
Predicting Mitochondrial Dynamic Behavior in Genetically Defined Neurodegenerative Diseases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
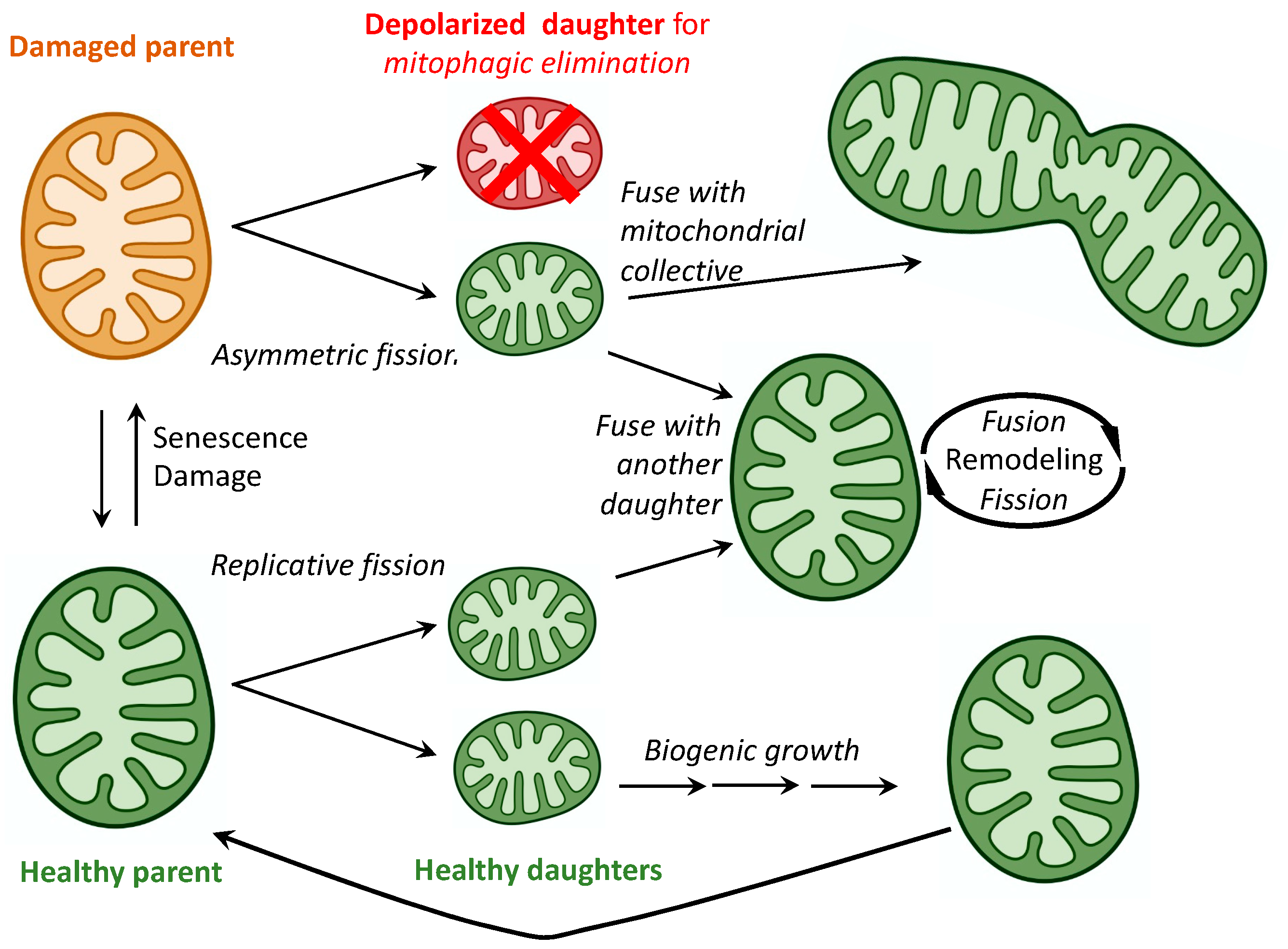
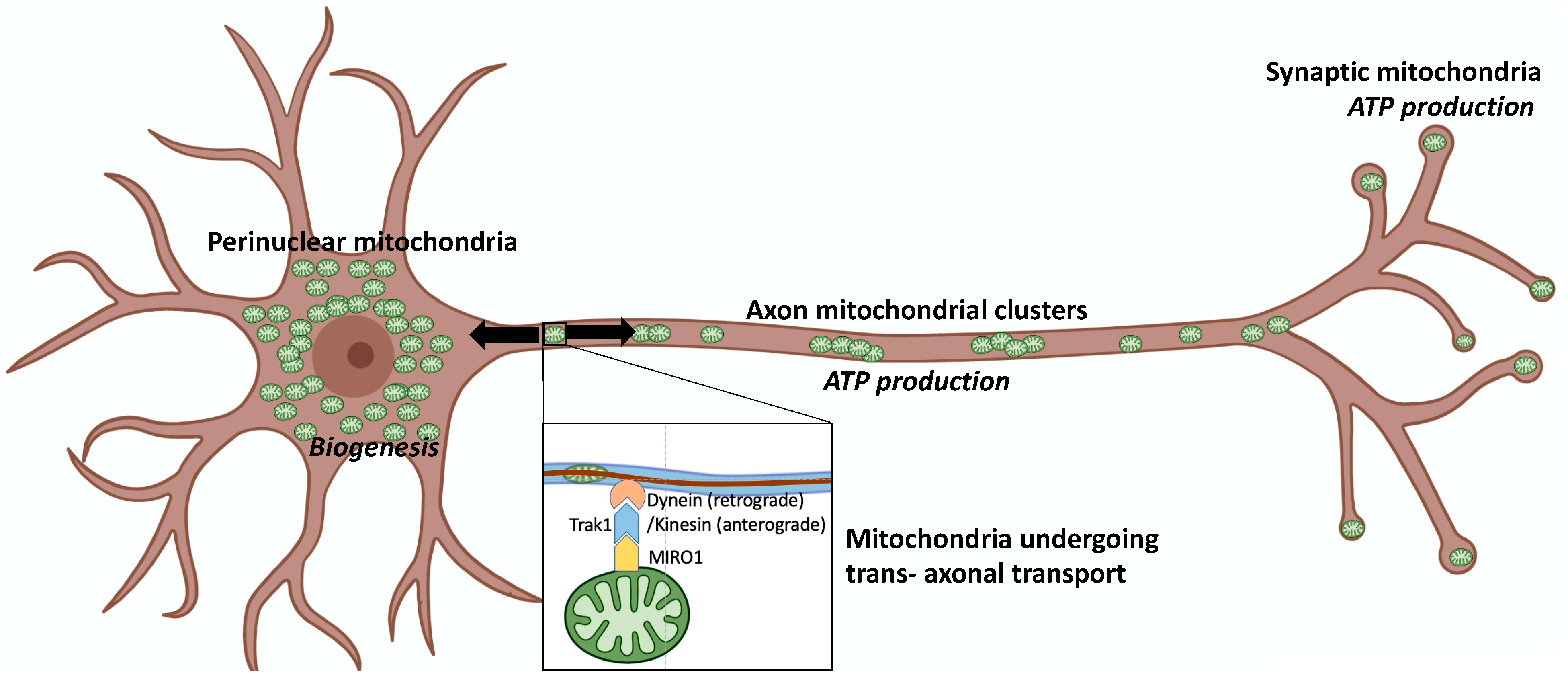
2. Mitochondrial Fusion, Fission, and Motility
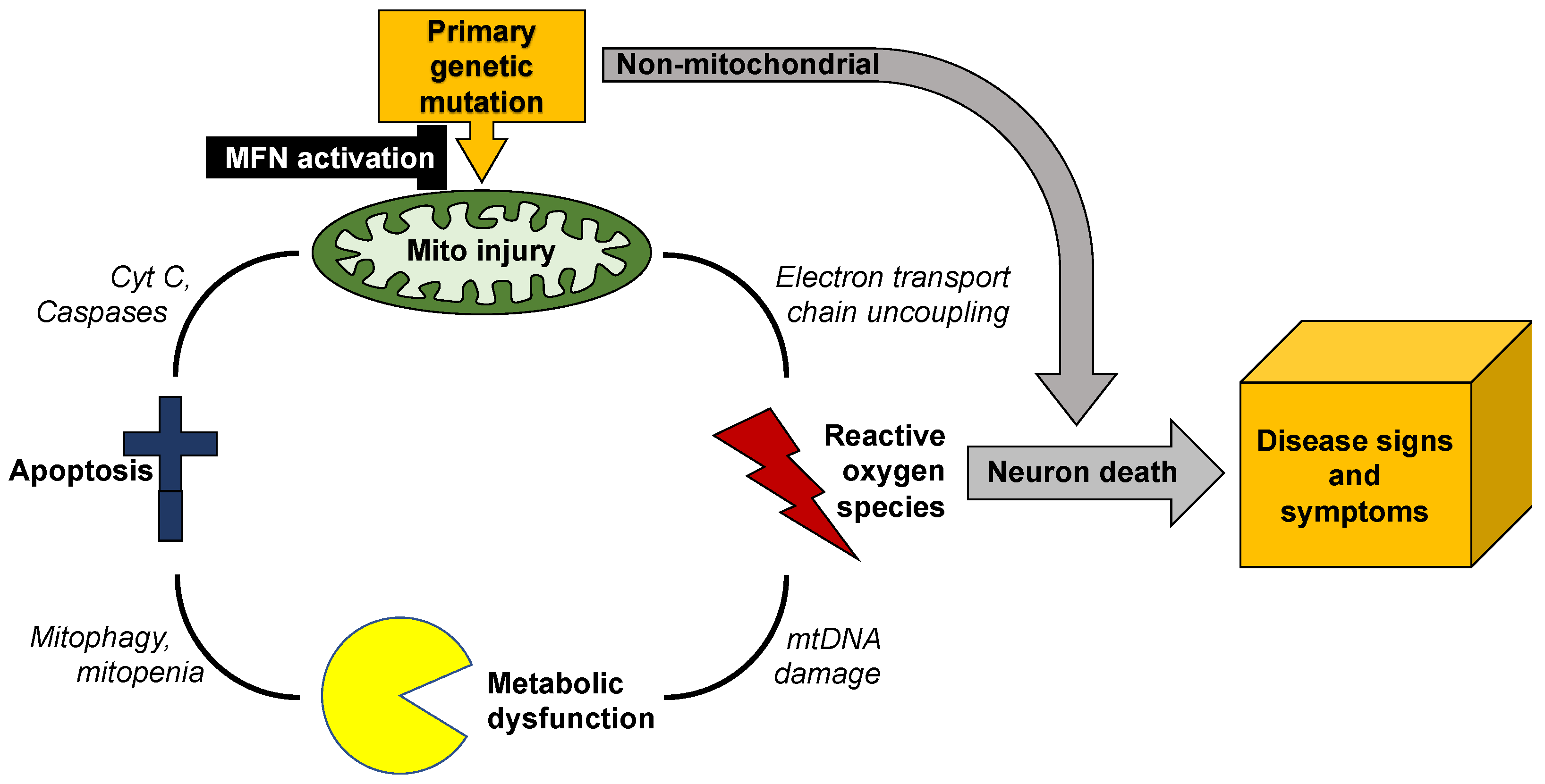
3. Mitochondrial Dysdynamism in Genetic Neurological Diseases
4. Evaluating Mitochondrial Dynamics
5. Patient-Derived Primary Fibroblasts Exhibit Disease-Related Imbalances in Mitochondrial Fission/Fusion
6. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lane, N.; Martin, W. The energetics of genome complexity. Nature 2010, 467, 929–934. [Google Scholar] [CrossRef]
- Gray, M.W. Mitochondrial evolution. Cold Spring Harb. Perspect. Biol. 2012, 4, a011403. [Google Scholar] [CrossRef] [Green Version]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Chan, D.C. Mitochondrial dynamics—Fusion, fission, movement, and mitophagy—In neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W., II. Evolving Concepts of Mitochondrial Dynamics. Annu. Rev. Physiol. 2019, 81, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Horbay, R.; Bilyy, R. Mitochondrial dynamics during cell cycling. Apoptosis 2016, 21, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Dorn, G.W., II. Mitoconfusion: Noncanonical functioning of dynamism factors in static mitochondria of the heart. Cell Metab. 2015, 21, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Dorn, G.W., II. Mitochondrial dynamism and heart disease: Changing shape and shaping change. EMBO Mol. Med. 2015, 7, 865–877. [Google Scholar] [CrossRef]
- MacAskill, A.F.; Kittler, J.T. Control of mitochondrial transport and localization in neurons. Trends Cell Biol. 2010, 20, 102–112. [Google Scholar] [CrossRef]
- Sheng, Z.H.; Cai, Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93. [Google Scholar] [CrossRef] [Green Version]
- Pareyson, D.; Piscosquito, G.; Moroni, I.; Salsano, E.; Zeviani, M. Peripheral neuropathy in mitochondrial disorders. Lancet Neurol. 2013, 12, 1011–1024. [Google Scholar] [CrossRef]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, T.J.; Gustafsson, C.M. Separating and Segregating the Human Mitochondrial Genome. Trends Biochem. Sci. 2018, 43, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Hyde, B.; Shirihai, O.S. Mitochondrial fusion, fission and autophagy as a quality control axis: The bioenergetic view. Biochim. Biophys. Acta 2008, 1777, 1092–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasca, A.; Legati, A.; Baruffini, E.; Nolli, C.; Moroni, I.; Ardissone, A.; Goffrini, P.; Ghezzi, D. Biallelic Mutations in DNM1L are Associated with a Slowly Progressive Infantile Encephalopathy. Hum. Mutat. 2016, 37, 898–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahrner, J.A.; Liu, R.; Perry, M.S.; Klein, J.; Chan, D.C. A novel de novo dominant negative mutation in DNM1L impairs mitochondrial fission and presents as childhood epileptic encephalopathy. Am. J. Med. Genet. A 2016, 170, 2002–2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandeleur, D.; Chen, C.V.; Huang, E.J.; Connolly, A.J.; Sanchez, H.; Moon-Grady, A.J. Novel and lethal case of cardiac involvement in DNM1L mitochondrial encephalopathy. Am. J. Med. Genet. A 2019, 179, 2486–2489. [Google Scholar] [CrossRef] [PubMed]
- Verrigni, D.; Di Nottia, M.; Ardissone, A.; Baruffini, E.; Nasca, A.; Legati, A.; Bellacchio, E.; Fagiolari, G.; Martinelli, D.; Fusco, L.; et al. Clinical-genetic features and peculiar muscle histopathology in infantile DNM1L-related mitochondrial epileptic encephalopathy. Hum. Mutat. 2019, 40, 601–618. [Google Scholar] [CrossRef] [PubMed]
- Gerber, S.; Charif, M.; Chevrollier, A.; Chaumette, T.; Angebault, C.; Kane, M.S.; Paris, A.; Alban, J.; Quiles, M.; Delettre, C.; et al. Mutations in DNM1L, as in OPA1, result in dominant optic atrophy despite opposite effects on mitochondrial fusion and fission. Brain 2017, 140, 2586–2596. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Gilkerson, R.W.; Schon, E.A.; Hernandez, E.; Davidson, M.M. Mitochondrial nucleoids maintain genetic autonomy but allow for functional complementation. J. Cell Biol. 2008, 181, 1117–1128. [Google Scholar] [CrossRef]
- Nakada, K.; Sato, A.; Hayashi, J. Mitochondrial functional complementation in mitochondrial DNA-based diseases. Int. J. Biochem. Cell Biol. 2009, 41, 1907–1913. [Google Scholar] [CrossRef]
- Detmer, S.A.; Chan, D.C. Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J. Cell Biol. 2007, 176, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Rocha, A.G.; Franco, A.; Krezel, A.M.; Rumsey, J.M.; Alberti, J.M.; Knight, W.C.; Biris, N.; Zacharioudakis, E.; Janetka, J.W.; Baloh, R.H.; et al. MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science 2018, 360, 336–341. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, T.L. Mitochondrial trafficking in neurons. Cold Spring Harb. Perspect. Biol. 2013, 5, a011304. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Z.H. Mitochondrial trafficking and anchoring in neurons: New insight and implications. J. Cell Biol. 2014, 204, 1087–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Schwarz, T.L. The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell 2009, 136, 163–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norkett, R.; Lesept, F.; Kittler, J.T. DISC1 Regulates Mitochondrial Trafficking in a Miro1-GTP-Dependent Manner. Front. Cell Dev. Biol. 2020, 8, 449. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.A.; Yalnizyan-Carson, A.; Fernández Busch, M.V.; van Zwieten, M.; Verhage, M.; Lohmann, C. Activity-dependent regulation of mitochondrial motility in developing cortical dendrites. eLife 2021, 10, e62091. [Google Scholar] [CrossRef] [PubMed]
- Frank, S. Dysregulation of mitochondrial fusion and fission: An emerging concept in neurodegeneration. Acta Neuropathol. 2006, 111, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Burte, F.; Carelli, V.; Chinnery, P.F.; Yu-Wai-Man, P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 2015, 11, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. 2020, 15, 235–259. [Google Scholar] [CrossRef] [Green Version]
- Dorn, G.W. Mitofusin activation enhances mitochondrial motility and promotes neuroregeneration in CMT2A. Neural Regen. Res. 2021, 16, 2201–2203. [Google Scholar] [CrossRef]
- Mink, J.W.; Blumenschine, R.J.; Adams, D.B. Ratio of central nervous system to body metabolism in vertebrates: Its constancy and functional basis. Am. J. Physiol. 1981, 241, R203–R212. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Ryan, T.A. Glucose metabolism in nerve terminals. Curr. Opin. Neurobiol. 2017, 45, 156–161. [Google Scholar] [CrossRef]
- Baloh, R.H.; Schmidt, R.E.; Pestronk, A.; Milbrandt, J. Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-Tooth disease from mitofusin 2 mutations. J. Neurosci. 2007, 27, 422–430. [Google Scholar] [CrossRef] [Green Version]
- De Vos, K.J.; Chapman, A.L.; Tennant, M.E.; Manser, C.; Tudor, E.L.; Lau, K.F.; Brownlees, J.; Ackerley, S.; Shaw, P.J.; McLoughlin, D.M.; et al. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet. 2007, 16, 2720–2728. [Google Scholar] [CrossRef]
- De Vos, K.J.; Grierson, A.J.; Ackerley, S.; Miller, C.C. Role of axonal transport in neurodegenerative diseases. Annu. Rev. Neurosci. 2008, 31, 151–173. [Google Scholar] [CrossRef]
- Knott, A.B.; Perkins, G.; Schwarzenbacher, R.; Bossy-Wetzel, E. Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 2008, 9, 505–518. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Disatnik, M.H.; Monbureau, M.; Shamloo, M.; Mochly-Rosen, D.; Qi, X. Inhibition of mitochondrial fragmentation diminishes Huntington’s disease-associated neurodegeneration. J. Clin. Investig. 2013, 123, 5371–5388. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.U.; Saw, N.L.; Shamloo, M.; Mochly-Rosen, D. Drp1/Fis1 interaction mediates mitochondrial dysfunction, bioenergetic failure and cognitive decline in Alzheimer’s disease. Oncotarget 2018, 9, 6128–6143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, A.U.; Saw, N.L.; Vogel, H.; Cunnigham, A.D.; Shamloo, M.; Mochly-Rosen, D. Inhibition of Drp1/Fis1 interaction slows progression of amyotrophic lateral sclerosis. EMBO Mol. Med. 2018, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, C.; Ciano, C.; Salsano, E.; Nanetti, L.; Milani, M.; Gellera, C.; Taroni, F.; Fabrizi, G.M.; Uncini, A.; Pareyson, D. Co-occurrence of amyotrophic lateral sclerosis and Charcot-Marie-Tooth disease type 2A in a patient with a novel mutation in the mitofusin-2 gene. Neuromuscul. Disord. 2011, 21, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, L.; Lin, W.L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, B.; Cabirol-Pol, M.J.; Miguel, L.; Whitworth, A.J.; Lecourtois, M.; Lievens, J.C. Enhancing Mitofusin/Marf ameliorates neuromuscular dysfunction in Drosophila models of TDP-43 proteinopathies. Neurobiol. Aging 2017, 54, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; Choe, Y.J.; Bannwarth, S.; Kim, J.; Maitra, S.; Dorn, G.W., II; Taylor, J.P.; Paquis-Flucklinger, V.; Kim, N.C. TDP-43 and PINK1 mediate CHCHD10(S59L) mutation-induced defects in Drosophila and in vitro. Nat. Commun. 2021, 12, 1924. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Dadon-Nachum, M.; Melamed, E.; Offen, D. The “dying-back” phenomenon of motor neurons in ALS. J. Mol. Neurosci. 2011, 43, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W., II. Mitofusin 2 Dysfunction and Disease in Mice and Men. Front. Physiol. 2020, 11, 782. [Google Scholar] [CrossRef] [PubMed]
- Moujalled, D.; Strasser, A.; Liddell, J.R. Molecular mechanisms of cell death in neurological diseases. Cell Death Differ. 2021, 28, 2029–2044. [Google Scholar] [CrossRef] [PubMed]
- Sah, E.; Krishnamurthy, S.; Ahmidouch, M.Y.; Gillispie, G.J.; Milligan, C.; Orr, M.E. The Cellular Senescence Stress Response in Post-Mitotic Brain Cells: Cell Survival at the Expense of Tissue Degeneration. Life 2021, 11, 229. [Google Scholar] [CrossRef]
- Züchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.A.; Rabideau, M.; Blevins, A.; Westbrook, M.J.; Ekstein, T.; Nykamp, K.; Deucher, A.; Harper, A.; Demmer, L. Autosomal recessive MFN2-related Charcot-Marie-Tooth disease with diaphragmatic weakness: Case report and literature review. Am. J. Med. Genet. A 2016, 170, 1580–1584. [Google Scholar] [CrossRef] [PubMed]
- Bombelli, F.; Stojkovic, T.; Dubourg, O.; Echaniz-Laguna, A.; Tardieu, S.; Larcher, K.; Amati-Bonneau, P.; Latour, P.; Vignal, O.; Cazeneuve, C.; et al. Charcot-Marie-Tooth disease type 2A: From typical to rare phenotypic and genotypic features. JAMA Neurol. 2014, 71, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Pipis, M.; Feely, S.M.E.; Polke, J.M.; Skorupinska, M.; Perez, L.; Shy, R.R.; Laura, M.; Morrow, J.M.; Moroni, I.; Pisciotta, C.; et al. Natural history of Charcot-Marie-Tooth disease type 2A: A large international multicentre study. Brain 2020, 143, 3589–3602. [Google Scholar] [CrossRef]
- Franco, A.; Dang, X.; Walton, E.K.; Ho, J.N.; Zablocka, B.; Ly, C.; Miller, T.M.; Baloh, R.H.; Shy, M.E.; Yoo, A.S.; et al. Burst mitofusin activation reverses neuromuscular dysfunction in murine CMT2A. eLife 2020, 9, e61119. [Google Scholar] [CrossRef]
- Zhou, Y.; Carmona, S.; Muhammad, A.; Bell, S.; Landeros, J.; Vazquez, M.; Ho, R.; Franco, A.; Lu, B.; Dorn, G.W., II; et al. Restoring mitofusin balance prevents axonal degeneration in a Charcot-Marie-Tooth type 2A model. J. Clin. Investig. 2021, 13, e147307. [Google Scholar] [CrossRef]
- Misko, A.; Jiang, S.; Wegorzewska, I.; Milbrandt, J.; Baloh, R. H, Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J. Neurosci. 2010, 30, 4232–4240. [Google Scholar] [CrossRef] [Green Version]
- Kong, J.; Xu, Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J. Neurosci. 1998, 18, 3241–3250. [Google Scholar] [CrossRef]
- Wang, H.; Lim, P.J.; Karbowski, M.; Monteiro, M.J. Effects of overexpression of huntingtin proteins on mitochondrial integrity. Hum. Mol. Genet. 2009, 18, 737–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renton, A.E.; Chiò, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Ligon, L.A.; LaMonte, B.H.; Wallace, K.E.; Weber, N.; Kalb, R.G.; Holzbaur, E.L. Mutant superoxide dismutase disrupts cytoplasmic dynein in motor neurons. Neuroreport 2005, 16, 533–536. [Google Scholar] [CrossRef]
- Sasaki, S.; Iwata, M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2007, 66, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Bates, G.P. History of genetic disease: The molecular genetics of Huntington disease—A history. Nat. Rev. Genet. 2005, 6, 766–773. [Google Scholar] [CrossRef]
- Polyzos, A.A.; McMurray, C.T. The chicken or the egg: Mitochondrial dysfunction as a cause or consequence of toxicity in Huntington’s disease. Mech. Ageing Dev. 2017, 161, 181–197. [Google Scholar] [CrossRef]
- Alexander, C.; Votruba, M.; Pesch, U.E.; Thiselton, D.L.; Mayer, S.; Moore, A.; Rodriguez, M.; Kellner, U.; Leo-Kottler, B.; Auburger, G.; et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 2000, 26, 211–215. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Chinnery, P.F. Dominant optic atrophy: Novel OPA1 mutations and revised prevalence estimates. Ophthalmology 2013, 120, 1712–1712.e1. [Google Scholar] [CrossRef] [PubMed]
- Lenaers, G.; Hamel, C.; Delettre, C.; Amati-Bonneau, P.; Procaccio, V.; Bonneau, D.; Reynier, P.; Milea, D. Dominant optic atrophy. Orphanet J. Rare Dis. 2012, 7, 46. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.; Alavi, M.V.; Kim, K.Y.; Kang, T.; Scott, R.T.; Noh, Y.H.; Lindsey, J.D.; Wissinger, B.; Ellisman, M.H.; Weinreb, R.N.; et al. A new vicious cycle involving glutamate excitotoxicity, oxidative stress and mitochondrial dynamics. Cell Death Dis. 2011, 2, e240. [Google Scholar] [CrossRef]
- Cretin, E.; Lopes, P.; Vimont, E.; Tatsuta, T.; Langer, T.; Gazi, A.; Sachse, M.; Yu-Wai-Man, P.; Reynier, P.; Wai, T. High-throughput screening identifies suppressors of mitochondrial fragmentation in OPA1 fibroblasts. EMBO Mol. Med. 2021, 13, e13579. [Google Scholar] [CrossRef]
- Zanna, C.; Ghelli, A.; Porcelli, A.M.; Karbowski, M.; Youle, R.J.; Schimpf, S.; Wissinger, B.; Pinti, M.; Cossarizza, A.; Vidoni, S.; et al. OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain 2008, 131, 352–367. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; LeVine, H., 3rd. Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta 2010, 1802, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Petrie, T.G.; Liu, Y.; Liu, J.; Fujioka, H.; Zhu, X. Parkinson’s disease-associated DJ-1 mutations impair mitochondrial dynamics and cause mitochondrial dysfunction. J. Neurochem. 2012, 121, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Gordon, D.M.; Pain, J.; Stemmler, T.L.; Dancis, A.; Pain, D. Frataxin directly stimulates mitochondrial cysteine desulfurase by exposing substrate-binding sites, and a mutant Fe-S cluster scaffold protein with frataxin-bypassing ability acts similarly. J. Biol. Chem. 2013, 288, 36773–36786. [Google Scholar] [CrossRef] [Green Version]
- Sahdeo, S.; Scott, B.D.; McMackin, M.Z.; Jasoliya, M.; Brown, B.; Wulff, H.; Perlman, S.L.; Pook, M.A.; Cortopassi, G.A. Dyclonine rescues frataxin deficiency in animal models and buccal cells of patients with Friedreich’s ataxia. Hum. Mol. Genet. 2014, 23, 6848–6862. [Google Scholar] [CrossRef] [Green Version]
- Stepanova, A.; Magrané, J. Mitochondrial dysfunction in neurons in Friedreich’s ataxia. Mol. Cell. Neurosci. 2020, 102, 103419. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, M.; Yano, M.; Namchai, C.; Yamamoto, S.; Ohtake, A.; Takayanagi, M.; Mori, M.; Niimi, H. Visualization of mitochondria with green fluorescent protein in cultured fibroblasts from patients with mitochondrial diseases. Biochem. Biophys. Res. Commun. 1997, 239, 580–584. [Google Scholar] [CrossRef]
- Pham, N.A.; Richardson, T.; Cameron, J.; Chue, B.; Robinson, B.H. Altered mitochondrial structure and motion dynamics in living cells with energy metabolism defects revealed by real time microscope imaging. Microsc. Microanal. 2004, 10, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, A.V.; Hermann, M.; Saks, V.; Hengster, P.; Margreiter, R. The cell-type specificity of mitochondrial dynamics. Int. J. Biochem. Cell Biol. 2009, 41, 1928–1939. [Google Scholar] [CrossRef]
- Franco, A.; Kitsis, R.N.; Fleischer, J.A.; Gavathiotis, E.; Kornfeld, O.S.; Gong, G.; Biris, N.; Benz, A.; Qvit, N.; Donnelly, S.K.; et al. Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature 2016, 540, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Pham, A.H.; McCaffery, J.M.; Chan, D.C. Mouse lines with photo-activatable mitochondria to study mitochondrial dynamics. Genesis 2012, 50, 833–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, A.H.; Chan, D.C. Analyzing mitochondrial dynamics in mouse organotypic slice cultures. Methods Enzymol. 2014, 547, 111–129. [Google Scholar]
- Saporta, M.A.; Dang, V.; Volfson, D.; Zou, B.; Xie, X.S.; Adebola, A.; Liem, R.K.; Shy, M.; Dimos, J.T. Axonal Charcot-Marie-Tooth disease patient-derived motor neurons demonstrate disease-specific phenotypes including abnormal electrophysiological properties. Exp. Neurol. 2015, 263, 190–199. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, F.; Ronchi, D.; Salani, S.; Nizzardo, M.; Fortunato, F.; Bordoni, A.; Stuppia, G.; Del Bo, R.; Piga, D.; Fato, R.; et al. Selective mitochondrial depletion, apoptosis resistance, and increased mitophagy in human Charcot-Marie-Tooth 2A motor neurons. Hum. Mol. Genet. 2016, 25, 4266–4281. [Google Scholar] [CrossRef]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Vermulst, M.; Wang, Y.E.; Chomyn, A.; Prolla, T.A.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 2010, 141, 280–289. [Google Scholar] [CrossRef] [Green Version]
- Cartoni, R.; Arnaud, E.; Médard, J.J.; Poirot, O.; Courvoisier, D.S.; Chrast, R.; Martinou, J.C. Expression of mitofusin 2(R94Q) in a transgenic mouse leads to Charcot-Marie-Tooth neuropathy type 2A. Brain 2010, 133, 1460–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, A.; Kontarakis, Z.; Gerri, C.; Nolte, H.; Hölper, S.; Krüger, M.; Stainier, D.Y. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 2015, 524, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Dang, X.; Walton, E.K.; Zablocka, B.; Baloh, R.H.; Shy, M.E.; Dorn, G.W., II. Mitochondrial Phenotypes in Genetically Diverse Neurodegenerative Diseases and their Response to Mitofusin Activation. Cells 2022, in press. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dorn, G.W., II; Dang, X. Predicting Mitochondrial Dynamic Behavior in Genetically Defined Neurodegenerative Diseases. Cells 2022, 11, 1049. https://doi.org/10.3390/cells11061049
Dorn GW II, Dang X. Predicting Mitochondrial Dynamic Behavior in Genetically Defined Neurodegenerative Diseases. Cells. 2022; 11(6):1049. https://doi.org/10.3390/cells11061049
Chicago/Turabian StyleDorn, Gerald W., II, and Xiawei Dang. 2022. "Predicting Mitochondrial Dynamic Behavior in Genetically Defined Neurodegenerative Diseases" Cells 11, no. 6: 1049. https://doi.org/10.3390/cells11061049
APA StyleDorn, G. W., II, & Dang, X. (2022). Predicting Mitochondrial Dynamic Behavior in Genetically Defined Neurodegenerative Diseases. Cells, 11(6), 1049. https://doi.org/10.3390/cells11061049