, J.; Mao, C.J.; Chechneva, O.; Chen, J.; Li, K.; Li, J.; Zhang, J.R.; Liu, C.F.; et al. Human iPSCs derived astrocytes rescue rotenone-induced mitochondrial dysfunction and dopaminergic neurodegeneration in vitro by donating functional mitochondria. Transl. Neurodegener. 2020, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Zhang, Z.; Lu, J.; Pei, G. Mitochondria Are Dynamically Transferring Between Human Neural Cells and Alexander Disease-Associated GFAP Mutations Impair the Astrocytic Transfer. Front. Cell Neurosci. 2019, 13, 316. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Pan, L.; Pembroke, W.G.; Rexach, J.E.; Godoy, M.I.; Condro, M.C.; Alvarado, A.G.; Harteni, M.; Chen, Y.W.; Stiles, L.; et al. Conservation and divergence of vulnerability and responses to stressors between human and mouse astrocytes. Nat Commun. 2021, 12, 3958. [Google Scholar] [CrossRef] [PubMed]
- Leventoux, N.; Morimoto, S.; Imaizumi, K.; Sato, Y.; Takahashi, S.; Mashima, K.; Ishikawa, M.; Sonn, I.; Kondo, T.; Watanabe, H.; et al. Human Astrocytes Model Derived from Induced Pluripotent Stem Cells. Cells 2020, 9, 2680. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takahashi, S. Metabolic Contribution and Cerebral Blood Flow Regulation by Astrocytes in the Neurovascular Unit. Cells 2022, 11, 813. https://doi.org/10.3390/cells11050813
Takahashi S. Metabolic Contribution and Cerebral Blood Flow Regulation by Astrocytes in the Neurovascular Unit. Cells. 2022; 11(5):813. https://doi.org/10.3390/cells11050813
Chicago/Turabian StyleTakahashi, Shinichi. 2022. "Metabolic Contribution and Cerebral Blood Flow Regulation by Astrocytes in the Neurovascular Unit" Cells 11, no. 5: 813. https://doi.org/10.3390/cells11050813
APA StyleTakahashi, S. (2022). Metabolic Contribution and Cerebral Blood Flow Regulation by Astrocytes in the Neurovascular Unit. Cells, 11(5), 813. https://doi.org/10.3390/cells11050813