1. Introduction
DNA sequence amplification is a phenomenon that occurs predictably at defined stages during normal development in
Xenopus,
Drosophila,
Sciara, and
Tetrahymena [
1,
2,
3,
4]. These amplifications affect specific DNA regions and appear during narrow windows of development [
4]. In normal mammalian cells, gene amplification remained disregarded for many years.
We published the first evidence of gene amplifications during differentiation in human neural progenitor cells [
5], mouse neural stem and progenitor cells [
6], mouse and human myoblasts [
7], and mesenchymal stem cells [
8]. Additional reports were published on gene amplification during human and mouse trophoblast differentiations [
9,
10]. So far, the mechanism for amplification, or the initiation of gene amplification, has yet to be elucidated in differentiating cells. Here, we set out to investigate whether rereplication is a possible mechanism leading to gene amplifications in human myoblasts following differentiation induction. It is likely that mammalian gene amplification during differentiation originates from mechanisms that have been described for chorion gene amplification in
Drosophila eggshell cells. Reports of multiple origin-firings were published by Osheim YN as early as 1988 [
11], and Alexander JL in 2015 [
12].
The replication factors CDT1 and CDC6 are essential for origin licensing and replication initiation. There are several mechanisms that ensure the initiation of origins only once per cell cycle, including the degradation of CDT1 and CDC6, as well as CDT1 inhibition by geminin (GMNN) [
13,
14]. A recent study on transgenic mouse embryo stem cells with inducible
CDC6 and
CDT1 expressions revealed rereplication when
CDT1 and
CDC6 were simultaneously overexpressed. Here, we analyze
CDC6,
CDT1, and
GMNN expressions in differentiating human skeletal myoblasts (HSkM) cells during a time window, where our studies detected rereplication by fiber combing.
2. Materials and Methods
2.1. Cell Culture and Differentiation
HSkM-S cells derived from primary normal human skeletal myoblasts were obtained from Thermo Fischer Scientific (Life Technologies, Carlsbad, CA, USA). For proliferation, HSkM cells were cultivated in Dulbecco’s Modified Eagle Medium (DMEM) (Life Technologies, Carlsbad, CA, USA), supplemented by 10% FCS (Fetal calf serum) (Biochrom, Berlin, Germany). For differentiation, the cells were cultivated for 24 h in Dulbecco’s Modified Eagle Medium (DMEM) supplemented by 2% Horse Serum (PAN Biotech, Aidenbach, Germany), according to the manufacturers’ instructions.
2.2. Thymidine Analogue Treatment
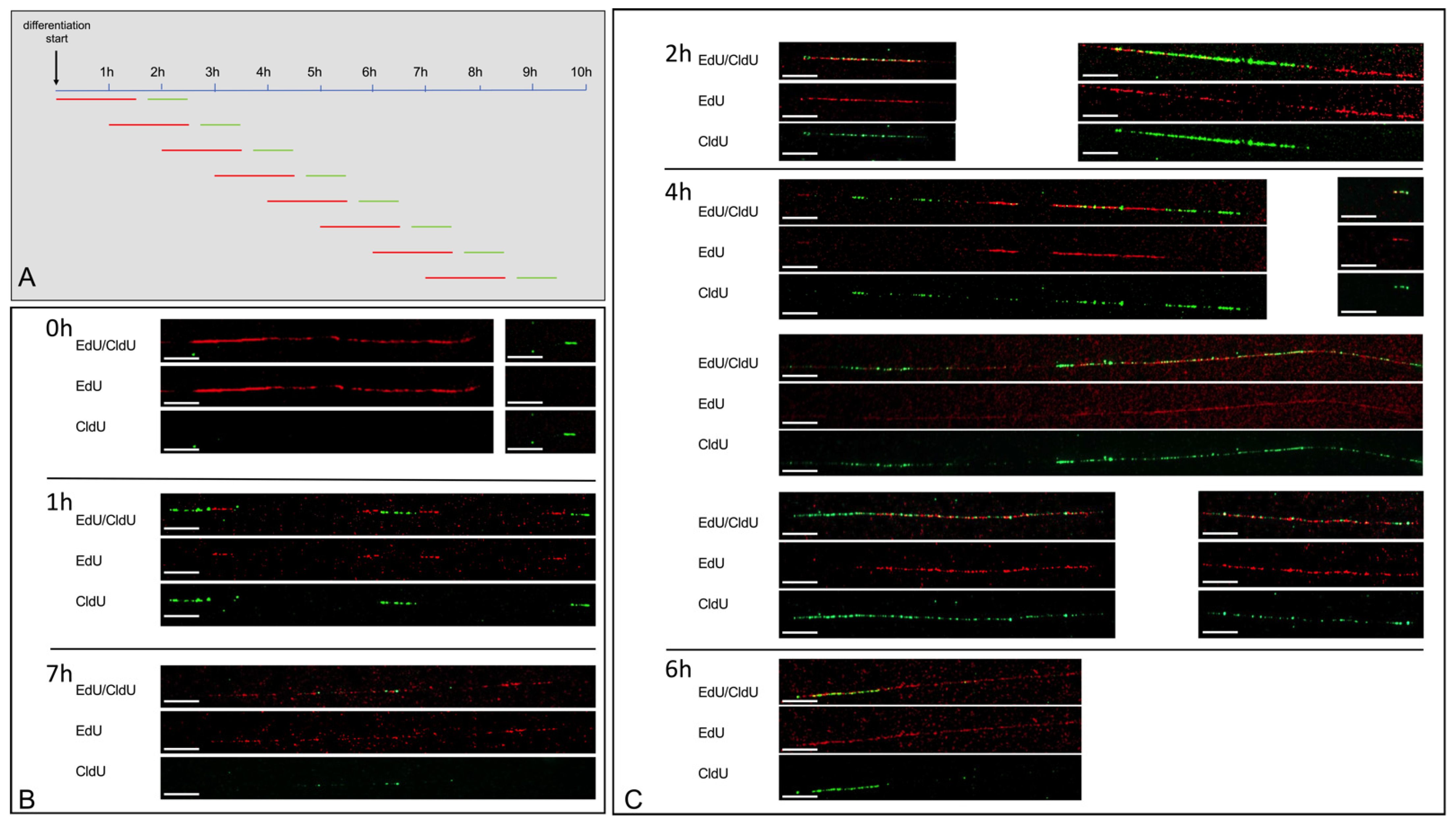
Cells were cultivated as described above. The media, that either supported proliferation or differentiation, were supplemented by thymidine analogues in two sequential pulses. A 90 min-long pulse with the first thymidine analogue was followed by a brief PBS wash step and a 15 min incubation in a medium without thymidine analogues, and was subsequently followed by a second 45 min pulse with a second, different, thymidine analogue. The thymidine analogue EdU (Life Technoligies, Carlsbad, CA, USA) was added to create a final concentration of 200 μM, CldU (Merck, Darmstadt, Germany) to create a final concentration of 250 µM, and IdU (Merck, Darmstadt, Germany) to create a final concentration of 30 µM. After all pulse labeling steps, the cells were harvested using Accutase, resuspended in PBS, and processed further.
2.3. Fiber Preparation for Molecular Combing
Harvested cells from the culture were diluted to 0.25 million cells/mL in PBS. The cells were then suspended in an agarose plug and allowed to digest overnight in an ESP buffer (containing proteinase K) at 50 °C. The plugs were then washed in a 1x TE buffer on an overhead rotator, and were subsequently digested overnight with agarase. The resulting DNA suspension was transferred to a combing reservoir obtained from the FiberPrep Kit (Genomic Vision, Bagneux, France) with a 0.1 M MES buffer at a pH of 6.5. The final DNA suspension in the FiberComb reservoirs were inserted into the FiberComb Molecular Combing Device, along with 1–2 silanized coverslips (Genomic Vision, Bagneux, France) per reservoir. The coverslips were then mechanically inserted into the DNA suspension and pulled out at a constant rate. This resulted in DNA fibers adhering to the silanized surface with a constant measurement of 2 kb/μm. Once the DNA was combed onto the coverslips, they were baked at 65 °C for two hours to fix the DNA onto the coverslip. They were then stored at −20 °C, or were processed further.
2.4. EdU Click-iT Reaction
Coverslips that were baked to fix the DNA were processed as described in the manual provided by Invitrogen™ (Thermo Fischer Scientific, Waltham, MA, USA). In brief, the coverslips were washed with 3% BSA and treated with the Click-iT™ reaction cocktail (containing Alexa-594), as detailed by Invitrogen™. This light-sensitive reaction was incubated for 30 min in a dark humid chamber at room temperature. After incubation, the coverslips were washed again in 3% BSA (Merck, Darmstadt, Germany). After washing, the solution was completely removed, and antibody immunostaining followed, as described below.
2.5. Fluorescent Immunostaining of Combed Fibers
Coverslips that were treated with the Click iT™ Imaging kit were then placed in a humid chamber with 10% goat serum diluted in PBS, and blocked for 30 min at 37 °C. This was followed by the rat anti BrdU (5-Bromo-2′-deoxyuridine) antibody for CldU (Abcam, Cambridge, UK), diluted 1:12.5 in 10% goat serum, and incubated for one hour in a humid chamber at 37 °C. The coverslips were subsequently washed in PBS, three times for 5 min each. Then, the corresponding secondary antibody solution (Alexa-488), diluted in 10% goat serum, was placed on the coverslips, which were incubated for 30 min in a humid chamber at room temperature. The coverslips were then washed again, dehydrated with an ethanol series, and mounted using SlowFade™ Gold (Invitrogen™, Thermo Fischer Scientific, Waltham, MA, USA). All coverslips were individually analyzed using the Olympus AX70 fluorescent microscope and the cellSens Dimension 1.8.1 Program from Olympus (Tokyo, Japan).
Cells with IdU/CldU pulse treatments for EasyComb (Genomic Vision) were harvested and embedded into agarose plugs using the Genomic Vision FiberPrep® Kit (Genomic Vision, Bagneux, France). Combing and immunostaining were performed according to the EasyComb procedures (Genomic Vision, Bagneux, France). Coverslips were scanned with the FiberVision® scanner and images were analyzed using Genomic Vision FiberStudio® software (Genomic Vision, Bagneux, France). Intact IdU (green) and CldU (red) replication tracks, flanked by counterstaining (blue), were selected and used for further validation.
2.6. Fluorescence In Situ Hybridization
The BAC clone RP11-611O2 (
MDM2 gene) is from the RP-11 (
http://www.chori.org/bacpac/, accessed on 1 October 2011) libraries of the Welcome Trust Sanger Institute and are available from SourceBioSciences, Germany. BAC-DNA (1 µg) was labeled with alexaFluor 488-dUTP (green fluorescence signals) using the FISH Tag DNA labeling Kit, according to the manufacturer’s instructions (Thermo Fischer Scientific, Waltham, MA, USA). Labeled probe DNA was precipitated in the presence of human Cot-1 DNA and resuspended in a hybridization mix (50% formamide, 20× SSPE, 20% dextrane sulfate, and 27% SDS).
HSkM cells were grown on glass slides and allowed to differentiate for 5 h. EdU (200 µM) was added for 45 min, and cells were briefly washed with PBS and supplied with a fresh differentiation medium for an additional 19 h. In addition, cells were differentiated for 2 h. Three hours after the differentiation induction, EdU was added for 3 h. Subsequently, cells were briefly washed with PBS and were supplied with a fresh differentiation medium for the remaining differentiation time (18 h). As a control experiment, HSkM cells were also cultured for 24 h in DMEM/10% FCS. Three hours after the proliferation start, EdU was added for 3 h. Again, cells were briefly washed with PBS and were supplied with a fresh DMEM/10% FCS medium for the remaining 18 h. Cells on glass slides were fixed in ice cold methanol for 15 min, washed in PBS for 5 min, and treated with 0.02% Tween-20 for 5 min. Slides were RNase-treated for 0.5 h at 37 °C and pepsin-treated for 10 min. Post-fixation was performed using 2.5% formaldehyde/1× PBS for 10 min at room temperature. Hybridization was performed over-night. After the post-hybridization washes, slides were treated according to the EdU Click-iT Reaction, as described above. Fluorescence images were captured with an Olympus AX70 microscope using cellSens software from Olympus.
2.7. RNA Isolation and qRT-PCR
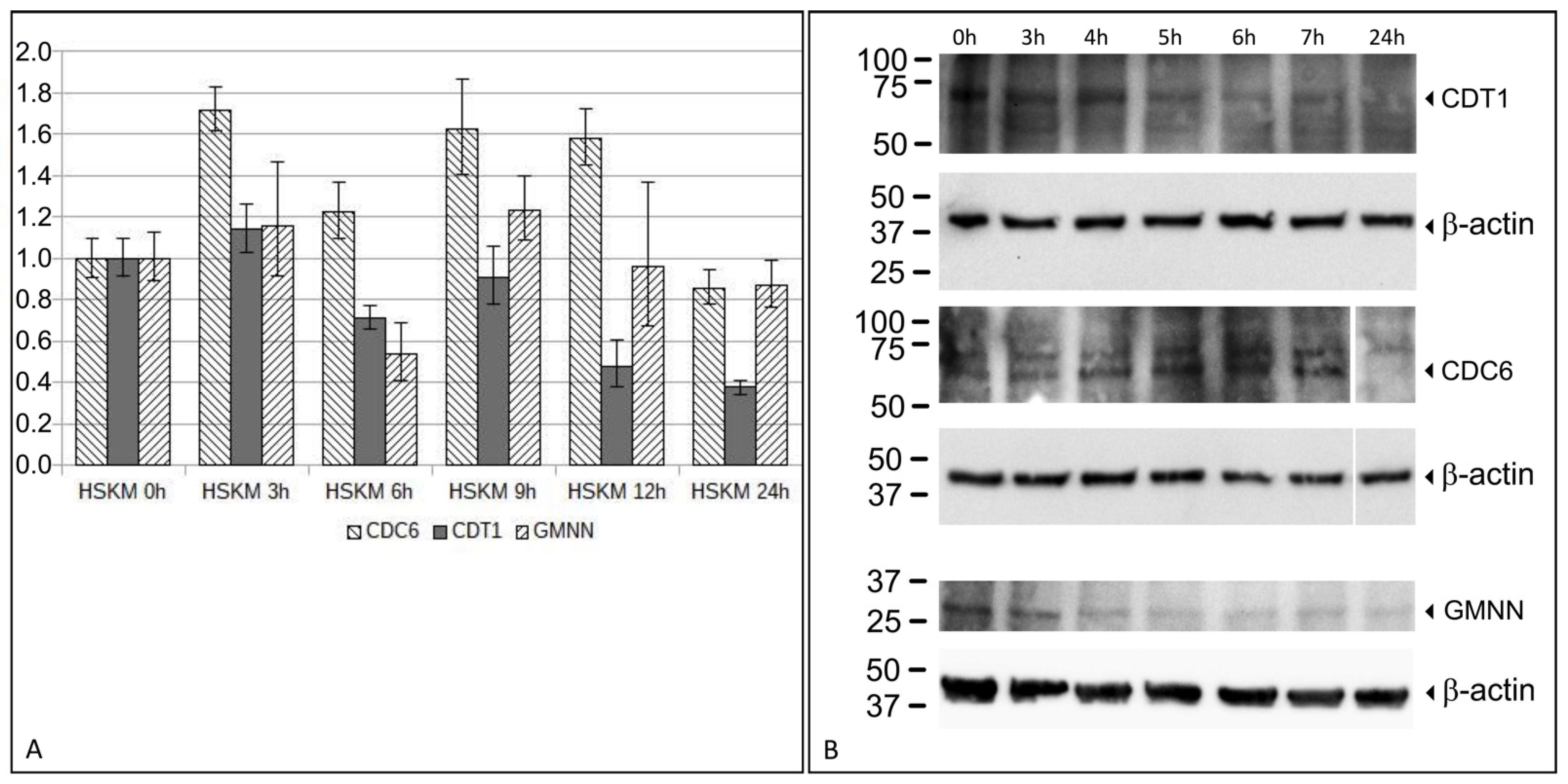
The total RNA from HSkM cells during 24 h of differentiation were isolated using the QIAGEN miRNeasy Mini Kit (QIAGEN, Hilden, Germany) after 3 h, 6 h, 9 h, 12 h, and 24 h, during the 24 h differentiation course. Undifferentiated HSkM cells served as a 0 h control. mRNA was converted to cDNA using the QuantiTect RT Kit (Qiagen GmbH, Hilden, Germany). qRT-PCR was performed using the SYBR® Green PCR Kit using 5 ng cDNA for qRT-PCR with QuantiTect Primer Assays. The Qiagen QuantiTect primer pairs are as follows: GAPDH (QT00079247), TBP (QT00000721), CDT1 (QT00020601), CDC6 (QT00065772), and GMNN (QT01019970). qRT-PCR was run on a StepOne™ Real-Time PCR System (Applied Biosystems™, Foster City, CA, USA). The relative expression was calculated with a ΔΔCt method with the endogenous controls GAPDH and TBP.
2.8. Protein Isolation and Western Blot Analysis
HSkM cells were harvested with Accutase, pelleted, and washed once with PBS. The pellet was resuspended in a RIPA buffer (Thermo Fisher Scientific) supplemented with a protease inhibitor (Sigma-Aldrich, St. Louis, MO, USA). The samples were sonicated at 20 joules for 2 s, incubated on ice for 30 min, and centrifuged at 14,000× g for 10 min. The supernatant was transferred to a new microcentrifuge tube. Twenty µg of total protein were denatured with a Laemmli buffer, separated by gel electrophoresis (SDS-PAGE) in a Mini-Protean® TGX Precast Gel (Bio-Rad Laboratories, Hercules, CA, USA), and subsequently transferred onto polyvinylidene difluoride (PVDF) membranes (Whatman, Maidstone, UK). Membranes/blots were blocked for 1 h at RT with TBS Blotto A (Santa Cruz Biotechnology, Dallas, TX, USA) and exposed to primary antibodies diluted in TBS Blotto A overnight at 4 °C with agitation. Membranes were subsequently washed three times with 1X-Tris buffered Saline with Tween-20 (TBS-T) (Santa Cruz Biotechnology) and exposed to the horseradish peroxidase (HRP)-conjugated secondary antibody diluted in 1X-TBS-T for 1 h. Membranes were then washed three times in 1X-TBS, developed with an enhanced chemiluminescence (ECL) reagent (Cell Signaling Technology, Danvers, MA, USA), and exposed to the ChemiDoc™ MP Imaging System to detect the chemiluminescence signals (Bio-Rad Laboratories). The primary antibodies used were rabbit anti-CDC6 (ab109315 at 1:2000, Abcam, Cambridge, UK), rabbit anti-CDT1 (ab202067 at 1:2000, Abcam), rabbit anti-GMNN (ab246509 at 1:1000, Abcam), and mouse anti-ß-Actin (5441, at 1:5000, Sigma-Aldrich, St. Louis, MO, USA) as a loading control.
4. Discussion
Replication normally occurs once per cell cycle [
17]. As demonstrated in Drosophila, this restriction can be circumvented to enable endoreplication during development. It is also known that during development, various cell types reveal polyploidy, including mammalian trophoblasts, mammalian megakaryocytes, and plant cells. Endocycles and endoreplications are postulated to account for this copy number gain [
18]. Several key regulators of replication control were reported to allow for rereplication when experimentally over-expressed [
14]. It has been reported that
CDT1 over-expression resulted in rereplication and gene amplification.
CDT1 is degraded during the mitotic cell cycle by E3 ubiquitinylation to prevent rereplication. Likewise,
CDC6 is degraded to prevent rereplication. In contrast, the repression or deletion of geminin (GMNN), an important regulator of replication control, resulted in rereplication [
19].
Studies by Munoz and colleagues revealed that
CDC6 overexpression was the limiting factor for origin relicensing, but it was not sufficient for rereplication induction. Likewise,
CDT1 overexpression alone was not sufficient for relicensing and rereplication [
14]. Only the simultaneous overexpression of
CDC6 and
CDT1 led to rereplication. Our expression analysis revealed both an increase in both
CDC6 and of
CDT1 expressions at 3 h after differentiation induction in HSkM cells. Protein expression levels of CDC6 and CDT1 were also elevated at specific time points. These observations are consistent with the idea that altered expression levels of CDC6 and CDT1 enable the observed rereplication in HSkM cells.
In addition, between 3 h and 6 h after differentiation induction, GMNN expression decreased. Accordingly, GMNN protein expression was reduced after 4 h and was not re-elevated to the initial expression level. These findings are consistent with the hypothesis that reduced or depleted GMNN expression contributes to the rereplication during this time window.
Although fiber combing is a very powerful technique for the visualization of rereplication, it requires rigid controls to exclude cross-detection between various thymidine analogues. Cross-detection may be due to antibody cross detection, fiber clumping, or label persistence.
As for antibody cross-detection, we used Edu and CldU as thymidine analogues that were identified with different detection systems, i.e., EdU was detected by a chemical Click-iT reaction, and CldU by an antibody binding. Accordingly, we did not find evidence for a cross detection between the thymidine analogues EdU and CldU. To further monitor repeated restarts in a given DNA sequence, we included a third thymidine analogue (IdU) pulse. Due to cross-detection of EdU and IdU, it was, however, not possible to use all three analogues (EdU, IdU, and CldU) in one experiment.
To monitor fiber clumping as a further potential cause of cross-detection, we counterstained the DNA with YOYO and ensured that multiple fibers were not overlapping.
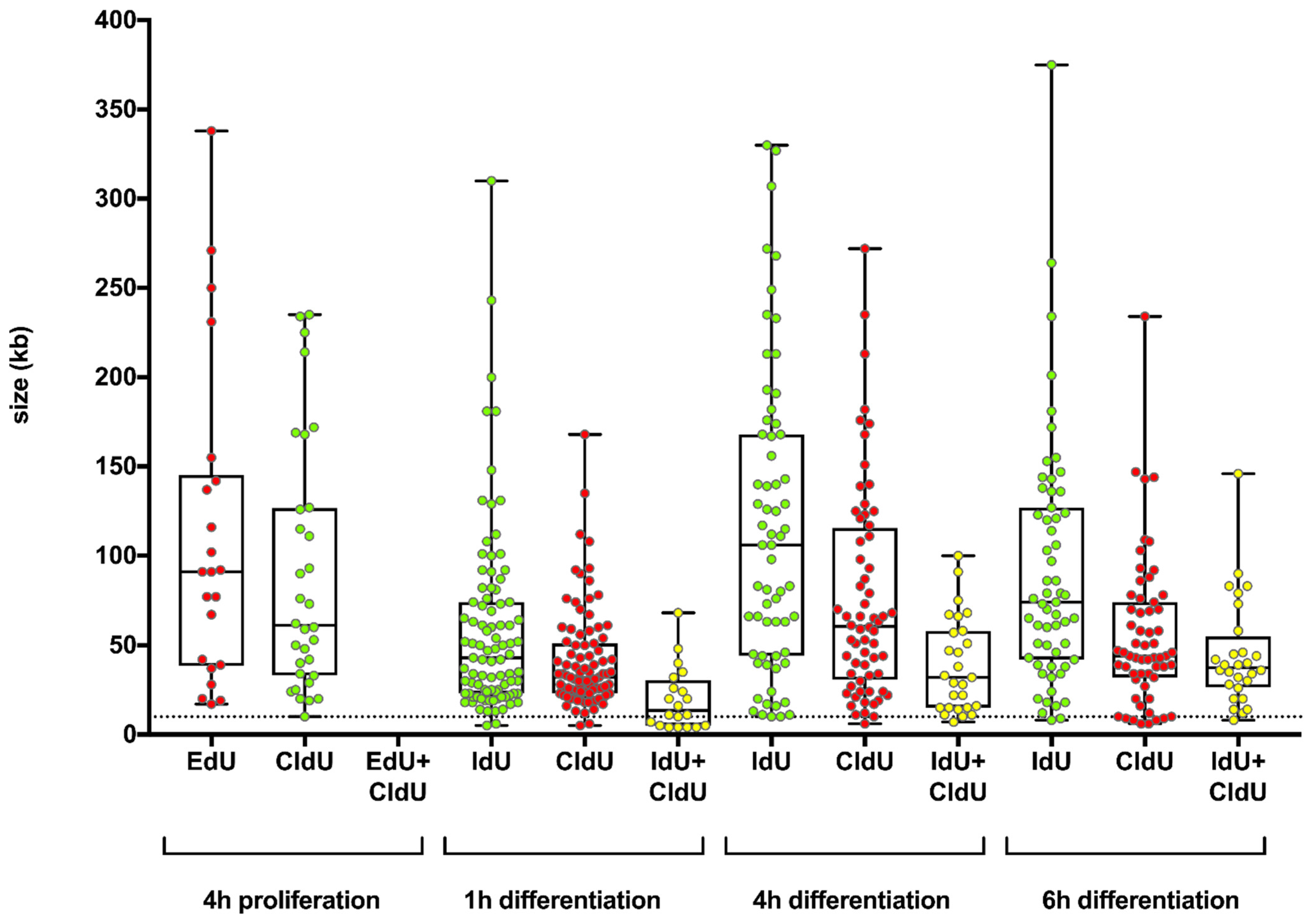
To counteract the potential persistence of labeling, we employed a PBS washing step after the first analogue pulse. The PBS washing step resulted in a reduced yellow staining, indicating a reduced analogue persistence due to the addition of a washing step. A further reduction in yellow staining was found after an additional incubation with an analogue free medium for 15 min. Nevertheless, we cannot completely rule out label persistence after the first pulse with the thymidine analogue. Towards a discrimination between rereplication and the potential label persistence, we only considered long uninterrupted yellow fibers. As a threshold, we chose a length of >10 kb.
Preliminary observations during our rereplication fiber-combing analysis, during differentiation, may indicate asymmetric replication bubbles (data not shown). Up until now, rereplication was mostly reported as a symmetric process described as an onion-like-shaped symmetric replication bubble. The
Drosophila chorion gene amplification, with symmetric replication bubbles, were shown as early as 1984 by Stark and Wahl [
20] and, more recently, in 2017 by Hua and Orr-Weaver [
21]. However, electron microscopy studies by Osheim 1988 suggested a lack of symmetry of the replication bubbles and replication forks during
Drosophila melanogaster chorion gene amplification [
11]. Further investigations with higher resolutions are required to clarify this issue.
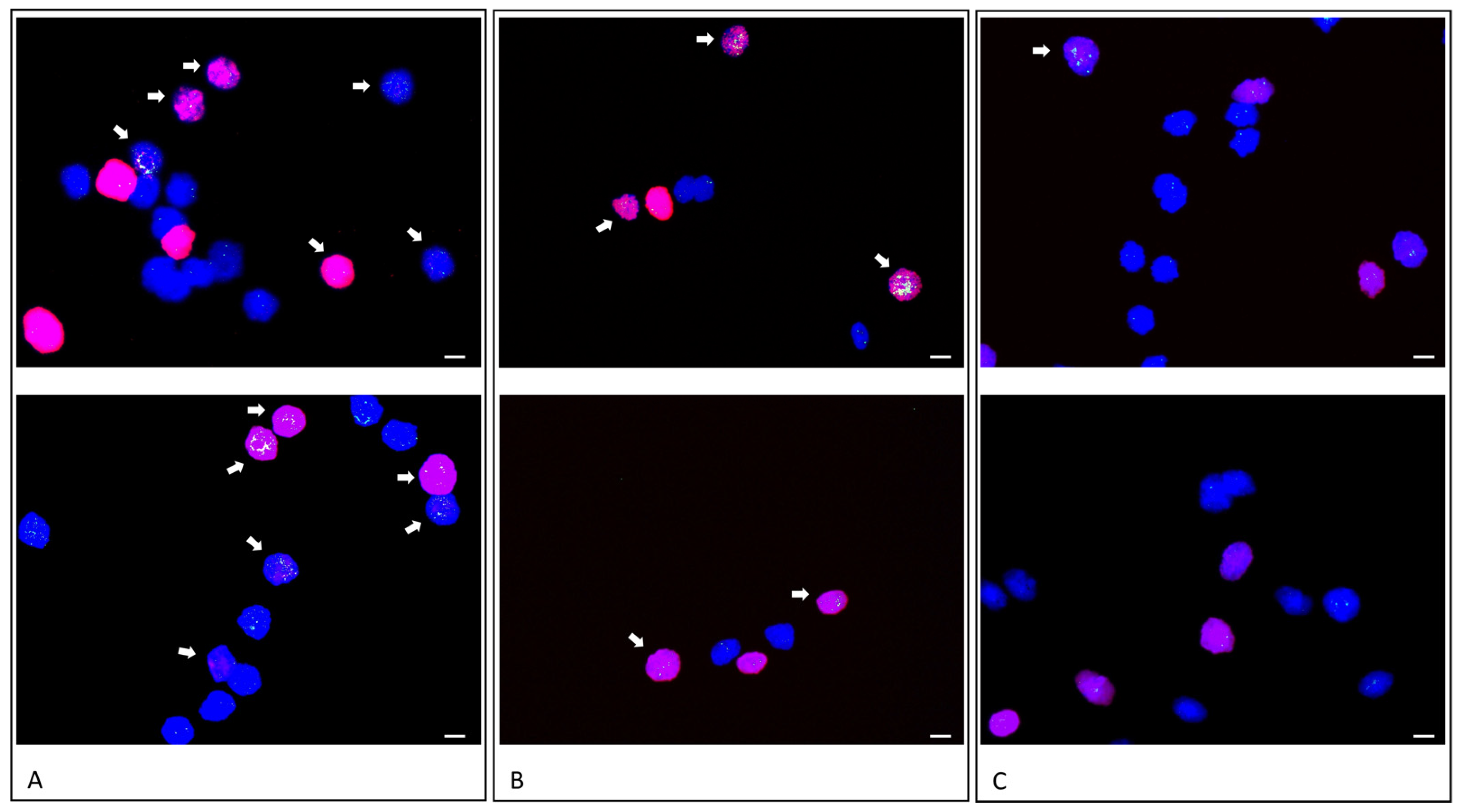
To correlate rereplication and gene amplification, we pulse treated differentiating HSkM cells with EdU for 3 h during the time window that showed a high rereplication frequency by fiber combing. We evidenced
MDM2 gene amplification by fluorescence in in situ hybridization. Multiple fluorescence signals for
MDM2, indicative of
MDM2 gene amplification, were colocalized with EdU incorporation in 13% of analyzed cell nuclei. In addition, 10% of analyzed nuclei revealed
MDM2 amplification without EdU incorporation. This observation is likely due to the amplification after EdU supplementation. Out of the analyzed nuclei, 21% revealed EdU incorporation with a normal diploid copy number of
MDM2. Previous amplification analyses revealed a heterogeneous pattern of gene amplifications during myogenic or neural differentiation [
6,
7], and it is very likely that other genes adjacent to
MDM2 were amplified during EdU incorporation. As a control-experiment, we pulse treated proliferating HSkM cells with EdU for 3 h during the same time window. The vast majority of nuclei revealed diploid
MDM2 fluorescence signals and no EdU incorporation (84%). Only 12% of nuclei revealed Edu incorporation with diploid
MDM2 fluorescence signals and only one nucleus (4%) revealed more than two
MDM2 fluorescence signals with EdU incorporation.
In conclusion, our study is the first to report rereplication in normal human cells during a restricted time window of early differentiation. During this short time window, we also found deregulated expression levels of genes involved in replication control. It remains to be investigated whether this time restriction is an essential component of controlled gene amplification during the differentiation processes in mammals. Additionally, it remains to be clarified as to what extent a temporal and spatial suspension of the time- and cell type-specific rereplication restriction contributes to uncontrolled gene amplifications during tumor development.


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}