
Microglial Endocannabinoid Signalling in AD

 ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Microglia
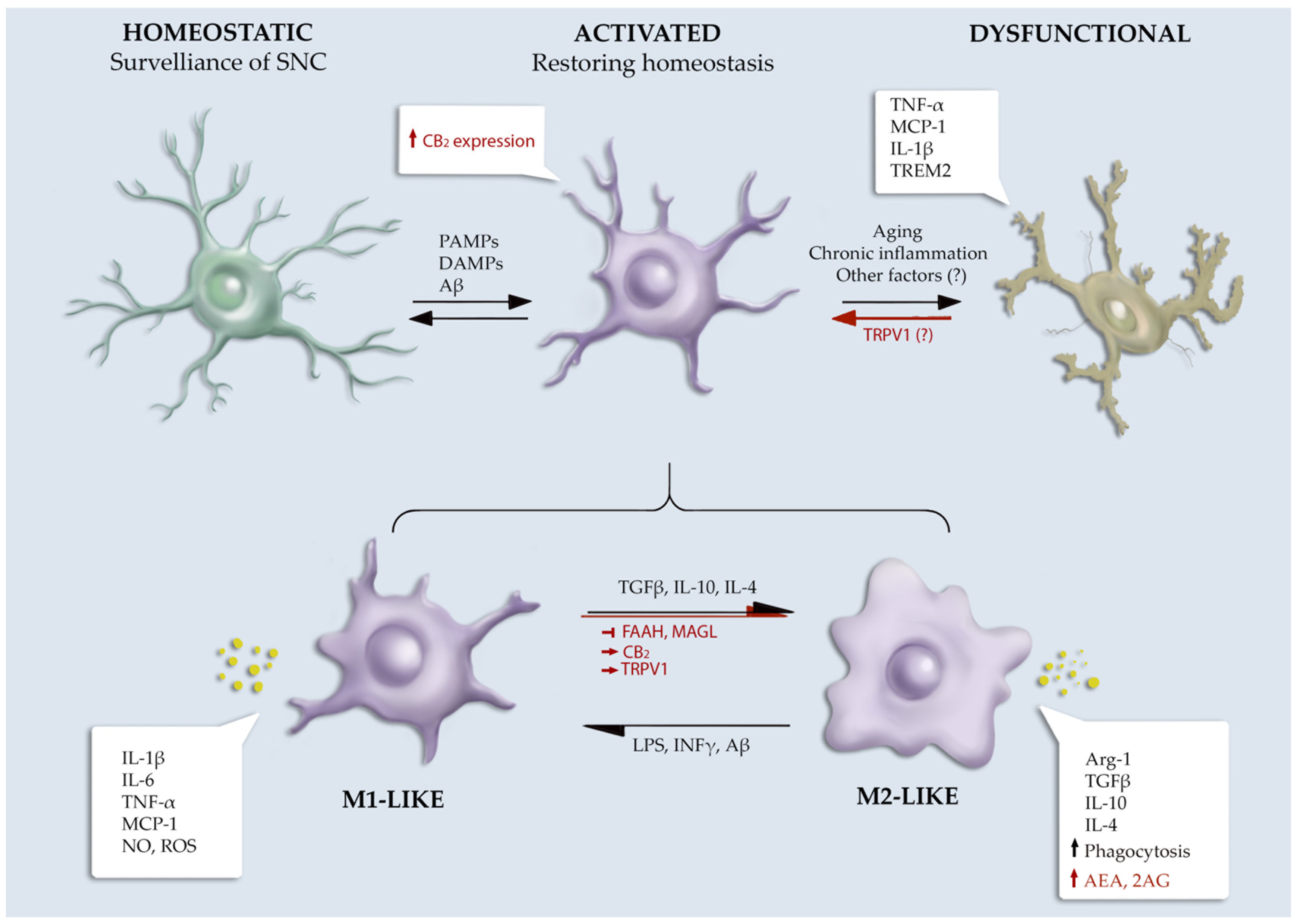
2.1. Microglial Functions and Phenotypes
2.1.1. Microglial Phenotypical Heterogeneity
Pro-Inflammatory-M1-like Microglia
Anti-Inflammatory-M2-like Microglia
2.2. Microglia and Alzheimer’s Disease
2.2.1. General Traits of Alzheimer’s Disease
2.2.2. The Involvement of Microglia in Alzheimer’s Disease
3. The Endocannabinoid System
3.1. eCBs Synthesis and Degradation
3.2. eCBs Receptors and Molecular Pathways
4. Microglial Endocannabinoid System in Alzheimer’s Disease
4.1. Role of the ECS in Microglial Functionality
4.1.1. eCBs Receptors
4.1.2. eCBs Metabolic Enzymes
4.2. Alteration of ECS in Alzheimer’s Disease
Human Studies
4.3. Preclinical Studies
4.4. Impact of Microglial Endocannabinoid Signalling in Alzheimer’s Disease
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Citron, M. Alzheimer’s disease: Strategies for disease modification. Nat. Rev. Drug Discov. 2010, 9, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Musiek, E.S.; Holtzman, D.M. Three dimensions of the amyloid hypothesis: Time, space and “wingmen”. Nat. Neurosci. 2015, 18, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.T. Microglial aging in the healthy CNS: Phenotypes, drivers, and rejuvenation. Front. Cell. Neurosci. 2013, 7, 800–806. [Google Scholar] [CrossRef] [Green Version]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef]
- Streit, W.J.; Xue, Q.-S.; Tischer, J.; Bechmann, I. Microglial pathology. Acta Neuropathol. Commun. 2014, 2, 142. [Google Scholar] [CrossRef]
- Chiurchiu, V.; Battistini, L.; Maccarrone, M. Endocannabinoid signalling in innate and adaptive immunity. Immunology 2015, 144, 352–364. [Google Scholar] [CrossRef]
- Stella, N. Endocannabinoid signaling in microglial cells. Neuropharmacology 2009, 56, 244–253. [Google Scholar] [CrossRef] [Green Version]
- Chiurchiù, V.; Leuti, A.; Maccarrone, M. Bioactive lipids and chronic inflammation: Managing the fire within. Front. Immunol. 2018, 9, 38. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2017, 18, 225–242. [Google Scholar] [CrossRef]
- Mecha, M.; Feliú, A.; Carrillo-Salinas, F.J.; Rueda-Zubiaurre, A.; Ortega-Gutiérrez, S.; de Sola, R.G.; Guaza, C. Endocannabinoids drive the acquisition of an alternative phenotype in microglia. Brain Behav. Immun. 2015, 49, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Chiang, N.; Dalli, J. New Pro-Resolving n-3 Mediators Bridge Resolution of Infectious Inflammation to Tissue Regeneration. Mol. Aspects Med. 2018, 64, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Young, A.P.; Denovan-Wright, E.M. The Dynamic Role of Microglia and the Endocannabinoid System in Neuroinflammation. Front. Pharmacol. 2022, 12, 4069. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Kigerl, K.A.; de Rivero Vaccari, J.P.; Dietrich, W.D.; Popovich, P.G.; Keane, R.W. Pattern recognition receptors and central nervous system repair. Exp. Neurol. 2014, 258, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Solé-Domènech, S.; Cruz, D.L.; Capetillo-Zarate, E.; Maxfield, F.R. The endocytic pathway in microglia during health, aging and Alzheimer’s disease. Ageing Res. Rev. 2016, 32, 89–103. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Wang, X.; Kalicki, C.; Menta, B.W.; Baumgardner, M.; Koppel, S.J.; Weidling, I.W.; Perez-Ortiz, J.; Wilkins, H.M.; Swerdlow, R.H. Effects of Microglial Cytokines on Alzheimer’s Disease-Related Phenomena. J. Alzheimers. Dis. 2019, 67, 1021–1034. [Google Scholar] [CrossRef]
- Guedes, J.R.; Lao, T.; Cardoso, A.L.; El Khoury, J. Roles of microglial and monocyte chemokines and their receptors in regulating Alzheimer’s disease-associated amyloid-β and tau pathologies. Front. Neurol. 2018, 9, 549. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet. Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.G.; Lue, L.F. Immune phenotypes of microglia in human neurodegenerative disease: Challenges to detecting microglial polarization in human brains. Alzheimers. Res. Ther. 2015, 7, 56. [Google Scholar] [CrossRef] [Green Version]
- Jurga, A.M.; Paleczna, M.; Kuter, K.Z. Overview of General and Discriminating Markers of Differential Microglia Phenotypes. Front. Cell. Neurosci. 2020, 14, 198. [Google Scholar] [CrossRef]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Investig. 2017, 127, 3240–3249. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Are “resting” microglia more “M2”? Front. Immunol. 2014, 5, 594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shui, X.; Sun, R.; Wan, L.; Zhang, B.; Xiao, B.; Luo, Z. Microglial Phenotypic Transition: Signaling Pathways and Influencing Modulators Involved in Regulation in Central Nervous System Diseases. Front. Cell. Neurosci. 2021, 15, 359. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.T.; Wu, W.F.; Deng, Y.H.; Ge, J.W. Modulators of microglia activation and polarization in ischemic stroke (Review). Mol. Med. Rep. 2020, 21, 2006–2018. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.K.; Mantovani, A. Orchestration of metabolism by macrophages. Cell Metab. 2012, 15, 432–437. [Google Scholar] [CrossRef] [Green Version]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflammation 2014, 11, 98. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization from M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 75. [Google Scholar] [CrossRef] [PubMed]
- Song, W.M.; Colonna, M. The identity and function of microglia in neurodegeneration. Nat. Immunol. 2018, 19, 1048–1058. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Faustino, J.; Woo, M.S.; Derugin, N.; Vexler, Z.S. Lack of the scavenger receptor CD36 alters microglial phenotypes after neonatal stroke. J. Neurochem. 2015, 135, 445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinaldi, M.; Thomas, L.; Mathieu, P.; Carabias, P.; Troncoso, M.F.; Pasquini, J.M.; Rabinovich, G.A.; Pasquini, L.A. Galectin-1 circumvents lysolecithin-induced demyelination through the modulation of microglial polarization/phagocytosis and oligodendroglial differentiation. Neurobiol. Dis. 2016, 96, 127–143. [Google Scholar] [CrossRef]
- Lampron, A.; Larochelle, A.; Laflamme, N.; Préfontaine, P.; Plante, M.M.; Sánchez, M.G.; Wee Yong, V.; Stys, P.K.; Tremblay, M.È.; Rivest, S. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J. Exp. Med. 2015, 212, 481–495. [Google Scholar] [CrossRef] [Green Version]
- Nizami, S.; Hall-Roberts, H.; Warrier, S.; Cowley, S.A.; Di Daniel, E. Microglial inflammation and phagocytosis in Alzheimer’s disease: Potential therapeutic targets. Br. J. Pharmacol. 2019, 176, 3515. [Google Scholar] [CrossRef] [Green Version]
- Tiberi, M.; Chiurchiù, V. Specialized Pro-resolving Lipid Mediators and Glial Cells: Emerging Candidates for Brain Homeostasis and Repair. Front. Cell. Neurosci. 2021, 15, 136. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, L.; Ye, X.; Hao, Q.; Zhang, T.; Cui, G.; Yu, M. Nrf2/ARE pathway inhibits ROS-induced NLRP3 inflammasome activation in BV2 cells after cerebral ischemia reperfusion. Inflamm. Res. 2018, 67, 57–65. [Google Scholar] [CrossRef]
- Dobri, A.M.; Dudău, M.; Enciu, A.M.; Hinescu, M.E. CD36 in Alzheimer’s Disease: An Overview of Molecular Mechanisms and Therapeutic Targeting. Neuroscience 2021, 453, 301–311. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Cai, W.; Dai, X.; Chen, J.; Zhao, J.; Xu, M.; Zhang, L.; Yang, B.; Zhang, W.; Rocha, M.; Nakao, T.; et al. STAT6/Arg1 promotes microglia/macrophage efferocytosis and inflammation resolution in stroke mice. JCI Insight 2019, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Fernández-Suárez, D. Alternatively activated microglia and macrophages in the central nervous system. Prog. Neurobiol. 2015, 131, 65–86. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2018 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2018, 14, 367–429. [Google Scholar] [CrossRef]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2015, 17, 22–35. [Google Scholar] [CrossRef]
- Okello, A.; Edison, M.P.; Archer, M.H.; Turkheimer, M.F.; Kennedy, J.; Bullock, M.R.; Walker, M.Z.; Kennedy, A.; Fox, N.; Rossor, M.; et al. Microglial activation and amyloid deposition in mild cognitive impairment A PET study. Neurology 2009, 72, 56–62. [Google Scholar] [CrossRef] [Green Version]
- Femminella, G.D.; Dani, M.; Wood, M.; Fan, Z.; Calsolaro, V.; Atkinson, R.; Edginton, T.; Hinz, R.; Brooks, D.J.; Edison, P. Microglial activation in early Alzheimer trajectory is associated with higher gray matter volume. Neurology 2019, 92, e1331–e1343. [Google Scholar] [CrossRef] [Green Version]
- Hanzel, C.E.; Pichet-Binette, A.; Pimentel, L.S.B.; Iulita, M.F.; Allard, S.; Ducatenzeiler, A.; Do Carmo, S.; Cuello, A.C. Neuronal driven pre-plaque inflammation in a transgenic rat model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2249–2262. [Google Scholar] [CrossRef]
- Philippens, I.H.; Ormel, P.R.; Baarends, G.; Johansson, M.; Remarque, E.J.; Doverskog, M. Acceleration of Amyloidosis by Inflammation in the Amyloid-Beta Marmoset Monkey Model of Alzheimer’s Disease. J. Alzheimers. Dis. 2017, 55, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Streit, W.J.; Braak, H.; Xue, Q.S.; Bechmann, I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 475. [Google Scholar] [CrossRef] [Green Version]
- Venegas, C.; Heneka, M.T. Danger-associated molecular patterns in Alzheimer’s disease. J. Leukoc. Biol. 2017, 101, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Maciuszek, M.; Cacace, A.; Brennan, E.; Godson, C.; Chapman, T.M. Recent advances in the design and development of formyl peptide receptor 2 (FPR2/ALX) agonists as pro-resolving agents with diverse therapeutic potential. Eur. J. Med. Chem. 2021, 213, 113167. [Google Scholar] [CrossRef] [PubMed]
- Plescher, M.; Seifert, G.; Hansen, J.N.; Bedner, P.; Steinhäuser, C.; Halle, A. Plaque-dependent morphological and electrophysiological heterogeneity of microglia in an Alzheimer’s disease mouse model. Glia 2018, 66, 1464–1480. [Google Scholar] [CrossRef]
- Bisht, K.; Sharma, K.P.; Lecours, C.; Gabriela Sánchez, M.; El Hajj, H.; Milior, G.; Olmos-Alonso, A.; Gómez-Nicola, D.; Luheshi, G.; Vallières, L.; et al. Dark microglia: A new phenotype predominantly associated with pathological states. Glia 2016, 64, 826–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristino, L.; Bisogno, T.; Di Marzo, V. Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat. Rev. Neurol. 2020, 16, 9–29. [Google Scholar] [CrossRef]
- Leuti, A.; Fazio, D.; Fava, M.; Piccoli, A.; Oddi, S.; Maccarrone, M. Bioactive lipids, inflammation and chronic diseases. Adv. Drug Deliv. Rev. 2020, 159, 133–169. [Google Scholar] [CrossRef]
- Lutz, B.; Marsicano, G.; Maldonado, R.; Hillard, C.J. The endocannabinoid system in guarding against fear, anxiety and stress. Nat. Rev. Neurosci. 2015, 16, 705. [Google Scholar] [CrossRef]
- Maccarrone, M.; Finazzi-Agró, A. The endocannabinoid system, anandamide and the regulation of mammalian cell apoptosis. Cell Death Differ. 2003, 10, 946–955. [Google Scholar] [CrossRef] [Green Version]
- Binte Mustafiz, S.S.; Uyama, T.; Morito, K.; Takahashi, N.; Kawai, K.; Hussain, Z.; Tsuboi, K.; Araki, N.; Yamamoto, K.; Tanaka, T.; et al. Intracellular Ca2+-dependent formation of N-acyl-phosphatidylethanolamines by human cytosolic phospholipase A2ε. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2019, 1864, 158515. [Google Scholar] [CrossRef]
- Fezza, F.; Bari, M.; Florio, R.; Talamonti, E.; Feole, M.; Maccarrone, M. Endocannabinoids, Related Compounds and Their Metabolic Routes. Molecules 2014, 19, 17078. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, L.; Harvey-White, J.; Osei-Hyiaman, D.; Razdan, R.; Gong, Q.; Chan, A.C.; Zhou, Z.; Huang, B.X.; Kim, H.Y.; et al. A biosynthetic pathway for anandamide. Proc. Natl. Acad. Sci. USA 2006, 103, 13345. [Google Scholar] [CrossRef] [Green Version]
- Stella, N.; Schweitzer, P.; Plomelli, D. A second endogenous cannabinoid that modulates long-term potentiation. Nature 1997, 388, 773–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisogno, T.; Howell, F.; Williams, G.; Minassi, A.; Cascio, M.G.; Ligresti, A.; Matias, I.; Schiano-Moriello, A.; Paul, P.; Williams, E.J.; et al. Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J. Cell Biol. 2003, 163, 463–468. [Google Scholar] [CrossRef]
- Hsu, K.L.; Tsuboi, K.; Adibekian, A.; Pugh, H.; Masuda, K.; Cravatt, B.F. DAGLβ inhibition perturbs a lipid network involved in macrophage inflammatory responses. Nat. Chem. Biol. 2012, 8, 999–1007. [Google Scholar] [CrossRef]
- Nakane, S.; Oka, S.; Arai, S.; Waku, K.; Ishima, Y.; Tokumura, A.; Sugiura, T. 2-Arachidonoyl-sn-glycero-3-phosphate, an arachidonic acid-containing lysophosphatidic acid: Occurrence and rapid enzymatic conversion to 2-arachidonoyl-sn-glycerol, a cannabinoid receptor ligand, in rat brain. Arch. Biochem. Biophys. 2002, 402, 51–58. [Google Scholar] [CrossRef]
- Higgs, H.N.; Glomset, J.A. Identification of a phosphatidic acid-preferring phospholipase A1 from bovine brain and testis. Proc. Natl. Acad. Sci. USA 1994, 91, 9574–9578. [Google Scholar] [CrossRef] [Green Version]
- Murataeva, N.; Straiker, A.; Mackie, K. Parsing the players: 2-arachidonoylglycerol synthesis and degradation in the CNS. Br. J. Pharmacol. 2014, 171, 1379–1391. [Google Scholar] [CrossRef] [Green Version]
- Araujo, D.J.; Prakash, N.; Mechoulam, R.; Saijo, K.; Tang, Y.; Tjoa, K.; Guaza, C. The Endocannabinoid System as a Window into Microglial Biology and Its Relationship to Autism. Front. Cell. Neurosci. 2019, 13, 424. [Google Scholar] [CrossRef]
- Cravatt, B.F.; Giang, D.K.; Mayfield, S.P.; Boger, D.L.; Lerner, R.A.; Gilula, N.B. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 1996, 384, 83–87. [Google Scholar] [CrossRef]
- Battista, N.; Di Tommaso, M.; Bari, M.; Maccarrone, M. The endocannabinoid system: An overview. Front. Behav. Neurosci. 2012, 6, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczocha, M.; Haj-Dahmane, S. Mechanisms of endocannabinoid transport in the brain. Br. J. Pharmacol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kano, M.; Ohno-Shosaku, T.; Hashimotodani, Y.; Uchigashima, M.; Watanabe, M. Endocannabinoid-mediated control of synaptic transmission. Physiol. Rev. 2009, 89, 309–380. [Google Scholar] [CrossRef]
- Blankman, J.L.; Simon, G.M.; Cravatt, B.F. A Comprehensive Profile of Brain Enzymes that Hydrolyze the Endocannabinoid 2-Arachidonoylglycerol. Chem. Biol. 2007, 14, 1347–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdeolivas, S.; Pazos, M.R.; Bisogno, T.; Piscitelli, F.; Iannotti, F.A.; Allarà, M.; Sagredo, O.; Di Marzo, V.; Fernández-Ruiz, J. The inhibition of 2-arachidonoyl-glycerol (2-AG) biosynthesis, rather than enhancing striatal damage, protects striatal neurons from malonate-induced death: A potential role of cyclooxygenase-2-dependent metabolism of 2-AG. Cell Death Dis. 2013, 4, e862. [Google Scholar] [CrossRef] [Green Version]
- Pertwee, R.G. Receptors and channels targeted by synthetic cannabinoid receptor agonists and antagonists. Curr. Med. Chem. 2010, 17, 1360–1381. [Google Scholar] [CrossRef] [Green Version]
- Duffy, S.S.; Hayes, J.P.; Fiore, N.T.; Moalem-Taylor, G. The cannabinoid system and microglia in health and disease. Neuropharmacology 2021, 190, 108555. [Google Scholar] [CrossRef]
- Klein, T.W. Cannabinoid-based drugs as anti-inflammatory therapeutics. Nat. Rev. Immunol. 2005, 5, 400–411. [Google Scholar] [CrossRef]
- Ibsen, M.S.; Connor, M.; Glass, M. Cannabinoid CB1 and CB2 Receptor Signaling and Bias. Cannabis Cannabinoid Res. 2017, 2, 48. [Google Scholar] [CrossRef] [Green Version]
- Connor, M.; Bagley, E.E.; Mitchell, V.A.; Ingram, S.L.; Christie, M.J.; Humphrey, P.P.A.; Vaughan, C.W. Cellular actions of somatostatin on rat periaqueductal grey neurons in vitro. Br. J. Pharmacol. 2004, 142, 1273. [Google Scholar] [CrossRef] [Green Version]
- Ativie, F.; Komorowska, J.A.; Beins, E.; Albayram, Ö.; Zimmer, T.; Zimmer, A.; Tejera, D.; Heneka, M.; Bilkei-Gorzo, A. Cannabinoid 1 Receptor Signaling on Hippocampal GABAergic Neurons Influences Microglial Activity. Front. Mol. Neurosci. 2018, 11, 295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, M.; Sackett, S.; Zhang, Y. Endocannabinoid Modulation of Microglial Phenotypes in Neuropathology. Front. Neurol. 2020, 11, 87. [Google Scholar] [CrossRef] [PubMed]
- Scotter, E.L.; Abood, M.E.; Glass, M. The endocannabinoid system as a target for the treatment of neurodegenerative disease. Br. J. Pharmacol. 2010, 160, 480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maccarrone, M.; Guzmán, M.; MacKie, K.; Doherty, P.; Harkany, T. Programming of neural cells by (endo)cannabinoids: From physiological rules to emerging therapies. Nat. Rev. Neurosci. 2014, 15, 786–801. [Google Scholar] [CrossRef] [Green Version]
- Sappington, R.M.; Calkins, D.J. Contribution of TRPV1 to microglia-derived IL-6 and NFkappaB translocation with elevated hydrostatic pressure. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3004–3017. [Google Scholar] [CrossRef] [Green Version]
- Schilling, J.; Kluge, A. Barriers to organizational learning: An integration of theory and research. Int. J. Manag. Rev. 2009, 11, 337–360. [Google Scholar] [CrossRef]
- Miyake, T.; Shirakawa, H.; Nakagawa, T.; Kaneko, S. Activation of Mitochondrial Transient Receptor Potential Vanilloid 1 Channel Contributes to Microglial Migration. Glia 2015, 63, 1870–1882. [Google Scholar] [CrossRef] [Green Version]
- Marrone, M.C.; Morabito, A.; Giustizieri, M.; Chiurchiù, V.; Leuti, A.; Mattioli, M.; Marinelli, S.; Riganti, L.; Lombardi, M.; Murana, E.; et al. TRPV1 channels are critical brain inflammation detectors and neuropathic pain biomarkers in mice. Nat. Commun. 2017, 8, 15292. [Google Scholar] [CrossRef]
- Friedman, D.; French, J.A.; Maccarrone, M. Safety, efficacy, and mechanisms of action of cannabinoids in neurological disorders. Lancet. Neurol. 2019, 18, 504–512. [Google Scholar] [CrossRef]
- Mecha, M.; Carrillo-Salinas, F.J.; Feliú, A.; Mestre, L.; Guaza, C. Microglia activation states and cannabinoid system: Therapeutic implications. Pharmacol. Ther. 2016, 166, 40–55. [Google Scholar] [CrossRef]
- Cabral, G.A.; Harmon, K.N.; Carlisle, S.J. Cannabinoid-mediated inhibition of inducible nitric oxide production by rat microglial cells: Evidence for cb1 receptor participation. Adv. Exp. Med. Biol. 2001, 493, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Cutando, L.; Busquets-Garcia, A.; Puighermanal, E.; Gomis-González, M.; Delgado-García, J.M.; Gruart, A.; Maldonado, R.; Ozaita, A. Microglial activation underlies cerebellar deficits produced by repeated cannabis exposure. J. Clin. Investig. 2013, 123, 2816–2831. [Google Scholar] [CrossRef]
- Lou, Z.Y.; Cheng, J.; Wang, X.R.; Zhao, Y.F.; Gan, J.; Zhou, G.Y.; Liu, Z.G.; Xiao, B.G. The inhibition of CB1 receptor accelerates the onset and development of EAE possibly by regulating microglia/macrophages polarization. J. Neuroimmunol. 2018, 317, 37–44. [Google Scholar] [CrossRef] [PubMed]
- De Meij, J.; Alfanek, Z.; Morel, L.; Decoeur, F.; Leyrolle, Q.; Picard, K.; Carrier, M.; Aubert, A.; Séré, A.; Lucas, C.; et al. Microglial Cannabinoid Type 1 Receptor Regulates Brain Inflammation in a Sex-Specific Manner. Cannabis Cannabinoid Res. 2021, 6, 488–507. [Google Scholar] [CrossRef] [PubMed]
- Benito, C.; Núñez, E.; Tolón, R.M.; Carrier, E.J.; Rábano, A.; Hillard, C.J.; Romero, J. Cannabinoid CB 2 Receptors and Fatty Acid Amide Hydrolase Are Selectively Overexpressed in Neuritic Plaque-Associated Glia in Alzheimer’s Disease Brains. J. Neurosci. 2003, 23, 11136–11141. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Gálvez, Y.; Palomo-Garo, C.; Fernández-Ruiz, J.; García, C. Potential of the cannabinoid CB(2) receptor as a pharmacological target against inflammation in Parkinson’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 64, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Yiangou, Y.; Facer, P.; Durrenberger, P.; Chessell, I.P.; Naylor, A.; Bountra, C.; Banati, R.R.; Anand, P. COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol. 2006, 6, 12. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Das, S.; Williams, E.A.; Moore, D.; Jones, J.D.; Zahm, D.S.; Ndengele, M.M.; Lechner, A.J.; Howlett, A.C. Lipopolysaccharide and cyclic AMP regulation of CB(2) cannabinoid receptor levels in rat brain and mouse RAW 264.7 macrophages. J. Neuroimmunol. 2006, 181, 82–92. [Google Scholar] [CrossRef]
- Bisogno, T.; Oddi, S.; Piccoli, A.; Fazio, D.; Maccarrone, M. Type-2 cannabinoid receptors in neurodegeneration. Pharmacol. Res. 2016, 111, 721–730. [Google Scholar] [CrossRef]
- Carrier, E.J.; Kearn, C.S.; Barkmeier, A.J.; Breese, N.M.; Yang, W.; Nithipatikom, K.; Pfister, S.L.; Campbell, W.B.; Hillard, C.J. Cultured Rat Microglial Cells Synthesize the Endocannabinoid 2-Arachidonylglycerol, Which Increases Proliferation via a CB 2 Receptor-Dependent Mechanism. Mol. Pharmacol. 2004, 65, 999–1007. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Jia, J.; Liu, X.; Bai, F.; Wang, Q.; Xiong, L. Activation of murine microglial N9 cells is attenuated through cannabinoid receptor CB2 signaling. Biochem. Biophys. Res. Commun. 2015, 458, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, B.G.; Blázquez, C.; del Pulgar, T.G.; Guzmán, M.; de Ceballos, M.L. Prevention of Alzheimer’s Disease Pathology by Cannabinoids: Neuroprotection Mediated by Blockade of Microglial Activation. J. Neurosci. 2005, 25, 1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrhart, J.; Obregon, D.; Mori, T.; Hou, H.; Sun, N.; Bai, Y.; Klein, T.; Fernandez, F.; Tan, J.; Shytle, D. Stimulation of cannabinoid receptor 2 (CB2) suppresses microglial activation. J. Neuroinflammation 2005, 2, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malek, N.; Popiolek-Barczyk, K.; Mika, J.; Przewlocka, B.; Starowicz, K. Anandamide, Acting via CB2 Receptors, Alleviates LPS-Induced Neuroinflammation in Rat Primary Microglial Cultures. Neural Plast. 2015, 2015, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correa, F.; Hernang Omez, M.; Mestre, L.; Lor, F.I.; Spagnolo, A.; Docagne, F.; Marzo, V.D.I.; Guaza, C. Anandamide Enhances IL-10 Production in Activated Microglia by Targeting CB 2 Receptors: Roles of ERK1/2, JNK, and NF-jB. Glia 2010, 58, 135–147. [Google Scholar] [CrossRef]
- Hernangómez, M.; Mestre, L.; Correa, F.G.; Loría, F.; Mecha, M.; Iñigo, P.M.; Docagne, F.; Williams, R.O.; Borrell, J.; Guaza, C. CD200-CD200R1 interaction contributes to neuroprotective effects of anandamide on experimentally induced inflammation. Glia 2012, 60, 1437–1450. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Hou, B.; Liang, P.; Lu, X.; Wu, Y.; Zhang, X.; Fan, Y.; Liu, Y.; Chen, T.; Liu, W.; et al. ARTICLE OPEN TRPV1 channel mediates NLRP3 inflammasome-dependent neuroinflammation in microglia. Cell Death Dis. 2021, 12, 1159. [Google Scholar] [CrossRef]
- Park, E.S.; Kim, S.R.; Jin, B.K. Transient receptor potential vanilloid subtype 1 contributes to mesencephalic dopaminergic neuronal survival by inhibiting microglia-originated oxidative stress. Brain Res. Bull. 2012, 89, 92–96. [Google Scholar] [CrossRef]
- Stella, N. Cannabinoid and cannabinoid-like receptors in microglia, astrocytes, and astrocytomas. Glia 2010, 58, 1017–1030. [Google Scholar] [CrossRef] [Green Version]
- Walter, L.; Franklin, A.; Witting, A.; Wade, C.; Xie, Y.; Kunos, G.; Mackie, K.; Stella, N. Nonpsychotropic Cannabinoid Receptors Regulate Microglial Cell Migration. J. Neurosci. 2003, 23, 1398–1405. [Google Scholar] [CrossRef] [Green Version]
- Muccioli, G.G.; Xu, C.; Odah, E.; Cudaback, E.; Cisneros, J.A.; Lambert, D.M.; Rodríguez, M.L.L.; Bajjalieh, S.; Stella, N. Identification of a novel endocannabinoid-hydrolyzing enzyme expressed by microglial cells. J. Neurosci. 2007, 27, 2883–2889. [Google Scholar] [CrossRef] [Green Version]
- Makara, J.K.; Mor, M.; Fegley, D.; Szabó, S.I.; Kathuria, S.; Astarita, G.; Duranti, A.; Tontini, A.; Tarzia, G.; Rivara, S.; et al. Selective inhibition of 2-AG hydrolysis enhances endocannabinoid signaling in hippocampus. Nat. Neurosci. 2005, 8, 1139–1141. [Google Scholar] [CrossRef]
- Fernández-Suárez, D.; Celorrio, M.; Riezu-Boj, J.I.; Ugarte, A.; Pacheco, R.; González, H.; Oyarzabal, J.; Hillard, C.J.; Franco, R.; Aymerich, M.S. The monoacylglycerol lipase inhibitor JZL184 is neuroprotective and alters glial cell phenotype in the chronic MPTP mouse model. Neurobiol. Aging 2014, 35, 2603–2616. [Google Scholar] [CrossRef] [PubMed]
- Mecha, M.; Yanguas-Casás, N.; Feliú, A.; Mestre, L.; Carrillo-Salinas, F.; Azcoitia, I.; Yong, V.W.; Guaza, C. The endocannabinoid 2-AG enhances spontaneous remyelination by targeting microglia. Brain. Behav. Immun. 2019, 77, 110–126. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Chico, A.; Canedo, M.; Manterola, A.; Victoria Sánchez-Gómez, M.; Pérez-Samartín, A.; Rodríguez-Puertas, R.; Matute, C.; Mato, S. Blockade of monoacylglycerol lipase inhibits oligodendrocyte excitotoxicity and prevents demyelination in vivo. Glia 2015, 63, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Nomura, D.K.; Morrison, B.E.; Blankman, J.L.; Long, J.Z.; Kinsey, S.G.; Marcondes, M.C.G.; Ward, A.M.; Hahn, Y.K.; Lichtman, A.H.; Conti, B.; et al. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science 2011, 334, 809–813. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.; Gao, F.; Chen, C.; Zhang, Y.; Liu, Q.-S. Endocannabinoid Metabolism and Traumatic Brain Injury. Cells 2021, 10, 2979. [Google Scholar] [CrossRef]
- Tchantchou, F.; Tucker, L.B.; Fu, A.H.; Bluett, R.J.; McCabe, J.T.; Patel, S.; Zhang, Y. The fatty acid amide hydrolase inhibitor PF-3845 promotes neuronal survival, attenuates inflammation and improves functional recovery in mice with traumatic brain injury. Neuropharmacology 2014, 85, 427–439. [Google Scholar] [CrossRef] [Green Version]
- Rivera, P.; Fernández-Arjona, M.d.M.; Silva-Peña, D.; Blanco, E.; Vargas, A.; López-Ávalos, M.D.; Grondona, J.M.; Serrano, A.; Pavón, F.J.; Rodríguez de Fonseca, F.; et al. Pharmacological blockade of fatty acid amide hydrolase (FAAH) by URB597 improves memory and changes the phenotype of hippocampal microglia despite ethanol exposure. Biochem. Pharmacol. 2018, 157, 244–257. [Google Scholar] [CrossRef]
- Ativie, F.; Albayram, O.; Bach, K.; Pradier, B.; Zimmer, A.; Bilkei-Gorzo, A. Enhanced microglial activity in FAAH−/− animals. Life Sci. 2015, 138, 52–56. [Google Scholar] [CrossRef]
- Murphy, N.; Cowley, T.R.; Blau, C.W.; Dempsey, C.N.; Noonan, J.; Gowran, A.; Tanveer, R.; Olango, W.M.; Finn, D.P.; Campbell, V.A.; et al. The fatty acid amide hydrolase inhibitor URB597 exerts anti-inflammatory effects in hippocampus of aged rats and restores an age-related deficit in long-term potentiation. J. Neuroinflammation 2012, 9, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tham, C.-S.; Whitaker, J.; Luo, L.; Webb, M. Inhibition of microglial fatty acid amide hydrolase modulates LPS stimulated release of inflammatory mediators. FEBS Lett. 2007, 581, 2899–2904. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Yagyu, K.; Sackett, S.; Zhang, Y. Anti-Inflammatory Effects by Pharmacological Inhibition or Knockdown of Fatty Acid Amide Hydrolase in BV2 Microglial Cells. Cells 2019, 8, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grieco, M.; De Caris, M.G.; Maggi, E.; Armeli, F.; Coccurello, R.; Bisogno, T.; D’Erme, M.; Maccarrone, M.; Mancini, P.; Businaro, R. Fatty Acid Amide Hydrolase (FAAH) Inhibition Modulates Amyloid-Beta-Induced Microglia Polarization. Int. J. Mol. Sci. 2021, 22, 7711. [Google Scholar] [CrossRef]
- Schmöle, A.-C.; Lundt, R.; Gennequin, B.; Schrage, H.; Beins, E.; Krämer, A.; Zimmer, T.; Limmer, A.; Zimmer, A.; Otte, D.-M. Expression Analysis of CB2-GFP BAC Transgenic Mice. PLoS ONE 2015, 10, e0138986. [Google Scholar] [CrossRef] [Green Version]
- Martín-Moreno, A.M.; Brera, B.; Spuch, C.; Carro, E.; García-García, L.; Delgado, M.; Pozo, M.A.; Innamorato, N.G.; Cuadrado, A.; de Ceballos, M.L. Prolonged oral cannabinoid administration prevents neuroinflammation, lowers β-amyloid levels and improves cognitive performance in Tg APP 2576 mice. J. Neuroinflammation 2012, 9, 8. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Zhou, W.; Dou, F.; Wang, C.; Yu, Z. TRPV1 sustains microglial metabolic reprogramming in Alzheimer’s disease. EMBO Rep. 2021, 22, e52013. [Google Scholar] [CrossRef]
- Wang, C.; Huang, W.; Lu, J.; Chen, H.; Yu, Z. TRPV1-Mediated Microglial Autophagy Attenuates Alzheimer’s Disease-Associated Pathology and Cognitive Decline. Front. Pharmacol. 2022, 12, 763866. [Google Scholar] [CrossRef]
- Pihlaja, R.; Takkinen, J.; Eskola, O.; Vasara, J.; López-Picón, F.R.; Haaparanta-Solin, M.; Rinne, J.O. Monoacylglycerol lipase inhibitor JZL184 reduces neuroinflammatory response in APdE9 mice and in adult mouse glial cells. J. Neuroinflammation 2015, 12, 81. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Farkas, S.; Nagy, K.; Palkovits, M.; Kovács, G.G.; Jia, Z.; Donohue, S.; Pike, V.; Halldin, C.; Máthé, D.; Harkany, T.; et al. [125I]SD-7015 reveals fine modalities of CB1 cannabinoid receptor density in the prefrontal cortex during progression of Alzheimer’s disease. Neurochem. Int. 2012, 60, 286–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manuel, I.; Lombardero, L.; LaFerla, F.M.; Giménez-Llort, L.; Rodríguez-Puertas, R. Activity of muscarinic, galanin and cannabinoid receptors in the prodromal and advanced stages in the triple transgenic mice model of Alzheimer’s disease. Neuroscience 2016, 329, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Solas, M.; Francis, P.T.; Franco, R.; Ramirez, M.J. CB2 receptor and amyloid pathology in frontal cortex of Alzheimer’s disease patients. Neurobiol. Aging 2013, 34, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Westlake, T.M.; Howlett, A.C.; Bonner, T.I.; Matsuda, L.A.; Herkenham, M. Cannabinoid receptor binding and messenger RNA expression in human brain: An in vitro receptor autoradiography and in situ hybridization histochemistry study of normal aged and Alzheimer’s brains. Neuroscience 1994, 63, 637–652. [Google Scholar] [CrossRef]
- Mulder, J.; Zilberter, M.; Pasquaré, S.J.; Alpár, A.; Schulte, G.; Ferreira, S.G.; Köfalvi, A.; Martín-Moreno, A.M.; Keimpema, E.; Tanila, H.; et al. Molecular reorganization of endocannabinoid signalling in Alzheimer’s disease. Brain 2011, 134, 1041–1060. [Google Scholar] [CrossRef] [PubMed]
- Halleskog, C.; Mulder, J.; Dahlström, J.; Mackie, K.; Hortobágyi, T.; Tanila, H.; Puli, L.K.; Färber, K.; Harkany, T.; Schulte, G. WNT Signaling in Activated Microglia Is Proinflammatory. Glia 2011, 59, 119–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual, A.C.; Martín-Moreno, A.M.; Giusto, N.M.; de Ceballos, M.L.; Pasquaré, S.J. Normal aging in rats and pathological aging in human Alzheimer’s disease decrease FAAH activity: Modulation by cannabinoid agonists. Exp. Gerontol. 2014, 60, 92–99. [Google Scholar] [CrossRef]
- Lee, Y.-J.; Bae Han, S.; Nam, S.-Y.; Oh, K.-W.; Tae Hong, J. Inflammation and Alzheimer’s Disease. Arch Pharm Res 2010, 33, 1539–1556. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Liss, L.; Horrocks, L.A. Neurochemical aspects of Alzheimer’s disease: Involvement of membrane phospholipids. Metab. Brain Dis. 1988, 3, 19–35. [Google Scholar] [CrossRef]
- Grünblatt, E.; Bartl, J.; Zehetmayer, S.; Ringel, T.M.; Bauer, P.; Riederer, P.; Jacob, C.P. Gene expression as peripheral biomarkers for sporadic Alzheimer’s disease. J. Alzheimers. Dis. 2009, 16, 627–634. [Google Scholar] [CrossRef] [Green Version]
- D’Addario, C.; Di Francesco, A.; Pucci, M.; Finazzi Agrò, A.; MacCarrone, M. Epigenetic mechanisms and endocannabinoid signalling. FEBS J. 2013, 280, 1905–1917. [Google Scholar] [CrossRef] [PubMed]
- Chiurchiù, V.; Scipioni, L.; Arosio, B.; Mari, D.; Oddi, S.; Maccarrone, M.; Grimm, W. Anti-Inflammatory Effects of Fatty Acid Amide Hydrolase Inhibition in Monocytes/Macrophages from Alzheimer’s Disease Patients. Biomolecules 2021, 11, 502. [Google Scholar] [CrossRef] [PubMed]
- Hall, S.; Constantinescu, R.; Andreasson, U.; Surova, Y.; Bostrom, F.; Nilsson, C.; kan Widner, H.; Decraemer, H.; Nägga, K.; Minthon, L.; et al. Accuracy of a Panel of 5 Cerebrospinal Fluid Biomarkers in the Differential Diagnosis of Patients with Dementia and/or Parkinsonian Disorders. Arch. Neurol. 2012, 69, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Jankowsky, J.L.; Zheng, H. Practical considerations for choosing a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 89. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M.; Totaro, A.; Leuti, A.; Giacovazzo, G.; Scipioni, L.; Mango, D.; Coccurello, R.; Nisticò, R.; Oddi, S. Early alteration of distribution and activity of hippocampal type-1 cannabinoid receptor in Alzheimer’s disease-like mice overexpressing the human mutant amyloid precursor protein. Pharmacol. Res. 2018, 130, 366–373. [Google Scholar] [CrossRef] [Green Version]
- Kalifa, S.; Polston, E.K.; Allard, J.S.; Manaye, K.F. Distribution patterns of cannabinoid CB1 receptors in the hippocampus of APPswe/PS1ΔE9 double transgenic mice. Brain Res. 2011, 1376, 94–100. [Google Scholar] [CrossRef]
- López, A.; Aparicio, N.; Pazos, M.R.; Grande, M.T.; Barreda-Manso, M.A.; Benito-Cuesta, I.; Vázquez, C.; Amores, M.; Ruiz-Pérez, G.; García-García, E.; et al. Cannabinoid CB 2 receptors in the mouse brain: Relevance for Alzheimer’s disease. J. Neuroinflammation 2018, 15, 158. [Google Scholar] [CrossRef] [Green Version]
- Aso, E.; Juvés, S.; Maldonado, R.; Ferrer, I. CB 2 Cannabinoid Receptor Agonist Ameliorates Alzheimer-Like Phenotype in APP/PS1 Mice. J. Alzheimer’s Dis. 2013, 35, 847–858. [Google Scholar] [CrossRef] [Green Version]
- Köfalvi, A.; Lemos, C.; Martín-Moreno, A.M.; Pinheiro, B.S.; García-García, L.; Pozo, M.A.; Valério-Fernandes, Â.; Beleza, R.O.; Agostinho, P.; Rodrigues, R.J.; et al. Stimulation of brain glucose uptake by cannabinoid CB2 receptors and its therapeutic potential in Alzheimer’s disease. Neuropharmacology 2016, 110, 519–529. [Google Scholar] [CrossRef] [Green Version]
- Piscitelli, F.; Coccurello, R.; Totaro, A.; Leuti, A.; Giacovazzo, G.; Verde, R.; Rossi, E.; Podaliri Vulpiani, M.; Ferri, N.; Giacominelli Stuffler, R.; et al. Targeted Lipidomics Investigation of N-acylethanolamines in a Transgenic Mouse Model of AD: A Longitudinal Study. Eur. J. Lipid Sci. Technol. 2019, 121, 1900015. [Google Scholar] [CrossRef]
- Piro, J.R.; Benjamin, D.I.; Duerr, J.M.; Pi, Y.; Gonzales, C.; Wood, K.M.; Schwartz, J.W.; Nomura, D.K.; Samad, T.A. A Dysregulated Endocannabinoid-Eicosanoid Network Supports Pathogenesis in a Mouse Model of Alzheimer’s Disease. Cell Rep. 2012, 1, 617–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aso, E.; Palomer, E.; Juvés, S.; Maldonado, R.; Muñoz, F.J.; Ferrer, I. CB 1 Agonist ACEA Protects Neurons and Reduces the Cognitive Impairment of APP/PS1 Mice. J. Alzheimer’s Dis. 2012, 30, 439–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakhfouri, G.; Ahmadiani, A.; Rahimian, R.; Grolla, A.A.; Moradi, F.; Haeri, A. WIN55212-2 attenuates amyloid-beta-induced neuroinflammation in rats through activation of cannabinoid receptors and PPAR-γ pathway. Neuropharmacology 2012, 63, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Schmöle, A.C.; Lundt, R.; Toporowski, G.; Hansen, J.N.; Beins, E.; Halle, A.; Zimmer, A. Cannabinoid Receptor 2-Deficiency Ameliorates Disease Symptoms in a Mouse Model with Alzheimer’s Disease-Like Pathology. J. Alzheimer’s Dis. 2018, 64, 379–392. [Google Scholar] [CrossRef]
- Li, C.; Shi, J.; Wang, B.; Li, J.; Jia, H. CB2 cannabinoid receptor agonist ameliorates novel object recognition but not spatial memory in transgenic APP/PS1 mice. Neurosci. Lett. 2019, 707, 134286. [Google Scholar] [CrossRef]
- Aparicio, N.; Grande, M.T.; Ruiz de Martín Esteban, S.; López, A.; Ruiz-Pérez, G.; Amores, M.; Vázquez, C.; Martínez-Relimpio, A.M.; Pazos, M.R.; Cravatt, B.F.; et al. Role of interleukin 1-beta in the inflammatory response in a fatty acid amide hydrolase-knockout mouse model of Alzheimer’s disease. Biochem. Pharmacol. 2018, 157, 202–209. [Google Scholar] [CrossRef]
- Ruiz-Pérez, G.; Ruiz de Martín Esteban, S.; Marqués, S.; Aparicio, N.; Grande, M.T.; Benito-Cuesta, I.; Martínez-Relimpio, A.M.; Arnanz, M.A.; Tolón, R.M.; Posada-Ayala, M.; et al. Potentiation of amyloid beta phagocytosis and amelioration of synaptic dysfunction upon FAAH deletion in a mouse model of Alzheimer’s disease. J. Neuroinflammation 2021, 18, 223. [Google Scholar] [CrossRef]
- Vázquez, C.; Tolón, R.M.; Grande, M.T.; Caraza, M.; Moreno, M.; Koester, E.C.; Villaescusa, B.; Ruiz-Valdepeñas, L.; Fernández-Sánchez, F.J.; Cravatt, B.F.; et al. Endocannabinoid regulation of amyloid-induced neuroinflammation. Neurobiol. Aging 2015, 36, 3008–3019. [Google Scholar] [CrossRef]
- Chen, R.; Zhang, J.; Wu, Y.; Wang, D.; Feng, G.; Tang, Y.-P.; Teng, Z.; Chen, C. Monoacylglycerol Lipase Is a Therapeutic Target for Alzheimer’s Disease. Cell Rep. 2012, 2, 1329–1339. [Google Scholar] [CrossRef] [Green Version]
- Koppel, J.; Vingtdeux, V.; Marambaud, P.; D’Abramo, C.; Jimenez, H.; Stauber, M.; Friedman, R.; Davies, P. CB2 Receptor Deficiency Increases Amyloid Pathology and Alters Tau Processing in a Transgenic Mouse Model of Alzheimer’s Disease. Mol. Med. 2013, 19, 29–36. [Google Scholar] [CrossRef]


{kind=link}
| ECS | Model | Pro-Anti-Inflammatory Challenge | Treatment | Effect on Microglia Function | Ref. |
|---|---|---|---|---|---|
| CB1 | BV2 | IFN-γ (100 U/mL) | SR141716A 1 µM | ↑IFN-γ, IL-1β, IL-6, TNF-α, NO ↓MCP-1, CX3CL1 | [93] |
| Rat primary microglia | IL-4 IL-13 10 ng/mL each (M2a) | AM251 1 µM | ↓Arg-1 immunostaining | [90] | |
| CB2 | Murine primary microglia | IFN-γ (100 U/mL) Aβ42 (1 µM) | JWH-015 5 µM | ↑phagocytosis of Aβ42 ↓TNFα, NO | [103] |
| LPS (100 ng/mL) IFNγ (20 ng/mL) | Constitutive KO | ↓IL-6, TNFα =phagocytosis of Aβ42 | [125] | ||
| IL-4 (10 ng/mL) IL-13 (10 ng/mL) | Constitutive KO | ↓phagocytosis | [90] | ||
| Basal | 2-AG (25 µM) | ↑migration | [110] | ||
| Rat primary microglia | Aβ40 soluble or fibrillar (500 nM) | HU-210, WIN55,212-2, JWH-133 (100 nM) | ↓microglia activation (morphology) ↓TNFα | [102] | |
| LPS (100 ng/mL) | AEA (1 µM) AM-630 (0.1–0.5 µM) | ↓NO ↓M1 phenotypic marker (mRNA TNFα, IL-β, IL-6, COX-2, iNOS) | [104] | ||
| IL-4 IL-13 (10 ng/mL each) | AM630 1 µM | ↓Arg-1 immunostaining | [90] | ||
| LPS (10 ng/mL) IFNγ (10 U/mL) | AM1241 (10 µM) | ↓IL-6, IL-β, iNOS ↑Arg-1, IL-10, BDNF, GDNF | [101] | ||
| APP/PS1 glioma cells | Aβ40 (5 μg/mL) | WIN55,212-2, JWH-133 (200 nM) | ↑Aβ transport through choroid plexus monolayers | [126] | |
| BV-2 | LPS (50 ng/mL) IFNγ (100 U/mL) | AEA 5–15 µM (SR2 1 µM) | ↑IL-10 | [105] | |
| TRPV1 | Murine primary microglia | LPS (10 ng/mL) Aβ oligomer (5 µM) | Capsaicin (10 µM) | ↑mTOR/AKT/HIF-1α pathway ↑phagocity of Aβ42 | [127] |
| LPS (100 ng/mL) ATP (5 mM) | Constitutive KO CPZ (10 µM) | ↓NLRP3 inflammasome ↓IL-1β =TNF-α | [107] | ||
| Basal | Capsaicin | ↑TNF-α ↓IL-10 | [88] | ||
| Constitutive KO | ↑IL-10 | ||||
| Capsaicin (10 µM) | ↑migration | [87] | |||
| Basal | Capsaicin (10 μM) | ↑phagocytosis Aβ | [128] | ||
| Basal | Constitutive KO | ↑phagocytosis Aβ | |||
| BV2 | basal | Capsaicin (10 μM) | ↑phagocytosis Aβ | ||
| BV2 | Phorbol myristate acetate (1 µM) | Capsazepine (50 µM) | ↓ROS | [86] | |
| MAGL | Microglia from adult brain | Aβ42 (10 μM) LPS (1 µg/mL) and IFN-γ (100 ng/mL) | JZL184 (1 μM) | ↓NO, IL-1β (stimulation with LPS/IFN-γ) ↓Iba1 (stimulation with Aβ42) | [129] |
| FAAH | Rat primary microglia | LPS (0.03 µg/mL) | URB597 (10 µM) | ↓COX-2, iNOS, PGE2 | [122] |
| BV2 | Aβ 25–35 (30 µM) | URB597 (5 µM) | ↑cell viability ↓basal migration ↑phagocytosis ↑mRNA TGF-β, IL-10, ARG1 ↓mRNA TNF-α, IL-1 β, iNOS | [124] | |
| LPS (100 ng/mL) | URB597(10 µM) PF3845 (10 µM) siRNA | PF3845 ↓mRNA COX-2, IL-1 β, MCP1 PGE2, TNF-α URB597 ↓mRNA PGE2, IL-1 β, MCP1, siRNA ↓mRNA TNF-α, il-6, IL-1 β, MCP1 ↓COX-2, iNOS | [123] |
| Model | ECS | Treatment | Molecular Effect | Behavioural Effect | Pre- Symptomatic | Early Symptomatic | Late Symptomatic | Ref. |
|---|---|---|---|---|---|---|---|---|
| APPSwe/PS1ΔE9 | CB2 | constitutive KO | =IL-6 ↓TNF-α and CCL2 ↓microgliosis, Iba1 in hipp ↓brain-infiltrating macrophage ↑ramified microglia around plaque ↓Aβ plaque in cx ↓Aβ plaque in hip | ↑MWM | ▼ | [125,154] | ||
| JWH-133 (0.2 mg/kg i.p.) 5 weeks | =Aβ burden in the cx =Aβ40 Aβ42 protein level | ↑V-maze ↑Active avoidance test | ▲ ▼ | [148] | ||||
| ↓TNF-α, IL-10, IL-6 IL-1β ↓microgliosis, Iba1 in cx (cells plaque associated) ↓tau phosphorylated ↓p38, GSK3β, SAPK/JNK ↓HNE =Aβ burden in the cx =Aβ40 Aβ42 protein level =Aβ plaque load | ↑V-maze =Active avoidance test | ▲ ▼ | ||||||
| JWH-015 (0.5 mg/kg i.p.) 8 weeks | ↓microgliosis, Iba1 in cx ↓mRNA TNF-α iNOS IL-6 = microgliosis, Iba1 in hipp =level of Plaque deposition | ↑NOR =MWM | ▲ | ▼ | [155] | |||
| TRPV1 | capsaicin (standard chow plus 0.01% capsaicin) 4 weeks | =Aβ40, Aβ42 soluble fraction ↓Aβ40, Aβ42 insoluble fraction ↑autophagy ↑clearance of Aβ via autophagy (colocalization of iba1/LC3) ↓IL-6, TNF-α | ↑MWM | ▲ ▼ | [127] | |||
| MAGL | Constitutive KO | ↓microglia, Iba1 ↓mRNA IL-1β, IL-6, TNF-α ↓Aβ plaques as well as the Aβ40, and Aβ42 amyloidogenic peptides | ▼ | [151] | ||||
| JZL184 (40 mg/kg i.p.) 2 weeks | ↓mRNA IL-1β, IL-6, TNF-α | ▲ ▼ | ||||||
| JZL184 (40 mg/kg i.p.) 4 weeks | ↓microgliosis, Iba1 in cx, hipp | ▲ ▼ | [129] | |||||
| CB1 | ACEA (1.5 mg/kg i.p.) 4 weeks | =microglia activation, Iba1 | ↑V-maze | ▲ ▼ | [152] | |||
| 5xFAD | FAAH | Constitutive KO | ↑M1/M2 ratio in (FAAH−/−) ↓microgliosis Iba1 ↑mRNA IL-1β, TNF-α ↓mRNA IL-10, IL-4 ↓soluble Aβ42 | =MWM | ▼ | [156] | ||
| ↑phagocytic Aβ by DAM | ▼ | [157] | ||||||
| Constitutive KO | ↑mRNA IL-1β, IL-6 =IL-6 ↑IL-1β in cx ↓microgliosis, Iba1 in hipp (cells plaque associated) ↓APP ↓Aβ42 and Aβ40 | ↑MWM | ▼ | [158] | ||||
| URB597 (3 mg/kg i.p.) 2 weeks | ↑mRNA IL-6 in hipp =mRNA IL-1β and TNF-α in hipp | =MWM | ▲ ▼ | |||||
| MAGL | JZL184 (12 mg/kg, i.p) 8 weeks | ↓Aβ40 and Aβ42 as well as APP c-terminal fragments (CTFa/b) ↓reactive microglia, CD11b in hip | ↑MWM | ▲ ▼ | [159] | |||
| J20 | CB2 | Constitutive KO | ↑microgliosis, Iba1 in hipp (cells plaque associated) =microgliosis, Iba1 in hipp ↑soluble Aβ ↑Aβ plaque load =soluble Aβ40 | ▼ | [160] | |||
| Tg2576 | JWH-133 (drinking water at a dose of 0.2 mg/kg) 16 weeks | ↓microgliosis, Iba1 in cx ↓COX-2 ↓CB2 protein ↓27% levels of Aβ40 ↓30% levels of Aβ42 | ↑NOR | ▲ | ▼ | [126] | ||
| CB1/2 | WIN 55,212-2 (drinking water at a dose of 0.2 mg/kg) 16 weeks | =microgliosis, Iba1 in cx =COX-2 ↓CB2 protein ↓30% levels of Aβ42 | =NOR | ▲ | ▼ | |||
| 3xTg | TRPV1 | Capsaicin (1 mg/kg i.p.) 4 weeks | ↑microgliosis, Iba1 ↑autophagy ↑activated microglia in hipp and cx | ↑Y Maze ↑MWM | ▲ ▼ | [128] | ||
| Rat (Aβ25-35 inj) | CB1/2 | WIN 55,212-2 (10 µg intracerebroventricular injection) 1 week | ↓microglia activation in cx | ↑MWM | [102] | |||
| Rat (Aβ42 inj) | ↓TNF-α ↓NF-kB | ↑MWM | [153] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scipioni, L.; Ciaramellano, F.; Carnicelli, V.; Leuti, A.; Lizzi, A.R.; De Dominicis, N.; Oddi, S.; Maccarrone, M. Microglial Endocannabinoid Signalling in AD. Cells 2022, 11, 1237. https://doi.org/10.3390/cells11071237
Scipioni L, Ciaramellano F, Carnicelli V, Leuti A, Lizzi AR, De Dominicis N, Oddi S, Maccarrone M. Microglial Endocannabinoid Signalling in AD. Cells. 2022; 11(7):1237. https://doi.org/10.3390/cells11071237
Chicago/Turabian StyleScipioni, Lucia, Francesca Ciaramellano, Veronica Carnicelli, Alessandro Leuti, Anna Rita Lizzi, Noemi De Dominicis, Sergio Oddi, and Mauro Maccarrone. 2022. "Microglial Endocannabinoid Signalling in AD" Cells 11, no. 7: 1237. https://doi.org/10.3390/cells11071237
APA StyleScipioni, L., Ciaramellano, F., Carnicelli, V., Leuti, A., Lizzi, A. R., De Dominicis, N., Oddi, S., & Maccarrone, M. (2022). Microglial Endocannabinoid Signalling in AD. Cells, 11(7), 1237. https://doi.org/10.3390/cells11071237